
Abstract
The use of digital pathology for the histomorphologic profiling of pathological specimens is expanding the precision and specificity of quantitative tissue analysis at an unprecedented scale; thus, enabling the discovery of new and functionally relevant histological features of both predictive and prognostic significance. In this study, we apply quantitative automated image processing and computational methods to profile the subcellular distribution of the multi-functional transcriptional regulator, Kaiso (ZBTB33), in the tumors of a large racially diverse breast cancer cohort from a designated health disparities region in the United States. Multiplex multivariate analysis of the association of Kaiso’s subcellular distribution with other breast cancer biomarkers reveals novel functional and predictive linkages between Kaiso and the autophagy-related proteins, LC3A/B, that are associated with features of the tumor immune microenvironment, survival, and race. These findings identify effective modalities of Kaiso biomarker assessment and uncover unanticipated insights into Kaiso’s role in breast cancer progression.
Similar content being viewed by others


Introduction
Last year in the United States there were over 260,000 new cases of invasive breast cancer and, by the year’s end, over 40,000 women diagnosed with breast cancer died from their disease1. Breast cancer is a very heterogeneous disease with various subtypes of diagnostic and prognostic significance that differ in distribution by both age and race2. A major tool in the diagnosis, management, and prevention of breast cancer is the identification and characterization of biomarkers that will predict survival, and guide treatment decisions with higher precision3. Traditional visual assessment of histopathological images in combination with antibody-based biomarker profiling has been the standard of practice. However, the recent application of algorithms for digital image analysis to augment image characterization and quantitative assessment has broadened the utility, application, and accuracy of antibody-based characterization, thus providing new insight into the functional roles of specific protein biomarkers, and spawning a new age of diagnostic, prognostic, and therapeutic innovation4,5,6,7. Here we apply multiple algorithms for digital image analysis to perform an antibody-based assessment of patient breast cancer tissues that quantitatively profiles the subcellular distribution of Kaiso (ZBTB33), a functional protein biomarker previously shown to be elevated in cancers of the breast, prostate, pancreas, and colon8,9,10.
Kaiso was originally identified as a transcription factor and member of the BTB/POZ (Broad complex, Tramtrak, Bric à Brac/Poxvirus zinc finger) subfamily of zinc finger proteins8,10. It contains a bimodal DNA-binding domain that recognizes both sequence-specific consensus sites and methylated CpG nucleotides through 3 C-terminal DNA-binding Kruppel-like CH2 zinc fingers8,10. In its nuclear capacity, it has been shown to drive transcriptional programs that increase breast cancer growth and metastasis8,10. Subsequent studies revealed that it binds to a variety of sequence motifs throughout the nucleus, the majority of which are unmethylated and located at loci with open chromatin8,11. Because of its role in transcriptional regulation, Kaiso has been predominantly investigated as a nuclear protein and multiple studies have shown that its nuclear accumulation is dynamically regulated by association with p120 (CTNND1)12, a catenin family member that regulates membrane-bound E-cadherin cell adhesion assemblies8. Although several studies have noted that the cytoplasmic localization of Kaiso is a common feature in human tissues and tumors9, the major emphasis placed on Kaiso, as a prognostic biomarker, has focused on its nuclear accumulation. In fact, the nuclear accumulation of Kaiso has been shown, in multiple studies, to be a predominant feature of more aggressive forms of breast cancer, including triple-negative breast cancer13,14. Moreover, in some studies, nuclear Kaiso was shown to be more predictive of poor survival in women of African heritage diagnosed with TNBC14,15. Nonetheless, there have been multiple reported observations of cytoplasmic or “non-nuclear” accumulation of Kaiso, implying potential functional roles for Kaiso outside of the nucleus9,16,17. These include interaction with centrosomes, assembly with RhoH and p120 at actin-containing cell protrusions, and regulation of Kaiso subcellular distribution by the EGF-19,18,19.
In this study, we leverage the analytic precision of automated image analysis algorithms to quantitatively profile the compartment-specific (nuclear versus cytoplasmic) distribution of Kaiso in a racially diverse cohort of breast cancer patients residing in a health disparities region of rural East North Carolina. These findings reveal that both nuclear and cytoplasmic Kaiso are associated with breast cancer outcome and each are independent predictors of overall breast cancer survival. Furthermore, compartment-specific profiling of Kaiso with multiple prognostic breast cancer biomarkers reveal new functional correlations that link the specific subcellular distribution of Kaiso with (1) the autophagy-related factor LC3A/B; (2) cellular phenotypes within the tumor immune microenvironment; and (3) overall breast cancer survival.
Results
The subcellular distribution of Kaiso is differentially correlated with breast cancer subtype and overall survival from breast cancer
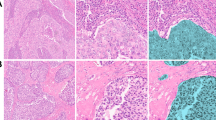
The protein expression of Kaiso was spatially profiled by automated analysis4,6 of immunohistochemically stained tissue microarrays (TMAs) (Fig. 1), containing 555 tumors from a cohort of racially diverse breast cancer patients (see Supplementary Fig. 1) residing in a designated health disparities catchment area of East North Carolina (median follow-up 8.5 years)6. By this analysis, staining intensity in the nucleus or cytoplasm of each cell in the annotated tumor regions is assigned one of four scores: from negative staining (0) to weak staining (+1), moderate staining (2+) or strong staining (3+). The percent of cells in the annotated regions demonstrating one of the 4 different intensities are then aggregated to derive an H-score (H-score= 3 × [%3+] + 2 × [%2+] + 1 [%1+]), thus generating a continuous score from 0 to 3004,6. The subcellular distribution of the staining intensity differs across many of the tumors, revealing various patterns of enrichment in the nucleus, cytoplasm, or both. (Fig. 1a). Notably, the cell segmentation algorithm-based quantitative profiling4,6 of Kaiso subcellular distribution reveals distinct differences in the cytoplasmic versus nuclear Kaiso patterns of distribution when examined in rank order (Fig. 1b, left). This Kaiso-specific difference in subcellular distributions is contrasted by the significant similarity in the nuclear and cytoplasmic distribution of a typical nuclear antigen that shuttles between the cytoplasm and nucleus like the androgen receptor (AR) (Fig. 1b, right), as well as the lower correlation between nuclear and cytoplasmic Kaiso, compared to AR (Supplementary Fig. 2). These stark differences suggest that the mechanisms governing the nuclear versus cytoplasmic localization of Kaiso are far more dynamic and complex than the androgen receptor whose intensity distribution in the nucleus reflects a more passive ligand-mediated nuclear-to-cytoplasmic distribution.

a Representative subcellular patterns of Kaiso expression in breast cancer tissues detected by anti -Kaiso immunohistochemical staining. Shown is a range of high (upper panels) versus low (bottom panel) nuclear and cytoplasmic protein expression. b Comparison of the distribution of digitally determined H-scores for nuclear versus cytoplasmic Kaiso enrichment. The difference in distribution is shown in contrast to the similarities in the distribution of nuclear versus cytoplasmic androgen receptor (AR). c Cytoplasmic Kaiso is differentially enriched in ER− breast cancers compared to nuclear Kaiso and is significantly enriched in the more aggressive breast cancer subtypes, LumB, HER2+, and TNBC. d Nuclear and cytoplasmic levels of Kaiso (H-score) do not show significant differences (t-test) based on race (also see Supplementary Fig. 1). e Kaiso (ZBTB33) mRNA abundance (median) is predictive of poor overall breast cancer survival as demonstrated in two independent publicly available breast cancer cohorts88. f Comparison of nuclear and cytoplasmic levels of Kaiso in breast cancer patients to levels detected by RNA-seq in (N = 131) patients demonstrates that Kaiso (ZBTB33) mRNA levels do not correlate with either nuclear or cytoplasmic levels of Kaiso. LumA Luminal A, LumB Luminal B, HER2+ human epidermal growth factor receptor 2 positive, TNBC triple-negative BC, ER estrogen-receptor status, NHW non-Hispanic White, NHB non-Hispanic White.
