
Abstract
While it is well known that the genome can affect social behavior, recent models posit that social lifestyles can, in turn, influence genome evolution. Here, we perform the most phylogenetically comprehensive comparative analysis of 16 bee genomes to date: incorporating two published and four new carpenter bee genomes (Apidae: Xylocopinae) for a first-ever genomic comparison with a monophyletic clade containing solitary through advanced eusocial taxa. We find that eusocial lineages have undergone more gene family expansions, feature more signatures of positive selection, and have higher counts of taxonomically restricted genes than solitary and weakly social lineages. Transcriptomic data reveal that caste-affiliated genes are deeply-conserved; gene regulatory and functional elements are more closely tied to social phenotype than phylogenetic lineage; and regulatory complexity increases steadily with social complexity. Overall, our study provides robust empirical evidence that social evolution can act as a major and surprisingly consistent driver of macroevolutionary genomic change.
Similar content being viewed by others


Introduction
Sociogenomics has provided important advances in our understanding of the molecular basis of social life1. Studies have repeatedly shown that the evolution of animal sociality is strongly influenced and accompanied by a variety of genomic changes2,3,4,5. An emerging theme in sociobiology is the observation that, in addition to genes affecting social behavior, social life itself may drive new patterns and processes in genome evolution6. For example, it has been proposed that changes in demography, levels of selection, and the novel demands of social life can drive rapid sequence evolution, changes in genome organization, and other forms of genomic change. To date, however, there remains a paucity of genomic information for multiple closely-related species that vary in levels of sociality, leaving these ideas without robust empirical support.
Eusocial organisms demonstrate the most complex form of social organization in nature: a single reproductive queen is supported by hundreds or thousands of sterile offspring, all cooperatively working together to rear additional generations of brood7,8. Eusociality has evolved only rarely but has emerged more often among bees than in any other group (as many as four times9,10,11). As is demonstrated by the obligate social nesting of advanced eusocial bees, the emergence of social life marks a pivot point from individual to group living, making it one of the most consequential evolutionary transitions in the history of biological complexity on Earth12. The progression from solitary to eusocial life among the socially diverse bees thus presents unique opportunities to address how the transition to sociality—and a new level of biological organization (i.e. superorganismal)—may influence genome evolution.
It is generally thought that the evolutionary change from ancestral solitary life to group living in bees could not have occurred in a single, abrupt evolutionary step. Rather, evidence indicates that bees have collectively reverted to solitary life at least nine times following an emergence of group living9,10,11. Additionally, many extant bees demonstrate social forms that are not eusocial, such as subsocial (i.e. extended parental care) or incipiently social taxa (i.e. rudimentary but totipotent division of labor13,14). As synthesized by the social ladder framework2, the solitary ancestors of eusocial species thus likely underwent multifaceted, incremental, and largely reversible augmentations in social complexity, with relatively few lineages experiencing enough sustained selective pressure to cross a ‘point of no return’ into the obligate eusocial state15,16,17,18,19.
One core evolutionary-developmental theory states that, at its evolutionary origin, the obligate reproductive (queen) and non-reproductive (worker) caste system of advanced eusocial bees (e.g. A. mellifera) was necessarily underpinned by a decoupling of ancestrally maternal foraging/provisioning from egg-laying behaviors at the molecular level (i.e. ovarian groundplan hypothesis14,18). Studies among other group living and eusocial bees, however, suggest that social dynamics may emerge antecedent to the molecular division between reproductive and non-reproductive activity. For example, in many species of incipiently or facultatively social carpenter bees (Apidae: Xylocopinae), newly eclosed females do not immediately disperse, but instead “wait” in their natal nest to succeed the older nestmate as the dominant reproductive and forager17,20. Using Bayesian trait mapping analysis among 16 allodapine bee species (Xylocopinae: Allodapini) collectively demonstrating subsocial through advanced eusocial biology, Schwarz et al.17 found that this non-reproductive wait strategy was the most likely ancestral state for the group. This suggests that (i) evolutionary trajectories towards derived sociality may be highly lineage-specific and (ii) molecular decoupling of maternal pathways may not be necessary for quantifiable sociality to emerge.
To date, comparative sociogenomic studies among bees have largely focused on eusocial corbiculates (Apidae: Apinae) to make important foundational inferences into the factors contributing to the evolutionary emergence and elaboration of insect sociality21,22. Empirical insights from these works, however, are effectively limited to one particularly derived bee lineage. Moving forward, as genomic resources continue to be developed, the field of sociogenomics will benefit enormously by expanding into bee systems representative of other social lineages and phenotypes. This project is a step along that route, incorporating genome data from six carpenter bee species (Apidae: Xylocopinae), collectively representative of an independent origin of sociality, and a first-ever monophyletic dataset incorporating solitary (i.e. ancestral) through advanced eusocial (i.e. derived) taxa11,17,23. This unique dataset also affords us an opportunity to begin empirically discerning the degree to which the evolution of eusocial traits may have been driven by environmental constraints (i.e. adaptive change) versus shared ancestry (i.e. phylogenetic inertia24,25)—a largely open question within the field of social evolution. Though often handled separately, the effects of adaptive change and phylogenetic inertia are not mutually exclusive24; and comparative assessment of the molecular impact of each on the evolution of social traits would greatly inform further theoretical and empirical approaches.