Unlike nuclear Kaiso, the levels of cytoplasmic Kaiso in this cohort were significantly different based on hormone receptor (ER) status and breast cancer subtype, where the cytoplasmic levels of Kaiso were distinctly higher in the subtypes of breast cancer known to be more aggressive, including triple-negative breast cancer (TNBC), human epidermal growth factor receptor 2 positive (HER2+), and Luminal B (LumB) (Fig. 1c). Although the levels of nuclear Kaiso have been previously reported to be higher in patients of African heritage15,20, we did not detect such differences in the current cohort although a minor trend for preferential distribution of high levels of cytoplasmic Kaiso in patients of African, compared to European descent, was observed (Fig. 1d). Interestingly, although the mRNA levels of Kaiso are highly predictive of poor breast cancer survival, as demonstrated in publicly available data sets (Fig. 1e); a direct comparison of available RNA-seq expression data, within a subset of this cohort (N = 134), demonstrates very little correlation between Kaiso mRNA and either nuclear Kaiso, cytoplasmic Kaiso, or their combined total (Fig. 1f).
Nuclear Kaiso, cytoplasmic Kaiso, and their combined score, defined as total Kaiso, are highly correlated with poor breast cancer survival (Fig. 2a–c). However, cytoplasmic Kaiso appears to be significantly more predictive with a hazard ratio (HR) of 16.29 (confidence interval (CI): 7.6–34.8; p-value 5.3E−13) for cytoplasmic Kaiso compared to HR: 2.83 (CI: 2.02–3.9; p-value 6.1E−11) for nuclear Kaiso, and HR: 7.86 (CI: 5.0–12.22; p-value 1.7E−18) for total Kaiso (Fig. 2a). These differences are also in good agreement with a non-digitally assisted expert pathologist’s assessment (Supplementary Fig. 4). In the multivariate setting, both nuclear and cytoplasmic Kaiso are independent predictors of overall breast cancer survival (Table 1). Finally, both nuclear and cytoplasmic Kaiso can stratify patients with lymph node-positive (high risk of recurrence) (Fig. 2b) or lymph node-negative breast cancer (low risk of recurrence) (Fig. 2c) into high versus low survival groups. However, in each case, although both nuclear Kaiso and cytoplasmic Kaiso are independent predictors of survival, cytoplasmic Kaiso is consistently more predictive of poor overall survival compared to nuclear Kaiso (Fig. 2a–c). Interestingly, total Kaiso appears to be the best predictor of overall survival in high risk (lymph node-positive) patients, with a hazard ratio of HR: 24.59 compared to HR: 19.58 (cytoplasmic Kaiso) or HR: 2.64 (nuclear Kaiso) (Fig. 2b).

a Analysis of the association between subcellular Kaiso distribution and survival demonstrates that high cytoplasmic Kaiso is much more predictive of poor survival compared to nuclear Kaiso. Nuclear, cytoplasmic, and total Kaiso H-scores predict survival in both b high risk (lymph node-positive) and c low risk (lymph node-negative) breast cancer patients, where total Kaiso score is most predictive of survival in both low and high-risk breast cancer patients. (HR could not be calculated for cytoplasmic Kaiso in low-risk patients because no deaths were recorded in that risk group). NHW non-Hispanic white; NHB non-Hispanic black. Optimized cut-offs were determined by the method of maximally selected rank statistics (Supplementary Fig. 3 and Supplementary Table 2).
Cytoplasmic Kaiso reveals a high correlation with TNBC and the autophagy-related protein LC3A/B
The high correlation between cytoplasmic Kaiso expression and more aggressive subtypes of breast cancer (Fig. 1c) implies that similar comparisons, between cytoplasmic Kaiso and other biomarkers that stratify aggressive forms of cancer, will provide deeper insights into the role of cytoplasmic Kaiso in poor breast cancer survival. To accomplish this goal, quantitative profiles comparing the relative enrichment of biomarkers recently implicated to be strongly associated with breast cancer progression21,22,23,24,25,26,27,28,29, were analyzed together by robust unsupervised hierarchical clustering (Fig. 3a). Biomarkers investigated included, estrogen-receptor ER30, the ER pioneer proteins FOXA131,32,33 and GATA334,35,36, HER2 membrane expression37,38, the protein-membrane adhesion molecule and tumor suppressor E-cadherin39,40,41, epithelial growth factor receptor EGFR37,38, and the autophagy-related factor LC3A/B (MAP1L3A and MAP1L3B), an autophagy-related biomarker recently implicated to be strongly associated with breast cancer progression21,22,23,24,25,26,27,28,29. A heatmap of clinical and pathological features of patients including survival, tumor subtype, and ER status is provided underneath for a direct comparison of biomarker expression with patient characteristics. Notably, patient clustering by these biomarkers in combination with cytoplasmic Kaiso expression identifies multiple breast cancer groups (A1-C2) with distinct survival differences (Fig. 3b and Supplementary Fig. 5).

a Unsupervised hierarchical clustering of the nuclear, membrane and cytoplasmic biomarker H-scores for each of (N = 555) patients is shown in correlation with patient clinicopathologic and demographic attributes (below). b Kaplan–Meier survival analysis of specific antigen expression clusters (identified by color code) in a demonstrating associations between cytoplasmic Kaiso, LC3A/B expression, and overall survival in TNBC. c Representative sample of LC3A/B immune-histochemical staining in breast cancer tissues. Arrowhead indicates subcellular puncta noted in the cytoplasm of multiple sections. d Kaplan–Meier survival analysis shows that high LC3A/B cytoplasmic staining is associated with poor overall breast cancer survival. e Like cytoplasmic Kaiso, LC3A/B staining is highly correlated with the more aggressive breast cancer subtypes LumB, HER2, and TNBC with the strongest association with TNBC. f LC3A/B shows a trend of higher expression in NHB versus NHW patients and is significantly more expressed in patients with ER- breast cancer. g Correlation between LC3A/B protein, nuclear Kaiso, cytoplasmic Kaiso, and the RNA levels for LC3A/B, (MAPL1LC3A, MAPL1LC3B), and Kaiso (ZBTB33) showing the strongest correlation between LC3A/B and MAPL1L3B RNA in addition to ZBTB33 RNA and MAPL1LC3B RNA. Spearman correlation is shown in red. p-value for Spearman correlation is shown in blue.
Of particular interest is the clustering of cytoplasmic Kaiso with the autophagy marker LC3A/B (Fig. 3a), which, in combination with the other biomarkers, stratifies TNBC patients into 3 different survival subgroups (B1, B2, and B3). The LC3A/B family of proteins has a major role in a variety of autophagy-related phagocytic and secretory processes including autophagy, phagocytosis, conventional secretion of cytokines, extracellular release of lysozymes, extracellular vesicle (EV) production, unconventional protein secretion, and LC3-dependent EV loading and secretion (LDELS)42,43,44,45. In each case, lipid conjugated LC3 has a major role in the loading of different cargoes, (organelles, protein, and nucleic acid) into membrane-bound structures destined for secretion or degradation. Collectively these processes are referred to as secretory autophagy43,44,46. Furthermore, each of these processes has the potential to influence tumor microenvironment47.
Remarkably, the clustering of low cytoplasmic Kaiso and low LC3A/B, in the context of the other biomarkers, identifies a class of TNBC patients that show favorable survival (compare B1 & B2, to B3) (Fig. 3b). Thus, low levels of cytoplasmic Kaiso combined with low levels of the autophagy-related factor LC3A/B, predict favorable survival in breast cancer patients with TNBC, implicating a significant role for this biomarker in tumor progression and survival. Representative LC3A/B IHC staining of patient tumors reveals a heterogeneous staining pattern in the cytoplasm with a mixture of diffuse and punctate cytoplasmic staining typical of LC3A/B27,48 (Fig. 3c). Quantitative analysis of the cytoplasmic staining shows that cytoplasmic LC3A/B is predictive of survival (HR: 2.5, CI: 1.77–3.68; p-value 5.9e−07) (Fig. 3d and Supplementary Fig. 3). Similar to cytoplasmic Kaiso, LC3A/B is (1) preferentially expressed in more aggressive forms of breast cancer including LumB, HER2, and TNBC (Fig. 3e); and (2) preferentially elevated in ER- compared to ER+ tumors with a trend toward higher expression in women of African compared to European ancestry (Fig. 3f). Analysis of the correlation between IHC-based protein expression and RNA expression (Fig. 3g and Supplementary Data 1) reveals very little correlation between Kaiso (ZBTB33) mRNA and Kaiso protein. However, there is a modest correlation between MAP1LC3B mRNA and LC3A/B with the highest correlation between ZBTB33, MAP1LC3B mRNA, and LC3A/B protein (Fig. 3g). By univariate survival analysis, LC3A/B expression is significantly predictive of poor survival but loses all significance in the multivariate setting, where only nuclear Kaiso, cytoplasmic Kaiso, and lymph node status are significant independent predictors of outcome (Table 1). This suggests that the survival predictive value of LC3A/B is closely associated with Kaiso protein expression.
Kaiso is required for the functional activation of LC3A/B
Given the close correlation between Kaiso and the LC3A/B autophagy-related proteins, we sought to further establish this relationship by employing a “bedside-to-bench” approach utilizing the human triple-negative breast cancer cell line, MDA-MB-231 depleted of Kaiso by RNA interference (RNAi) (Fig. 4a and Supplementary Fig. 6). The analysis of genes that are significantly differentially expressed (p-values < 0.001) in WT versus Kaiso-depleted cells reveals a large overlap (142 of 518) with a list of autophagy-associated genes compiled from the MSigDbase and the Autophagy Database49,50 (Fig. 4a) (also see Supplementary Data 2). This overlap is supported further by GSEA revealing a large and substantial enrichment in six (6) different autophagy-related gene sets (Fig. 4b) (also see Supplementary Data 3).

a Differential gene expression analysis of wild type TNBC cell line MDA-MB-231 versus MDA-MB-231 cells depleted of Kaiso by RNAi shows a significant overlap of differentially expressed genes with an autophagy-related gene list (also see Supplementary Fig. 6, and Supplementary Table 3) (p-value for the significance of the overlap is provided by the hypergeometric test). b Gene set enrichment analysis shows significant enrichment for autophagy terms in MDA-MB-231 cells that differentially express Kaiso (Supplementary Data 4). c Analysis of autophagocytic flux in WT versus MDA-MB-231 depleted of Kaiso by RNAi reveal a significant defect in autophagy demonstrated by decreased autophagocytic puncta formation in cells depleted of Kaiso. d Quantitative GFP-LC3 immunofluorescent analysis of LC3 puncta formation (average puncta per cell before (left) and after (right) addition of chloroquine in MDA-MB-231 cells expressing GFP-LC3). e Immunoblot analysis of LC3A/B lipidation (GFP-LC3ii formation) in MDA-MB-231 transfected with scramble control RNAi versus 3 different short hairpins against Kaiso. f Quantitative densitometer scan (N = 4 biological replicates each with N = 3 technical replicates). (*) = p-value < 0.05, (***) = p-value < 0.001 (t-test) for each unique Kaiso RNAi hairpin. g Immunofluorescent colocalization of Kaiso (green) and LC3A/B (red) in MCF-7 and MDA-MB-231 cells. h Quantitative profiling of Kaiso and LC3A/B colocalization in both the cytoplasm and nucleus (overall) of MCF-7 and MDA-MB-231 cells.
A hallmark feature of autophagy-related processes is the conjugation of LC3A/B to phosphatidylethanolamine (PE) followed by its incorporation into intracellular membrane vesicles within the endocytic vesicle system44,46,51. The lipid conjugation and membrane incorporation of LC3 protein enable it to load specific cargo into various endocytic trafficking vesicles including autophagosomes and extracellular vesicles44,45,46. A commonly used method to examine autophagocytic flux is to visualize the incorporation of GFP-labeled LC3A/B into autophagocytic membrane structures that appear as intracytoplasmic puncta (Fig. 4c)52,53. As demonstrated in Fig. 4c, MDA-MB-231 cells depleted of Kaiso show a significant absence (p < 0.0001) of puncta both in the presence or absence of autophagosome stabilization by the lysosome inhibitor chloroquine (CQ) (Fig. 4d). LC3A/B maturation can be followed by immunoblot analysis through the detection of changes in LC3A/B phosphatidylethanolamine (PE) lipid conjugation by the demonstration of a change in mobility, where the lipid conjugated LC3A/B (LC3ii) migrates with faster mobility than the unconjugated form (LC3i) (Fig. 4e). This difference in LC3 conjugation can be amplified by the inhibition of LC3 lysosomal degradation by chloroquine (CQ) (Fig. 4e, f). As shown in Fig. 4f, compared to the non-targeting short hairpin, the addition of 3 different RNAi short hairpins targeting Kaiso, results in a significant reduction of GFP-LC3 conjugation. Similar results are observed with the ER+ cell line MCF-7 (Supplementary Fig. 7). Finally, Kaiso and LC3A/B immunofluorescence show significant colocalization in both ER+ (MCF-7) and TNBC (MDA-MB-231) cell lines in both the cytoplasm and nucleus (Fig. 4g, left and right, respectively), with quantitative profiling in Fig. 4h (also see Supplementary Fig. 8).
Kaiso and LC3A/B show significant colocalization in patient tumors
Consistent with the observations in breast cancer cell lines, there is also significant cytoplasmic colocalization of Kaiso and LC3A/B in patient tumors with substantial variation by subtype (Fig. 5a, b) Thus, consistent with the co-enrichment of cytoplasmic Kaiso with LC3A/B in patient samples (Fig. 3a) and their colocalization in breast cancer cell lines (Fig. 4g), both proteins also show a substantial colocalization in patient tumors that varies by subtype (Fig. 5a, b).