Owing to their global distribution and rich social diversity, carpenter bees have long been the focus of illuminating phylogenetic and behavioral ecological research (e.g.17,23,26,27,28). To date, published genomic and transcriptomic resources for the Xylocopinae have been limited but highly informative resources for comparative studies of early social evolution29,30,31,32,33,34,35. Here we present four newly sequenced xylocopine genomes and transcriptomes and combine these with published genomic and transcriptomic data from 12 additional bee species to address three main questions. (1) Do genomes of independently evolved social bee lineages (Apinae and Xylocopinae) undergo parallel molecular changes during various stages of social evolution? As articulated by the social ladder framework2 and supported by previous comparative genomic research (e.g.4,21,36), we hypothesize that patterns of genome evolution (e.g. gene family expansions, rate of birth of novel genes, gene regulatory complexity) will be similar between lineages of comparable social evolutionary complexity despite phylogenetic distance. (2) How do rates of molecular evolution vary by social complexity and social lineage? We hypothesize, based on both theoretical15 and empirical support37,38, that rates of protein evolution will be(i) higher across socially derived lineages, and (ii) elevated among genes associated with caste roles18. (3) Are there regulatory elements associated with social traits that are conserved across evolutionary lineages of bees; and, do these elements allow for a disentangling of the influences of phylogenetic inertia from adaptive change on the emergence of social phenotypes? We hypothesize that while shared ancestry undoubtedly plays a role in the likelihood of social trait emergence within a given lineage, subsequent elaborations in the social form are likely attributable to environmental pressures acting consistently across lineages18,24,25,39. Covering 16 genomes and two well-represented and independent social lineages, this study represents the most comprehensive comparative analysis of social evolution in bees to date, and a “way forward” to investigate the tractability of the social ladder framework2. Further, this dataset provides an exciting opportunity to explore how adaptive change and phylogenetic inertia may influence the evolution of insect social complexity.
Results and discussion
Genome evolution
Sociality shapes gene family expansions and taxonomically restricted genes
The estimated genome sizes of our de novo assemblies (via SSpace, Trinity, and Gapfiller40,41,42) ranged from 280 Mb (C. japonica) to 460 Mb (E. robusta) with final assembly N50s ranging from 54.9 kb (C. japonica) to 452 kb (E. robusta). These genomes are thus within the expected size range given previously published bee genome data (Data S1–S3). Analysis of Benchmarking Universal Single-Copy Orthologs (BUSCOs43) revealed that each assembly is highly complete, containing at least 95.9% complete Arthropod genes (Data S3; Fig. S5). A combination of RNA sequencing, de novo assembly and corrective editing via the MAKER2 pipeline44 yielded predicted gene counts consistent with previously published bee genomes (Data S2, S3).
A total of 10,355 orthologous gene families were identified among our 16 lineages (Fig. 1a) during CAFE analysis45, of which 2,036 were found to be significantly expanded in at least one lineage (Data S4, S5). Overall, counts of significantly expanded gene families increased dramatically with social complexity, from 60 groups uniquely expanded across our solitary species to 510 uniquely expanded among our advanced eusocial taxa (Data S4, S7; Fig. 2a). Evolutionary derivations in social complexity are expected to be accompanied by functional elaborations and expansions among gene families as pleiotropic constraints are removed during the emergence of castes2,15. Our results provide support for this prediction and corroborate previous comparative genomic analyses4. Among notable gene families that followed this trend were the 7 transmembrane (tm) and 7tm Odorant receptor domain-containing genes. Chemosensory capability is critical for navigation and resource acquisition among insects46, and plays an important role in the continuous, caste-based communication of socially complex Hymenoptera47,48.

a All species included in study with available genome data; asterisks identify species for which genome data were generated de novo during current study; hashed box identifies Xylocopine species which were also assessed via gene expression analyses. Divergence time in millions of years (mya) among lineages is provided. b Xylocopine species with social trait mapping. Lineage sociality and worker phenotypes are indicated in colored boxes as per legend.