a Immunofluorescent staining of Kaiso (green) and LC3A/B (red) in TNBC breast cancer (upper panel) and Luminal B breast cancer (lower panel). b Colocalization maps showing relative colocalization of Kaiso and LC3A/B in patient tumors.
Patients stratified by nuclear and cytoplasmic Kaiso are variably enriched in cellular stress and immune response pathways and differentially predict the overall outcome based on genetic ancestry
A comparison of the gene expression enrichment patterns of patients stratified by nuclear versus cytoplasmic Kaiso shows concordant (red) and discordant (blue) expression of genes linked to cellular differentiation, metabolism, immune modulation, and cell-microenvironment interactions (Fig. 6a, b). This observation is corroborated by GSEA profiles revealing opposing enrichment for inflammatory response pathways in nuclear versus cytoplasmic Kaiso (Fig. 6a, b, right) with notable negative enrichment for allograft rejection pathways in patients over-expressing cytoplasmic Kaiso (also see Supplementary Data 4 and 5).

a Volcano plot of differential gene expression of patients stratified by nuclear Kaiso. (Right) Gene set enrichment analysis of patients stratified by cytoplasmic Kaiso (Supplementary Data 5 and 6). b Volcano plot of differential gene expression of patients stratified by cytoplasmic Kaiso (right) Gene set enrichment analysis of patients stratified by nuclear Kaiso. c Population-specific composition of a representative portion (N = 131) of the breast cancer cohort, by genetic ancestry-based ancestry informative markers extracted from the RNA-seq data. Indicated colors reflect the percent admixture of each genetic population. d Forest plot analysis of overall survival hazard-based optimized cut-off for cytoplasmic Kaiso for the total population, NHW patients, NHB patients, and median cut-off for the total population. e Forest plot analysis of overall survival hazard-based optimized cut-off for LC3A/B in the total population, NHW patients, NHB patients, and the median cut-off for the total population. f Forest plot analysis of overall survival hazard based on optimized cut-off for nuclear Kaiso in the total population, NHW patients, NHB patients, and the median cut-off for the total population. g Forest plot analysis of overall survival hazard based on optimized cut-off for nuclear Kaiso in TNBC patients using optimized cut-off for NHW patients, NHB patients, and the median cut-off for the total TNBC population.
As discussed earlier, nuclear Kaiso expression has been linked previously to racial differences in breast cancer outcome where nuclear Kaiso was found to be higher and more predictive of poor outcome in women of African Heritage (non-Hispanic black (NHB)) diagnosed with TNBC compared to their European (non-Hispanic white (NHW)) counterparts8,14,15. Most notably this distinction seemed to be greater depending on the degree of African ancestry15. To profile the degree of racial admixture in our study cohort, ancestral informative markers (AIMs) were extracted from patient (N = 131) tumor RNA-seq data (>23% of the study cohort)54,55. Each patient in this group was then assigned a percent ancestry based on five genetic populations (African, European, East Asian, South Asian, and Admixed Native American) Fig. (6c). Among the 69 patients that self-identified as NHB, all but 1 had >50% African ancestry. Of the 62 patients that self-identified as NHW, 3 patients showed greater than 80% admixed Native American ancestry (Fig. 6c). Notably, forest plot analysis of the Cox proportional-hazards model for overall survival, optimized by Race, reveals that cytoplasmic Kaiso is more predictive of survival in women of African ancestry (Fig. 6d), consistent with prior indications of a differential survival risk based on Kaiso and African genetic background8,14. Moreover, even within admixed populations, and consistent with previous reports, there is a greater survival risk associated with nuclear Kaiso in patients of African ancestry diagnosed with TNBC (Fig. 6g). However, neither LC3A/B nor Nuclear Kaiso shows significant racial differences in survival hazard in the total breast cancer cohort (Fig. 6e, f).
Cytoplasmic Kaiso levels and LC3A/B are associated with an immune-suppressed tumor microenvironment in breast cancer tissues
The immune tumor microenvironment has been found to have a broad prognostic and predictive role in breast cancer56,57,58,59,60,61. With respect to breast cancer racial health disparities, there is a wide consensus supporting a deterministic role for race-based genetic variation in the immune response in influencing racial survival disparities in breast cancer62,63,64,65,66,67. Given the observed influence of elevated levels of cytoplasmic and nuclear Kaiso on immune regulatory pathways (Fig. 6a, b), and the extensive potential role for secretory autophagy in the immune response43,44,46, we sought to define the linkage between LC3A/B, the subcellular distribution of Kaiso, and immune properties of the tumor microenvironment (Fig. 7). This analysis was conducted through a nearest-neighbor analysis of TMA tissues co-stained for pan-cytokeratin (tumor), CD8 (killer T-cells), CD68 (macrophages), and PD-L1 (immune checkpoint regulator) (Fig. 7a, b). The x and y-coordinates (Fig. 7b) of the resultant combinations of the tumor and immune phenotypes (Supplementary Figs. 9 and 10) were then mapped and the frequency distribution of distances between each cellular phenotype was profiled (Fig. 7c). Notably, nearest-neighbor profiling showed significant coordination between cytoplasmic Kaiso and LC3A/B, and the proximity of PD-L1-positive CD8 cells near tumor compared to insignificant association with nuclear Kaiso and Race (Fig. 7c). Similarly, elevated levels of both cytoplasmic Kaiso and LC3A/B were associated with increased proximity of PD-L1-positive CD68 cells near tumor compared to insignificant association with nuclear Kaiso and Race (Fig. 7c). In a similar fashion nearest-neighbor profiling of total CD8 cells in proximity to PD-L1-positive tumor showed a significant association between elevated cytoplasmic Kaiso, LC3A/B and Race compared to the insignificant association with nuclear Kaiso (Fig. 7c). Finally, proximity profiling of total CD68 cells near PD-L1-positive tumor cells similarly shows a significant association with elevated cytoplasmic Kaiso and LC3A/B compared to nuclear Kaiso and Race (Fig. 7c). Given the known immunosuppressive role of PD-L1 expression in both immune cells and tumor59,60,61,68,69, these findings reveal a strong association between LC3A/B and cytoplasmic Kaiso expression and the potential for an immune-suppressive tumor microenvironment. Notably, all associations between Kaiso and LC3A/B are specific to PDL1-positive cells as the trends described above are not significant when comparing total CD8 nor CD68 cells (Supplementary Fig. 11).

a Representative multi-spectral quantitative immunofluorescence (mQIF) of the tumor microenvironment of a breast cancer TMA core, stained for pan-cytokeratin (Cy7, cyan); PDL-1 (Cy5, red); CD8 (FITC, green); CD68 (TRITC, carmine). b Coordinate map for nearest-neighbor analysis of tumor and stromal immunophenotypes. c Nearest-neighbor analysis showing the frequency distribution of immune cell proximities to tumor-associated with expression quartiles (Q1–Q4) of cytoplasmic Kaiso, LC3A/B, nuclear Kaiso, and race (white versus black).
Discussion
In this work, we utilized a novel application of computational digital analysis and a unique cohort of breast cancer patients to define new relationships between biomarkers based on their differential subcellular distribution to define strong prognostic markers of breast cancer survival. This “bedside-to-bench” approach not only reveals biomarkers correlated with overall breast cancer survival and an immune-suppressed tumor microenvironment but provides new functional and mechanistic insights into the cellular processes linked to the biomarkers. The functional linkage of cytoplasmic Kaiso to LC3A/B and the tumor microenvironment uncovers a new area of investigation into the role and mechanism of Kaiso in breast cancer progression. Kaiso could have multiple roles in promoting tumor progression through both modulations of transcription and autophagy-related events including (1) autophagy-mediated degradation pathways; and (2) secretory autophagy pathways dependent on LC3 conjugation. The precise mechanism of how Kaiso influences LC3 maturation will require further investigation. One possibility is that it may act as a scaffold to facilitate enzymatic lipid conjugation of LC3 proteins in the cytoplasm. Such a possibility is supported by the computational identification of a predicted LC3 interaction domain (LIR)70 in the C-terminal region of Kaiso adjacent to the DNA-binding zinc fingers. The possible dual role for Kaiso in autophagy and LC3-mediated secretion are not necessarily mutually exclusive since recent studies indicate that LDELS can occur independently of autophagy based on the observation that gene deletions that impair autophagy initiation do not block LDELS44. Other observations that will require further investigation arise from the morphological studies of cell lines and tumors. Notably, immunofluorescent staining for Kaiso reveals staining on tubulin (Fig. 4g and Supplementary Fig. 8) consistent with prior reports of the association of Kaiso with centrosomes19. Other observations include the accumulation of Kaiso at focal adhesion-like structures or assemblies reminiscent of actin-containing cell protrusion sites18 in MCF-7 (Supplementary Fig. 8). Such associations have not been previously described, but suggest a linkage between autophagy-related processes and the cycling of cell-matrix adhesion machinery71. Another interesting finding is the detection of race-specific differences in the tumor environment where there is a trend to a more suppressed immune microenvironment in women of African ancestry, particularly when considering CD8 cells near PD-L1-positive tumor (Fig. 7c). The differential contribution of race in the association of nuclear and cytoplasmic Kaiso with overall breast cancer survival (Fig. 6d, g) may contribute in part to this trend.
The general findings presented in this study, including the discovery of the prognostic significance of Kaiso subcellular partitioning and its linkage to immune-suppressive features of the tumor microenvironment, highlight its potential as a predictive biomarker to guide future treatment decisions, particularly in the use of immune checkpoint inhibitors. These results provide support for future applications in prospective studies where profiles of nuclear and cytoplasmic Kaiso are evaluated in clinical trials as both a predictive and prognostic breast cancer biomarker72. Furthermore, because the predictive value of nuclear and cytoplasmic Kaiso varies across racial groups, these findings further emphasize the need for the inclusion of diverse racial and ethnic groups in clinical trials.
Methods
Study population, tissue microarray construction, and analysis
Following IRB approval from East Carolina University and the National Institutes of Health intramural research program, de-identified formalin-fixed and paraffin-embedded (FFPE) tissue samples and de-identified clinical information abstracted from the medical records were requisitioned and initially procured for 733 breast cancer patients who underwent surgery for Stage 0 to Stage IV breast cancer between 2001 and 2010 at Pitt County Memorial Hospital (now Vidant Medical Center), Greenville, NC. Race, ethnicity, or “ancestry” was self-reported at the initial visit and captured in the medical record. Survival was recorded retrospectively from the medical records and the cancer registry. All patient samples and data obtained were de-identified and approved by the East Carolina University Institutional Review Board as a human subject exempt project, for which no informed consent is needed. The study was conducted in accordance with the Declaration of Helsinki. Race and/or ethnicity was self-reported at the initial visit and captured in the medical record. Survival was recorded retrospectively from the medical records and the cancer registry. The median follow-up is 8.5 years. 588 patient tumor blocks from this cohort were found suitable for use in the construction of a tissue microarray. Replicate tissue microarrays were constructed using 1 mm cores per previously described methods73,74, with a representation inclusive of 555 patients. Detailed methods for IHC, scoring, and the assignment of clinical variables are provided in the Supplementary Methods.
Patient tumor RNA-seq analysis
RNA-seq analysis was performed on RNA extracted from FFPE tissue blocks (Total N = 126; EA N = 61; AA N = 65, Other = 1)6. Following a review of H&E stained slides, areas for tumors with >80% nuclei were circled, and 2.5 × 2–3 mm tissue cores were extracted from the corresponding regions of FFPE tissue blocks. Cores were shipped to the Beijing Genomics Institute (BGI) (Beijing, China) where RNA was extracted and sequenced (60M paired-end reads per sample) as previously described75,76. Detailed methods are provided in the data supplement.
Immunofluorescence
Cells were grown on 22-mm-glass coverslips in 6-well plates to ~80% confluency before exposure to appropriate drugs or vehicular controls. After washing with PBS, cells were fixed with 4% formaldehyde in PBS at room temperature (RT) for 10 min. Cells were permeabilized with 0.1% Triton X-100 for 10 min, then incubated with blocking medium (PBS, 0.1% Tween-20, 10% normal goat serum) for 30 min in a humidified chamber at RT. Blocked samples were then stained with mouse anti-Kaiso (Abcam, ab12723; 1:1000) and rabbit anti-LC3A/B (Abcam, ab128025; 1:1000) in antibody diluent (PBS, 0.1% Tween-20, 1% BSA) for 1 h at RT in a humidified chamber. Anti-LC3A/B antibody recognizes both LC3A and LC3B proteins. After appropriate PBS washing, cells were stained with anti-mouse Alexa Fluor 488 (Invitrogen, A-11001; 1:2000) and anti-rabbit Alexa Fluor 594 (Invitrogen, A-11002; 1:1000) secondary antibodies in a humidity chamber in the dark for 1 h at RT. Coverslips were washed and then mounted on slides using ProLong Gold antifade reagent (Molecular Probes, P36934). MDA-MB-231 and MCF-7 used in this study were obtained from ATCC and identify was validated by STRS profiling. All cells were tested and found to be free of mycobacterial contamination.
Colocalization analysis
Z-stack images for each channel were captured at ×100 magnification with Immersol 518F (Zeiss) oil immersion using a Zeiss Axiovert 200M fluorescent microscope running AxioVision software. Slices with the clearest resolution were selected for further analysis. The background was removed by subtracting the mean gray value for each channel in an area containing no cells. Colocalization analysis was performed using the ImageJ plug-in, “JaCoP”, according to the developer’s instructions. Negative controls were achieved by rotating one channel at least 90° and re-running the analysis. Positive controls were achieved by running colocalization analysis on two of the same channels. All graphs are plotted as the mean Pearson’s coefficient of at least three independent experiments with error bars representing the standard error. Each MCF-7 image contains ~15–20 cells, and MDA-MB-231 images contain ~5–10 cells per image.