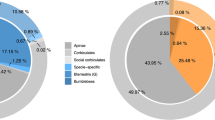
Dots and lines in bottom right indicate unique or shared membership of orthogroups among social groups; vertical columns indicate total orthogroup counts for those categories. Colored lateral columns indicate total counts of orthogroups by sociality from solitary (orange) through advanced eusocial (dark blue); simple (green), complex (purple), and all social forms (gray) are also specified. a Counts and uniqueness of orthologous gene families experiencing significant expansion (p < 0.05) both increase dramatically with derivations in social complexity. b Similarly, counts and uniqueness of orthogroups under significant positive selection (dN/dS > 0; p < 0.05) increase with derivations in social complexity.
An additional 247 families were uniquely expanded among our six xylocopine species (Ntotal = 2283). Within the Xylocopinae, a total of 19 gene families were significantly expanded across all Xylocopinae except the solitary Ct. terminalis, including gene families for reverse transcriptases, homeobox transcription factors, and two Immunoglobulin I-sets (Data S6). Two families, the cytochrome P450 and cadherin domains, were expanded specifically in our eusocial xylocopine species. Cytochrome P450 and cadherin domains likely play important roles in detoxification, chemical communication, and immune function49; and affiliated genes have been repeatedly and consistently implicated in the operation of derived forms of social nesting across Hymenoptera50. Capacities for chemical communication and immune resistance are critical among advanced eusocial Apinae51. As has been noted across ants and other eusocial lineages5, these data suggest that affiliated expansions in the P450 and related gene families may also be important during the evolution of social complexity in the Xylocopinae.
Positive selection
Rates of protein evolution are tied to social complexity rather than phylogenetic lineage
Phylogenetic Analysis by Maximum Likelihood (PAML v 4.952) was used to determine whether gene orthogroups may be undergoing positive selection (i.e. elevated non-synonymous over synonymous mutations; dN/dS > 1, p < 0.05) at either the lineage or social phenotypic levels and thus likely operating with some evolutionary consequence. We identified a total of 1460 orthogroups experiencing significant positive selection by lineage (Data S20–S31) and 1302 by sociality (Fig. 2b; Data S32–S36). Here, we discuss PAML results with regard only to branches under positive selection. At the family level, Apidae contained the greatest number of taxonomically restricted orthogroups under positive selection (N = 313, Fig. 3a, S7; Data S23–S25), and the highest average rates of protein evolution (dN/dS values) of any family considered (Data S45). Notably, functional enrichment among these orthogroups including both aromatic and organic cyclic compound metabolism, suggests additional support for the role of chemical communication within this family (Data S40; 48). Within Apidae, the two subfamilies containing advanced eusocial taxa also featured both the largest numbers of orthogroups under positive selection (Xylocopinae, N = 293, Data S27; and Apinae, N = 196, Data S26) and comparable rates of protein evolution (Wilcoxon test, Z = 0.26, p = 0.7892; Fig. 3a, S7), both of which were significantly higher than non-eusocial subfamilies (Data S45). Evidence of increased protein evolution was also observed within Xylocopinae. Tribe Allodapini featured both the greatest counts of orthogroups under positive selection (N = 246, Figs. 3a, S7; Data S30) and significantly elevated overall rates of protein evolution compared to both sister tribe Ceratinini (Wilcoxon test, Z = 8.01, p = 1.10E−15) and all remaining species (Data S45). Regardless of lineage, increases in social complexity accounted for greater numbers of novel orthogroups under positive selection (unique OGs, Nsolitary = 77 through Nadvanced eusocial = 171; χ2 = 74.33, df = 4, p = 0; Fig. 2b; Data S39) and higher rates of protein evolution (dN/dS values; Wilcoxon test, Z = −4.314, p < 0.0001; Fig. 3b; Data S45). Interestingly, orthogroups under positive selection in eusocial lineages were uniquely functionally enriched for oxidoreductase activity (Data S40). Mitigation of oxidative damage is likely a critical component of caste longevity across eusocial insects, which typically feature exceptionally long-lived queens53. Taken together, our results provide clear evidence of both quantitatively and qualitatively greater measures of positive selection on larger sets of taxonomically restricted genes across two independent origins of eusociality2. They also present additional empirical support for the theory that positive selection will operate with increasing intensity as lineages become more socially complex4,15. Notably, our results also reveal that positive selection appears to operate with considerable consistency both within and across independent social lineages.

Dots are jittered for visualization; vertical black line indicates global median dN/dS value; larger colored dots indicate group-specific median values (full lists of dN/dS values can be found in Datas S23-S38). a At every phylogenetic level inspected, lineages containing highly social lines (e.g. Apidae; Apinae and Xylocopinae; Allodapini) feature significantly higher rates of protein evolution (Data S45). b Orthogroups associated with more complex forms of sociality (i.e. Primitive and Advanced Eusociality) experience significantly higher rates of protein evolution than less derived forms.