Multi-spectral fluorescent imaging and nearest-neighbor analysis
We used the Ultivue UltiMapper I/O PD-L1 assay to collect the qmIF data. This kit uses the following antigens: CD8, CD68, PD-L1, pan-cytokeratin (panCK), and DAPI (DNA marker). The raw image data is collected at 20×. The fluorescent dye intensities are normalized to 0–255. Image analysis was performed using a commercial software package (HALO, Indica Labs) at full magnification. The TMA spots were decomposed into individual analysis regions using the TMA module, with an invalidation threshold of about 80–90% empty space. A coordinate system was established for each spot with the origin being the bottom-left corner of the square TMA boundary. A unit coordinate is equivalent to one pixel or 0.5 microns. Watershed nuclear identification was performed on the DAPI channel with a nuclear contrast threshold of 0.5, and nuclear segmentation aggressiveness of 92%. Nuclei are required to be between 10 and 250 μm2 in size. A cytoplasmic region was grown from the nuclear boundary up to a radius of 4.2 μm. Cells were required to be <500 μm2. The average stain intensity within the cytoplasmic region was measured, and the positive-dye status for the antigen was defined as follows: CD8 (15) CD68 (8), panCK (10), and PD-L1 (13). Overall, we observed low backgrounds and strong signals. Phenotypes are defined using the coincidence/anti-coincidence logic of the positive-dye status. The logical combination for the main cell types are stromal (not panCK), T-cell (CD8), macrophage (CD68), and tumor (panCK and not CD8 and not CD68). These four cell types have 3 sub-phenotypes, inclusive and PD-L1+ (and PD-L1) or PD-L1- (and not PD-L1). The result of the phenotyping analysis is a text file for each tissue sample consisting of entries listing information about each cell location, including the manual phenotyping result and raw staining intensities using the defined coordinate system. The cell-point location was taken as the center of the rectangle which fully bounds the cell.
Statistics and reproducibility
The nearest-neighbor algorithm was implemented as follows: for a given pair of phenotypes P1, P2, each composed of cells (detected with thresholds on their staining intensities) with two coordinates, k in [1,2], we compute, for a given cell Ci belonging to P1, the Euclidean distances to all cells belonging to P2, excluding those whose distance is less than 0.05 microns to prevent cell-overlap (Supplementary Figs. 9 and 10). We keep the minimum distance value among those, which we call the nearest-neighbor distance, and repeat this process for each cell in P1 to form a distribution of nearest-neighbor distances, di. Measures of central tendencies for di were recorded as well as a histogram of frequencies of di using a bin width of 2 pixels up to 120 pixels. A counting error was assigned to each bin as being the square root of the number of entries. The normalization was chosen to be the total number of cells in the sample core, such that the integral of the histogram is equal to the density of the base phenotype being considered.
Population statistics or the average histogram shape were obtained by computing the mean value for each bin given a sub-population sample. The counting error was propagated and summed in quadrature with the standard error of the mean. The combined error is shown in the shaded band. To establish a test of statistical significance between two different histograms, we first define a test statistic as being the summed log-likelihood that each bin in the distribution has the same mean between two sub-populations. The natural log of the p-value, or “likelihood”, from a t-test between the individual bin values is taken. If the hypothesis sub-population mean is larger, this likelihood is defined to be positive, else it is negative. Schematically, large positive likelihoods represent significant upward fluctuations while large negative likelihoods represent significant downward fluctuations. These likelihoods are then summed across all bins. This forms the observed (hypothesis) statistic. The summed log-likelihood was then recomputed for 1k iterations using randomly assigned sub-populations, which have the same number of patients as the test sub-population. This forms the null distribution for the test statistic. The final p-value reported is the one-sided integral of the resultant null distribution from the observed value (Supplementary Fig. 10). This method overestimates the p-value since real differences in the sub-population can be double-counted when building the null distribution. However, this method treats bin-to-bin correlations correctly since it samples from real data.
Patient baseline characteristics and disease factors were summarized using descriptive statistics. Categorical variables were compared using the two-sided Pearson χ2 test. A comparison of IHC scoring was performed by a two-sided t-test and plotted as previously described77. Univariate and multivariate Cox proportional-hazards model was used to test the independent and combined prognostic values of proteins of interest with/without the presence of selected clinical variables. Spearman rank correlations were used to assess the relationship between protein H-score and gene expression (RPKM) values78. The significance of individual hazard ratios was estimated by Wald’s test. Optimal cut-off points for H-score were determined as previously described6,79 (Supplementary Fig. 3). The solid lines and histogram present data for samples with levels higher (red) or lower (blue); the dashed lines present data for samples divided into two groups (higher-red or lower-blue) based on the “optimal cut-off” algorithm79. Unsupervised hierarchical clustering of IHC protein score from all breast cancer samples was performed using complete linkage and distance correlations with the number of bootstrap replications (n = 1000) using the ‘pvclust’ R package80. The estimated clustering stability is measured by AU (approximately unbiased) (red) p-value and BP (bootstrap probability) (green) value for each cluster in a dendrogram80 (Supplementary Fig. 12). To explore the expression value together with clinical-pathological information, a heatmap was drawn where patients were arranged based on the order of the hierarchical clustering outcome.
Gene set enrichment analysis of patient RNA-Seq data
The median cut-off of protein data was used to classify patients into two groups based on H-scores (e.g., low versus high Kaiso cytoplasmic) and mRNA abundance (RNA-seq). A two-sided t-test was performed, and all available genes were ranked according to p-value (lowest to highest). The p-value ranked gene list was used for functional correlation using the GSEA software (http://software.broadinstitute.org/gsea/index.jsp).
Gene set enrichment analysis of MDA-MB-231 cell line gene expression
Four (N = 4) RNA samples each from WT MBA-MB-231 and RNAi Kaiso-depleted MBA-MD-231 cells were analyzed on the Affymetrix Human Gene 2.0 ST microarray and CEL files generated were normalized to produce gene-level expression values with the Robust Multiarray Average from the affy package to pre-process arrays and the limma package for identifying differentially expressed genes81,82. Relative fold depression of Kaiso (ZBTB33) mRNA was a 6-fold reduction. The hypergeometric test and Gene Set Enrichment Analysis (GSEA)83 was used to identify enriched signatures using the different pathway collections in the MSigDB database84. The GSEA pre-ranked method from GSEA was applied for this analysis. Human Gene 2.0 ST microarray of WT versus RNAi depleted MDA-MB-231 shows significant concordance with scrambled hairpin RNAi expressing MDA-MB-231 cells compared to Kaiso-depleted cell lines analyzed on the Nanostring DGE platform (Supplementary Fig. 6 and Supplementary Table 7).
Genetic admixture analysis
For admixture analysis, RNA-Seq reads from 136 breast cancer patients were aligned to hg19 using STAR v2.5.2b85 with subsequent variant calling completed using GATK (v3.8) HaplotypeCaller54,86. After variant calling, Admixture v1.3.055 was used to estimate ancestry proportions based on reference populations from the 1000 Genomes Project phase 387 super populations. Rare variants (i.e., <5% across all phase 3 1000 genomes), all INDELs, and any SNPs that were not biallelic were removed before analysis.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
RNA-seq data are available at SRA archives https://www.ncbi.nlm.nih.gov/sraSRP158272. Proteomic and immunofluorescent intensity (x,y) coordinate point cloud data for nearest-neighbor analysis (Figs. 1–7) has been uploaded to figshare https://figshare.com/s/b5652eb7712fa83cf8bc. Additional supplemental data for Figs. 1–6 are also provided as Supplementary Data 1–7. Additional clinical source data and custom program code used to generate figures will be made available upon request (contact: SK Singhal).
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34 (2019).
DeSantis, C. E. & Jemal, A. Re: Black-White breast cancer incidence trends: effects of ethnicity. J. Natl Cancer Inst. 111, 99–100 (2019).