Comparative transcriptomics
Elevated protein evolution specifically among genes associated with caste-like roles
Differentially expressed genes (DEGs) are expected to play a major role in the emergence and elaboration of social traits3 and thus to become the targets of positive selection as social traits establish15. Within Xylocopinae, we found that the majority of DEGs under significant positive selection showed overexpression in non-reproductive individuals (Nreproductive = 10 vs Nnon-reproductive = 22, 69%) and foragers (Nforaging = 20 vs Nwaiting = 12; Data S8–S11; Fig. S8). We also found that significantly more non-reproductive DEGs were under positive selection than expected by chance in both of the eusocial allodapine species (Data S39). Across all 16 bee genomes, the number of genes under positive selection also increased with species social complexity (Fig. 2b). These results provide further empirical support from outside Apinae for the role of elevated protein change specifically among DEGs associated with non-reproductive and/or foraging roles4,36,38.
Differentially expressed genes associated with carpenter bee sociality are ancient
Phylostrata analysis (via phylostratR v 0.2054) was used to assign a total of 20,405 orthologous gene groups to twenty levels of taxonomic constraint based on orthogroup evolutionary age (Data S18). Across lineages, most orthogroups were assigned to older levels (i.e., cellular organisms through Insecta, 65%). Although the overall majority of differentially expressed genes were also ancient, they were significantly overrepresented at older levels in our ceratinine species (cellular to Insecta vs Hymenoptera to tribe; χ2Ceratinini = 20.63, df = 1, p = 5.57e−6; Fig. S6; Data S8–11; S19). It thus appears that while taxonomically restricted genes are thought to play an important role in the expression of derived sociality among some lineages (e.g., A. mellifera55; Formicidae5) our data indicate that this may not be the case among social Xylocopinae.
Gene expression, enrichment, and regulatory consistencies by social phenotype rather than shared lineage
The toolkit hypothesis suggests that conserved differentially expressed genes likely play consistent underlying roles in the emergence and expression of similar social traits across taxa18. Accordingly, the 396 differentially expressed genes identified across our xylocopine taxa (Data S8–S12) included notable homologs (determined by BLASTn with shared gene identity ≥ 70% and p < 1.0E-5) expressed in phenotypically consistent contexts across other lineages (Fig. S9; Data S14–17). For example, DEGs associated with queens or workers of advanced eusocial E. tridentata (e.g. Troponin C) were also differentially regulated in comparable roles (i.e. foragers) among other advanced eusocial bees (e.g. A. mellifera, Fig. S9) and ants (e.g. T. longispinosus, S. invicta; Data S17). These results signal additional support for the role of differentially expressed and deeply conserved genes in the regulation of insect social traits18,28,56.
Despite occupying separate phylogenetic lineages within Xylocopinae, there were consistencies in gene ontological (GO) enrichment among our ceratinine and allodapine taxa by whether workers waited on or foraged for the nest (Fig. 4; Data S41). For example, non-reproductive females of C. australensis and E. robusta were significantly enriched for reproductive activity (e.g. reproduction), directly corroborating the “workers wait” strategy of attempting to lay eggs and eventually superseding as the reproductive dominant of the nest57,58. By contrast, the foraging-focused workers of C. japonica and E. tridentata were instead enriched for immune (e.g., regulation of Toll signaling pathway) and neural functions, processes that are likely important for individuals that spend most of their time foraging for the nest along the lines of other derived worker castes26,27,59.

Circle overlap sizes are equivalent to relative proportions of total GO terms uniquely associated with each phenotype considered; images are illustrative of foraging and guarding/waiting behavior by queens and workers. Enrichment for reproduction was detected in all groups except for foraging workers, which instead featured more enrichment for immune activity. The full list of GO terms can be found in Data S41.
In comparing predicted regulatory elements related to each taxon’s DEGs, we found a trend of increased overall counts with increasing social complexity; i.e. 88 transcription factors (TFs) in incipiently social C. australensis (44 of which were unique to C. australensis) to 396 TFs in advanced eusocial E. tridentata (of which 304 were unique; Data S42). Exoneurella tridentata and E. robusta, both eusocial, were also enriched for significantly more TFs than expected given DEG counts (NDEGs vs NTFBS by Species; χ2-test, χ2 = 117.70, d.f. = 3, p < 0.00001). Comparing TFs enriched in common among taxa, significantly more were shared among non-reproductive females that demonstrated similar social phenotypes than among those that shared a lineage (NPhenotype = 18 vs NLineage = 4 vs NNotShared; χ2 = 8.09, df = 1, p = 0.004; Fig. 5; Data S42, S43). However, this was not found among reproductive females (NPhenotype = 15 vs NLineage = 13 vs NNotShared; χ2 = 0.123, df = 1, p = 0.73). TFs enriched among non-reproductives that wait on the nest were associated primarily with development (e.g. D, tll) and included those which were also enriched among the reproductive individuals of C. japonica and E. tridentata (e.g. gt, prd, and z). Gt, prd, and z are functionally associated with neural development (including chemosensation) and epigenetic regulation of gene expression and have all been previously associated with guarding behavior in C. calcarata34. Non-foraging individuals often act as nest guards, either while waiting to supersede the nest, or to ensure the survival of their own brood28,57,58. As such, gt, prd, and z may play conserved regulatory roles in the induction of guarding behavior among social lineages3. By contrast, workers that forage shared more functionally diverse regulatory enrichment, including TFs involved in development (e.g. NKX3-1; TFAP2a), learning, circadian rhythm, and memory (e.g. Egr1, NFYA, ZEB1), and immunity (e.g. GATA3; Tal1_Gata1; Fig. 5). Further, six TFs from this set were previously associated with pre-reproductive foraging in C. calcarata34 including Egr1, GATA3, NFYA, and Tal1_Gata1, associated with learning, memory, and immune function. Of particular note from this set is early growth response protein 1 (Egr1), previously found to have a widely-conserved role in socially responsive gene regulation60 including a critical role in honey bee foraging61, and recently proposed as a candidate TF for tasks involving time-memory62. Taken together, these results support the suggestion that regulatory networks underlying social behavioral phenotypes may be broadly convergent across lineages5,36. Our data also corroborate previous observations that regulatory network scale and complexity tend to increase as lineages evolve greater degrees of sociality4,5,63, reinforcing the importance of regulatory expansion and elaboration during the evolution of sociality.

Motif names and broad regulatory involvement are provided. Enrichment counts for each motif in the upregulation of genes associated with each phenotype is then indicated by color intensity (see legend: gray—no enrichment, blue—enriched in both species, with darker blues indicating greater enrichment).
Conclusions
In this study, we present newly sequenced genomes and transcriptomes of four carpenter bees (Apidae: Xylocopinae) and combine these data with published resources from 12 additional bee species to perform the most comprehensive comparative assessment of social evolution in bees to date. Our data provide a chance to carefully compare two independently evolved and ecologically distinct social bee lineages; and an unprecedented opportunity to examine mechanisms of social evolution within an understudied and socially diverse eusocial lineage (Xylocopinae). Ultimately, our study finds clear empirical support for the predictions of the social ladder framework: gene family expansions, protein evolution, and regulatory element assortment are consistently extended among increasingly complex social lineages2,19. Differentially expressed genes are deeply conserved and evolutionarily ancient; and gene regulatory and functional elements appear to play highly conserved roles in the expression of particular social phenotypes (e.g. foraging behavior) across lineages5,18,64. More broadly, despite independent origins of eusociality, members of at least two different bee lineages appear to have similar evolutionary signatures of social complexity as a result of gene family expansions and increasingly strong positive selection on key proteins, differentially expressed genes, and regulatory elements. It therefore appears that sociality itself, more than phylogenetic inertia, shapes the evolutionary trajectory of social lineages. At present, available data offer abundant evidence in support of the applicability of the social ladder framework and highlight the importance of social evolution as a major and surprisingly consistent sculptor of genomic change among bees1,2,6. Future studies across additional independent origins of sociality that consider the great diversity of social taxa are necessary to further test the ubiquity and importance of what appear to be key molecular mechanisms of evolutionary change towards group living.
Methods
Sample collection and preparation
Four bee species were collected for new genomes and transcriptome analyses. The primitively eusocial Exoneura robusta57 and advanced eusocial Exoneurella tridentata59 were collected from the Dandenong Ranges and Lake Giles, Australia respectively. The primitively eusocial Ceratina japonica26,27 was collected from Sapporo, Japan, and solitary Ctenoplectra terminalis was collected in Kakamega, Kenya. Details on sampling and preservation protocols can be found in supplementary materials.
Genome sequencing and analysis
Whole body genomic DNA was extracted using phenol-chloroform extraction and submitted to Genome Quebec for cleanup, library preparation, and Illumina shotgun sequencing. To improve genome assembly, DNA samples were also used to construct 150 bp mate pair and 100 bp single strand libraries and sequenced on an Illumina HiSeq 2500. Before filtering, genome sequencing produced a total of 139 Gb of raw sequence data across our six species (with an average of 34.8 GB, 39.7 million reads at 33x coverage per species). Prior to assembly, filtering removed low quality reads, reads with a high proportion of Ns or poly-A sections, and reads for which mate pair ends overlapped or were merged. Each genome was then assembled and annotated before being assessed for completeness in relation to the A. mellifera genome. All newly generated genomic data can be found using NCBI BioProject numbers PRJNA413373, 526224, 413974, and 526241 (Data S1). De novo genome data were then combined with published genomic data from twelve additional bee species (Data S2, Fig. 1, S4) and aligned for comprehensive comparative analyses of gene family expansions (CAFE, Figs. S1–3; 45), evidence of molecular evolution (PAML52), and gene ages (phylostratR54). Additional test details are provided in supplementary methods.