Braden, A. M., Stankowski, R. V., M. Engel, M. J. & Onitilo, A. A. Breast cancer biomarkers: risk assessment, diagnosis, prognosis, prediction of treatment efficacy and toxicity, and recurrence. Curr. Pharm. Des. 20, 4879–4898 (2014).
Di, L. J. et al. Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat. Commun. 4, 1449 (2013).
Acs, B. et al. An open source automated tumor infiltrating lymphocyte algorithm for prognosis in melanoma. Nat. Commun. 10, 5440 (2019).
Byun, J. S. et al. Racial differences in the association between luminal master regulator gene expression levels and breast cancer survival. Clin. Cancer Res. 26, 1905–1914 (2020).
Davis, M. et al. Identification of distinct heterogenic subtypes and molecular signatures associated with african ancestry in triple negative breast cancer using quantified genetic ancestry models in admixed race populations. Cancers 12, https://doi.org/10.3390/cancers12051220 (2020).
Pierre, C. C., Hercules, S. M., Yates, C. & Daniel, J. M. Dancing from bottoms up - roles of the POZ-ZF transcription factor Kaiso in cancer. Biochim. Biophys. Acta Rev. Cancer 1871, 64–74 (2019).
Soubry, A. et al. Expression and nuclear location of the transcriptional repressor Kaiso is regulated by the tumor microenvironment. Cancer Res. 65, 2224–2233 (2005).
Daniel, J. M. Dancing in and out of the nucleus: p120(ctn) and the transcription factor Kaiso. Biochim. Biophys. Acta 1773, 59–68 (2007).
Blattler, A. et al. ZBTB33 binds unmethylated regions of the genome associated with actively expressed genes. Epigenet. Chromatin 6, 13 (2013).
Prokhortchouk, A. et al. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 15, 1613–1618 (2001).
Vermeulen, J. F. et al. Nuclear Kaiso expression is associated with high grade and triple-negative invasive breast cancer. PLoS ONE 7, e37864 (2012).
Jones, J. et al. Nuclear localization of Kaiso promotes the poorly differentiated phenotype and EMT in infiltrating ductal carcinomas. Clin. Exp. Metastasis 31, 497–510 (2014).
Bassey-Archibong, B. I. et al. Kaiso is highly expressed in TNBC tissues of women of African ancestry compared to Caucasian women. Cancer Causes Control 28, 1295–1304 (2017).
Shumskaya, V. S., Zhigalova, N. A., Prokhorchouk, A. V. & Prokhorchouk, E. B. Distribution of Kaiso protein in mouse tissues. Histochem. Cell Biol. 143, 29–43 (2015).
Dai, S. D. et al. Cytoplasmic Kaiso is associated with poor prognosis in non-small cell lung cancer. BMC Cancer 9, 178 (2009).
Mino, A. et al. RhoH participates in a multi-protein complex with the zinc finger protein kaiso that regulates both cytoskeletal structures and chemokine-induced T cells. Small GTPases 9, 260–273 (2018).
Kantidze, O. L., Kamalyukova, I. M. & Razin, S. V. Association of the mammalian transcriptional regulator kaiso with centrosomes and the midbody. Cell Cycle 8, 2303–2304 (2009).
Jones, J. et al. African Americans with pancreatic ductal adenocarcinoma exhibit gender differences in Kaiso expression. Cancer Lett. 380, 513–522 (2016).
Onorati, A. V., Dyczynski, M., Ojha, R. & Amaravadi, R. K. Targeting autophagy in cancer. Cancer 124, 3307–3318 (2018).
Clarke, R. et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 72, 1321–1331 (2012).
Debnath, J. The multifaceted roles of autophagy in tumors-implications for breast cancer. J. Mammary Gland Biol. Neoplasia 16, 173–187 (2011).
Cook, K. L., Shajahan, A. N. & Clarke, R. Autophagy and endocrine resistance in breast cancer. Expert Rev. Anticancer Ther. 11, 1283–1294 (2011).
Galluzzi, L. et al. Autophagy in malignant transformation and cancer progression. EMBO J. 34, 856–880 (2015).
Towers, C. G., Wodetzki, D. & Thorburn, A. Autophagy and cancer: modulation of cell death pathways and cancer cell adaptations. J. Cell Biol. 219, https://doi.org/10.1083/jcb.201909033 (2020).
Schlafli, A. M. et al. Prognostic value of the autophagy markers LC3 and p62/SQSTM1 in early-stage non-small cell lung cancer. Oncotarget 7, 39544–39555 (2016).
Schlafli, A. M., Berezowska, S., Adams, O., Langer, R. & Tschan, M. P. Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry. Eur. J. Histochem. 59, 2481 (2015).
Othman, E. Q., Kaur, G., Mutee, A. F., Muhammad, T. S. & Tan, M. L. Immunohistochemical expression of MAP1LC3A and MAP1LC3B protein in breast carcinoma tissues. J. Clin. Lab. Anal. 23, 249–258 (2009).
Siersbaek, R., Kumar, S. & Carroll, J. S. Signaling pathways and steroid receptors modulating estrogen receptor alpha function in breast cancer. Genes Dev. 32, 1141–1154 (2018).
Fu, X. et al. FOXA1 upregulation promotes enhancer and transcriptional reprogramming in endocrine-resistant breast cancer. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.1911584116 (2019).
Fu, X. et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc. Natl Acad. Sci. USA 113, E6600–e6609 (2016).
Meyer, K. B. & Carroll, J. S. FOXA1 and breast cancer risk. Nat. Genet. 44, 1176–1177 (2012).
Takaku, M. et al. GATA3 zinc finger 2 mutations reprogram the breast cancer transcriptional network. Nat. Commun. 9, 1059 (2018).
Fararjeh, A. S. et al. The impact of the effectiveness of GATA3 as a prognostic factor in breast cancer. Hum. Pathol. 80, 219–230 (2018).
Mehra, R. et al. Identification of GATA3 as a breast cancer prognostic marker by global gene expression meta-analysis. Cancer Res. 65, 11259–11264 (2005).
Hsu, J. L. & Hung, M. C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 35, 575–588 (2016).
Macrinici, V. & Romond, E. Clinical updates on EGFR/HER targeted agents in early-stage breast cancer. Clin. Breast Cancer 10(Suppl 1), E38–46 (2010).
Corso, G. et al. Hereditary lobular breast cancer with an emphasis on E-cadherin genetic defect. J. Med Genet. 55, 431–441 (2018).
Fearon, E. R. Connecting estrogen receptor function, transcriptional repression, and E-cadherin expression in breast cancer. Cancer Cell 3, 307–310 (2003).
Berx, G. & Van Roy, F. The E-cadherin/catenin complex: an important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Cancer Res. 3, 289–293 (2001).
Kaur, J. & Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 16, 461–472 (2015).
Ponpuak, M. et al. Secretory autophagy. Curr. Opin. Cell Biol. 35, 106–116 (2015).
Leidal, A. M. et al. The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat. Cell Biol. https://doi.org/10.1038/s41556-019-0450-y (2020).
Romao, S. & Munz, C. LC3-associated phagocytosis. Autophagy 10, 526–528 (2014).
Rak, J. L(C3)icensing of exosomes for RNA export. Nat. Cell Biol. https://doi.org/10.1038/s41556-020-0466-3 (2020).
Liu, J. & Debnath, J. The evolving, multifaceted roles of autophagy in cancer. Adv. Cancer Res 130, 1–53 (2016).