Transcriptome sequencing and analysis
RNA was extracted from whole heads of queens and workers of C. japonica, E. tridentata, and E. robusta, and solitary females of Ct. terminalis and submitted for library prep and paired-end Illumina HiSeq 2500 sequencing (Genome Quebec). Read data were aligned to species genomes before being used for analysis (accessible under PRJNA413373, 526224, 413974, and 526241; Data S1). Significantly differentially expressed genes (DEGs; adjusted p-value < 0.05) were identified using DESeq65 and corroborated by DESeq266. Results of DEG analysis were then used to inform analyses of gene ontology (GO) term (topGO v3.767) and transcription factor binding site (TFBS) motif enrichment (cis-Metalysis pipeline68,69), and comparative analyses of biological contexts of differential gene expression between newly sequenced xylocopine species and 24 additional studies (Data S32–36).
Additional details on all methods employed for transcriptome analysis can be found in supplementary materials.
Data availability
All newly generated genomic and transcriptomic data used in this study can be freely accessed via NCBI BioProject numbers PRJNA413373, 412093, 526224, 413974, and 526241.
References
Robinson, G. E., Grozinger, C. M. & Whitfield, C. W. Sociogenomics: social life in molecular terms. Nat. Rev. Genet. 6, 257–270 (2005).
Rehan, S. M. & Toth, A. L. Climbing the social ladder: the molecular evolution of sociality. Trends Ecol. Evolution 30, 426–433 (2015).
West-Eberhard, M. J Developmental Plasticity and Evolution.(Oxford University Press: 2003.
Kapheim, K. M. et al. Genomic signatures of evolutionary transitions from solitary to group living. Science 348, 1139–1143 (2015).
Simola, D. F. et al. Social insect genomes exhibits dramatic evolution in gene composition and regulation while preserving regulatory features linked to sociality. Genome Res. 23, 1235–1247 (2013).
Rubinstein, D. R. et al. Coevolution of genome architecture and social behavior. Trends Ecol. Evolution 34, 844–855 (2019).
Michener, C. D. Comparative social behavior of bees. Annu. Rev. Entomol. 14, 299–342 (1969).
Wilson, E. O The Insect Societies. (Belknap Press of Harvard University Press: 1971.
Cardinal, S. & Danforth, B. N. The antiquity and evolutionary history of social behavior in bees. PLOS ONE 6, e21086 (2011).
Gibbs, J., Brady, S. G., Kanda, K. & Danforth, B. N. Phylogeny of halictine bees supports a shared origin of eusociality for Halictus and Lasioglossum (Apoidea: Anthophila: Halictidae). Mol. Phylogenet. Evol. 65, 926–939 (2012).
Rehan, S. M., Leys, R. & Schwarz, M. P. A mid-Cretaceous origin of sociality in Xylocopine bees with only two origins of true worker castes indicates severe barriers to eusociality. PLoS ONE 7, e34690 (2012).
Szathmary, E. & Smith, J. M. The major evolutionary transitions. Nature 374, 227–232 (1995).
Michener, C. D The Bees of the World. 2nd edn., ((Johns Hopkins University Press: 2007.
West-Eberhard, M. J. in Natural History and Evolution of Paper Wasps (eds Turillazzi, S., West-Eberhard, M.J.) 291–317 (Oxford University Press, 1996).
Gadagkar, R. The evolution of caste polymorphism in social insects: genetic release followed by diversifying evolution. J. Genet. 76, 167–179 (1997).
Wilson, E. O. & Hölldobler, B. Eusociality: origin and consequences. Proc. Natl Acad. Sci. USA 102, 13367–13371 (2005).
Schwarz, M. P., Tierney, S. M., Rehan, S. M., Chenoweth, L. B. & Cooper, S. J. B. The evolution of eusociality in allodapine bees: workers began by waiting. Biol. Lett. 7, 277–280 (2011).
Toth, A. L. & Robinson, G. E. Evo-devo and the evolution of social behavior. Trends Genet. 23, 334–341 (2007).
Toth, A. L. & Rehan, S. M. Molecular evolution of insect sociality: an eco-evo-devo perspective. Annu. Rev. Entomol. 62, 419–442 (2017).
Mikát, M., Franchino, C. & Rehan, S. M. Sociodemographic variation in foraging behavior and the adaptive significance of worker production in the facultatively social small carpenter bee, Ceratina calcarata. Behav. Ecol. Sociobiol. 71, 135 (2017).
Woodard, S. H., Fischman, B. J., Venkat, A., Hudson, M. E. & Varala, K. Genes involved in convergent evolution of eusociality in bees. Proc. Natl Acad. Sci. 108, 7472–7477 (2011).
Sadd, B. M. et al. The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol. 16, 76 (2015).
Schwarz, M. P., Richards, M. H. & Danforth, B. N. Changing paradigms in insect social evolution: insights from halictine and allodapine bees. Annu. Rev. Entomol. 52, 127–150 (2007).
Blomberg, S. P. & Garland, T. Tempo and mode in evolution: phylogenetic inertia, adaptation and comparative methods. J. Evol. Biol. 15, 899–910 (2002).
Hansen, T. F. & Orzack, S. H. Assessing current adaptation and phylogenetic inertia as explanations of trait evolution: the need for controlled comparisons. Evolution 59, 2063–2072 (2005).
Sakagami, S. F. & Maeta, Y. Multifemale nests and rudimentary castes in the normally solitary bee Ceratina japonica (Hymenoptera: Xylocopinae). J. Kans. Entomol. Soc. 57, 639–656 (1984).
Sakagami, S. F. & Maeta, Y. Multifemale nests and rudimentary castes of an “almost” solitary bee Ceratina flavipes, with additional observation on multifemale nests of Ceratina japonica (Hymenoptera, Apoidea). Entomological Soc. Jpn. 55, 391–409 (1987).
Rehan, S. M. et al. Conserved genes underlie phenotypic plasticity in an incipiently social bee. Genome Biol. Evol. 10, 2749–2758 (2018).
Durant, D. R., Berens, A. J., Toth, A. L. & Rehan, S. M. Transcriptional profiling of overwintering gene expression in the small carpenter bee, Ceratina calcarata. Apidologie 47, 572–582 (2016).
Rehan, S. M., Berens, A. J. & Toth, A. L. At the brink of eusociality: transcriptomic correlates of worker behaviour in a small carpenter bee. BMC Evolut. Biol. 14, 260 (2014).
Rehan, S. M., Glastad, K. M., Lawson, S. P. & Hunt, B. G. The genome and methylome of a subsocial small carpenter bee, Ceratina calcarata. Genome Biol. Evol. 8, 1401–1410 (2016).
Withee, J. R. & Rehan, S. M. Social aggression, experience, and brain gene expression in a subsocial bee. Integr. Comp. Biol. 57, 640–648 (2017).
Shell, W. A. & Rehan, S. M. The price of insurance: costs and benefits of worker production in a facultatively social bee. Behav. Ecol. 29, 204–211 (2018).
Shell, W. A. & Rehan, S. M. Social modularity: conserved genes and regulatory elements underlie caste-antecedent behavioural states in an incipiently social bee. Proc. R. Soc. B 286, 20191815 (2019).
Steffen, M. A. & Rehan, S. M. Genetic signatures of dominance hierarchies reveal conserved cis-regulatory and brain gene expression underlying aggression in a facultatively social bee. Genes Brain Behav. 19, e12597 (2020).
Dogantzis, K. A. et al. Insects with similar social complexity show convergent patterns of adaptive molecular evolution. Sci. Rep. 8, 10388 (2018).
Kent, C. F., Minaei, S., Harpur, B. A. & Zayed, A. Recombination is associated with the evolution of genome structure and worker behavior in honey bees. Proc. Natl Acad. Sci. USA 109, 18012–18017 (2012).
Harpur, B. A. et al. Population genomics of the honey bee reveals strong signatures of positive selection on worker traits. Proc. Natl Acad. Sci. USA 111, 2614–2619 (2014).
Chandrasekaran, S. et al. Behavior-specific changes in transcriptional modules lead to distinct and predictable neurogenomic states. Proc. Natl Acad. Sci. 108, 18020–18025 (2011).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Boetzer, M. & Pirvano, W. Toward almost closed genomes with GapFiller. Genome Biol. 13, R56 (2012).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494 (2013).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12, 491 (2011).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Sánchez-Garcia, A., Vieira, F. G. & Rozas, J. Molecular evolution of the major chemosensory gene families in insects. Heredity 103, 208–216 (2009).
Wittwer, B. et al. Solitary bees reduce investment in communication compared with their social relatives. Proc. Natl Acad. Sci. USA 114, 6569–6574 (2017).
Zhou, X. et al. Chemoreceptor evolution in Hymenoptera and its implications for the evolution of eusociality. Genome Biol. Evol. 7, 2407–2416 (2015).
Scott, J. G. & Wen, Z. Cytochromes P450 of insects: the tip of the iceberg. Pest Manag. Sci. 57, 958–967 (2001).
Hoffmann, K., Gowin, J., Hartfelder, K. & Korb, J. The scent of royalty: a P450 gene signals reproductive status in a social insect. Mol. Biol. Evol. 31, 2689–2696 (2014).
Cremer, S., Pull, C. D. & Fürst, M. A. Social immunity: emergence and evolution of colony-level disease protection. Annu. Rev. Entomol. 63, 105–123 (2019).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Li-Byarlay, H. & Cleare, X. Current trends in the oxidative stress and ageing of social hymenopterans. Adv. In Insect Phys. 59, 43–69 (2020).