Berezowska, S. & Galvan, J. A. Immunohistochemical detection of the autophagy markers LC3 and p62/SQSTM1 in formalin-fixed and paraffin-embedded tissue. Methods Mol. Biol. 1560, 189–194 (2017).
Homma, K., Suzuki, K. & Sugawara, H. The Autophagy Database: an all-inclusive information resource on autophagy that provides nourishment for research. Nucleic Acids Res. 39, D986–990 (2011).
Liberzon, A. A description of the Molecular Signatures Database (MSigDB) web site. Methods Mol. Biol. 1150, 153–160 (2014).
New, J. & Thomas, S. M. Autophagy-dependent secretion: mechanism, factors secreted, and disease implications. Autophagy 15, 1682–1693 (2019).
Bhutia, S. K. et al. Monitoring and measuring mammalian autophagy. Methods Mol. Biol. 1854, 209–222 (2019).
du Toit, A., Hofmeyr, J. S., Gniadek, T. J. & Loos, B. Measuring autophagosome flux. Autophagy 14, 1060–1071 (2018).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Li, X. et al. Infiltration of CD8(+) T cells into tumor cell clusters in triple-negative breast cancer. Proc. Natl Acad. Sci. USA 116, 3678–3687 (2019).
Kaur, H. B. et al. Association of tumor-infiltrating T-cell density with molecular subtype, racial ancestry and clinical outcomes in prostate cancer. Mod. Pathol. 31, 1539–1552 (2018).
Savas, P. et al. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat. Rev. Clin. Oncol. 13, 228–241 (2016).
Roux, C. et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc. Natl Acad. Sci. USA 116, 4326–4335 (2019).
Wang, Y. et al. Regulation of PD-L1: emerging routes for targeting tumor immune evasion. Front. Pharmacol. 9, 536 (2018).
Brochez, L. et al. Challenging PD-L1 expressing cytotoxic T cells as a predictor for response to immunotherapy in melanoma. Nat. Commun. 9, 2921 (2018).
Yao, S. et al. Genetic ancestry and population differences in levels of inflammatory cytokines in women: role for evolutionary selection and environmental factors. PLoS Genet. 14, e1007368 (2018).
Hong, C. C. et al. Genetic variants in immune-related pathways and breast cancer risk in African American women in the AMBER Consortium. Cancer Epidemiol. Biomark. Prev. 27, 321–330 (2018).
Deschamps, M. et al. Genomic signatures of selective pressures and introgression from archaic hominins at human innate immunity genes. Am. J. Hum. Genet. 98, 5–21 (2016).
Quan, L. et al. Cytokine and cytokine receptor genes of the adaptive immune response are differentially associated with breast cancer risk in American women of African and European ancestry. Int J. Cancer 134, 1408–1421 (2014).
Karlsson, E. K., Kwiatkowski, D. P. & Sabeti, P. C. Natural selection and infectious disease in human populations. Nat. Rev. Genet. 15, 379–393 (2014).
Ness, R. B., Haggerty, C. L., Harger, G. & Ferrell, R. Differential distribution of allelic variants in cytokine genes among African Americans and White Americans. Am. J. Epidemiol. 160, 1033–1038 (2004).
Lee, J., Kim, D. M. & Lee, A. Prognostic role and clinical association of tumor-infiltrating lymphocyte, programmed death ligand-1 expression with neutrophil-lymphocyte ratio in locally advanced triple-negative breast cancer. Cancer Res. Treat. 51, 649–663 (2019).
Park, I. H. et al. Prognostic implications of tumor-infiltrating lymphocytes in association with programmed death ligand 1 expression in early-stage breast cancer. Clin. Breast Cancer 16, 51–58 (2016).
Kalvari, I. et al. iLIR: A web resource for prediction of Atg8-family interacting proteins. Autophagy 10, 913–925 (2014).
Sharifi, M. N. et al. Autophagy promotes focal adhesion disassembly and cell motility of metastatic tumor cells through the direct interaction of paxillin with LC3. Cell Rep. 15, 1660–1672 (2016).
Jacquelot, N. et al. Predictors of responses to immune checkpoint blockade in advanced melanoma. Nat. Commun. 8, 592 (2017).
Hewitt, S. M. The application of tissue microarrays in the validation of microarray results. Methods Enzymol. 410, 400–415 (2006).
Khoury, T. et al. Ki-67 expression in breast cancer tissue microarrays: assessing tumor heterogeneity, concordance with full section, and scoring methods. Am. J. Clin. Pathol. 148, 108–118 (2017).
Jia, W. et al. SOAPfuse: an algorithm for identifying fusion transcripts from paired-end RNA-Seq data. Genome Biol. 14, R12 (2013).
Peng, Z. et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 30, 253 https://www.nature.com/articles/nbt.2122#supplementary-information (2012).
Hintze, J. L. & Nelson, R. D. Violin plots: A box plot-density trace synergism. Am. Stat. 52, 181–184 (1998).
Myers, J. L., Well, A. & Lorch, R. F. Research Design and Statistical Analysis 3rd edn (Routledge, 2010).
Hothorn, T. & Lausen, B. On the exact distribution of maximally selected rank statistics. Comput. Stat. Data Anal. 43, 121–137 (2003).
Suzuki, R. & Shimodaira, H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542 (2006).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Gautier, L., Cope, L., Bolstad, B. M. & Irizarry, R. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Ringner, M., Fredlund, E., Hakkinen, J., Borg, A. & Staaf, J. GOBO: gene expression-based outcome for breast cancer online. PLoS ONE 6, e17911 (2011).
Acknowledgements
This work was supported in part by the intramural research programs of NCI and the National Institute on Minority Health and Health Disparities, Bethesda Maryland, 20892, the NIH/NCI Cancer Center Support Grant P30CA013696 (K.G.), R01 1R01CA253368 (K.G.), and U54-MD007585-26 NIH/NIMHD (C.Y.) and U54 CA118623 (NIH/NCI) (C.Y.), via Department of Defense Grant (PC170315P1, W81XWH-18-1-0589 (C.Y)), the Brody School of Medicine Department of Oncology Cancer Research and Education Fund and National Institute of General Medical Sciences of the National Institutes of Health Award Number U54GM128729. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations, or imply endorsement by the US Government.
Author information
Authors and Affiliations
Contributions
S.K.S., J.S.B., and K.G. conceived of and performed experiments, designed figures, and helped write the manuscript. S.P., T.Y., R.Y., A.C., S.G.H., S.M.H., H.B., S.J., M.B., M.S.U.A., J.W., C.Y., A.A., R.V., R.B., R.L., P.L.F., A.D.E., A.M.N., and N.V. conceived of and performed experiments, designed figures, and helped in manuscript preparation. S.K.S., J.S.B., and K.G. conceived of experimental design and organized and composed the manuscript.
Corresponding author
Ethics declarations
Competing interests
C.Y. received consulting from QED Therapeutics and Riptide Biosciences. C.Y. owns stock in Riptide Biosciences. The remainder of authors declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singhal, S.K., Byun, J.S., Park, S. et al. Kaiso (ZBTB33) subcellular partitioning functionally links LC3A/B, the tumor microenvironment, and breast cancer survival. Commun Biol 4, 150 (2021). https://doi.org/10.1038/s42003-021-01651-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-01651-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