Arendsee, Z. et al. phylostratr: A framework for phylostratigraphy. Bioinformatics 35, 3617–3627 (2019).
Johnson, B. R. & Tsutsui, N. D. Taxonomically restricted genes are associated with the evolution of sociality in the honey bee. BMC Genomics 12, 164 (2011).
Behl, S., Wu, T., Chernyshova, A. M. & Thompson, G. J. Caste-biased genes in a subterranean termite are taxonomically restricted: implications for novel gene recruitment during termite caste evolution. Insectes Sociaux 65, 593–599 (2018).
Cronin, A. L. & Schwarz, M. P. Latitudinal variation in the life cycle of allodapine bees (Hymenoptera; Apidae). Can. J. Zool. 77, 857–864 (1999).
Rehan, S. M., Richards, M. H. & Schwarz, M. P. Social polymorphism in the Australian small carpenter bee. Ceratina (Neoceratina) australensis. Insect Soc. 57, 403–412 (2010).
Hurst, P. S. Social biology of Exoneurella tridentata, an allodapine bee with morphological castes and perennial colonies. Unpublished D. Phil. Thesis, Flinders University (2001).
Robinson, G. E., Fernald, R. D. & Clayton, D. F. Genes and social behavior. Science 322, 896–900 (2011).
Singh, A. S., Shah, A. & Brockmann, A. Honey bee foraging induces upregulation of early growth response protein 1, hormone receptor 38 and candidate downstream genes of the ecdysteroid signaling pathway. Insect Mol. Biol. 27, 90–98 (2018).
Shah, A., Jain, R. & Brockmann, A. Egr-1: a candidate transcription factor involved in molecular processes underlying time-memory. Front. Psychol. 9, 865 (2018).
Molodtsova, D., Harpur, B. A., Kent, C. F., Seevananthan, K. & Zayed, A. Pleiotropy constrains the evolution of protein but not regulatory sequences in a transcription regulatory network influencing complex social behaviors. Front. Genet. 5, 431 (2014).
Berens, A. J., Hunt, J. H. & Toth, A. L. Comparative transcriptomics of convergent evolution: different genes but conserved pathways underlie caste phenotypes across lienages of eusocial insects. Mol. Biol. Evol. 32, 690–703 (2014).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Alexa, A. & Rahnenfuhrer, J. topGO: enrichment analysis for gene ontology. R package version 2.28.0. CRAN (2016).
Sinha, S., Liang, Y. & Siggia, E. Stubb: a program for discovery and analysis of cis-regulatory modules. Nucleic Acids Res. 34, W555–W559 (2006).
Ament, S. A. et al. New meta-analysis tools reveal common transcriptional regulatory basis for multiple determinants of behavior. Proc. Natl Acad. Sci. USA 109, E1801–E1810 (2012).
Bossert, S. et al. Combining transcriptomes and ultraconserved elements to illuminate the phylogeny of Apidae. Mol. Phylogenet. Evol. 130, 121–131 (2019).
Acknowledgements
The authors thank Scott Groom, Minna Mathiasson, Erika Tucker for assistance with field collections, Michael Schwarz for providing allodapine bee specimens. This work was supported by funding from National Geographic Society Explorer’s Grants 9659-15 and 9917-16 to S.M.R., National Science Foundation Graduate Research Fellowship 1450271 to W.A.S, NSF IOS-1456283 to A.L.T and IOS-1456296 to S.M.R.
Author information
Authors and Affiliations
Contributions
S.M.R. conceived, managed and coordinated the project; M.A.S. performed CAFE analyses; A.S.-S. performed PAML and Phylostrata analyses; H.K.P. and W.A.S. performed DESeq analyses; W.A.S. performed DNA and RNA sample preparation, gene ontology, regulatory enrichment, and comparative transcriptomic analyses; A.J.S. provided bioinformatic support; W.A.S., S.M.R., and A.L.T. drafted and wrote the manuscript; W.A.S., A.S.-S., and M.S. wrote and organized the Supplementary Information; W.A.S. and S.M.R. prepared figures for the manuscript. All authors read, corrected and/or commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shell, W.A., Steffen, M.A., Pare, H.K. et al. Sociality sculpts similar patterns of molecular evolution in two independently evolved lineages of eusocial bees. Commun Biol 4, 253 (2021). https://doi.org/10.1038/s42003-021-01770-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-01770-6
This article is cited by
-
Eusociality is not a major evolutionary transition, and why that matters
Insectes Sociaux (2024)
-
Putting hornets on the genomic map
Scientific Reports (2023)
-
Social complexity, life-history and lineage influence the molecular basis of castes in vespid wasps
Nature Communications (2023)
-
Molecular underpinnings of division of labour among workers in a socially complex termite
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
