
Abstract
Domestication begins with the selection of animals showing less fear of humans. In most domesticates, selection signals for tameness have been superimposed by intensive breeding for economical or other desirable traits. Old World camels, conversely, have maintained high genetic variation and lack secondary bottlenecks associated with breed development. By re-sequencing multiple genomes from dromedaries, Bactrian camels, and their endangered wild relatives, here we show that positive selection for candidate genes underlying traits collectively referred to as ‘domestication syndrome’ is consistent with neural crest deficiencies and altered thyroid hormone-based signaling. Comparing our results with other domestic species, we postulate that the core set of domestication genes is considerably smaller than the pan-domestication set – and overlapping genes are likely a result of chance and redundancy. These results, along with the extensive genomic resources provided, are an important contribution to understanding the evolutionary history of camels and the genomic features of their domestication.
Similar content being viewed by others

Introduction
The birth and ascent of human civilization can largely be attributed to the habituation and cultivation of wild plants and animals. By providing a more reliable stream of resources such as food and clothing, this process of domestication facilitated the shift from hunter-gatherer subsistence to that of agriculture. In animals, domestication likely occurred via multistage processes depending on the anthropophily of the wild ancestor (commensal pathway) and/or needs of humans (prey or directed pathways)1. Whether initiated by the wild animal ancestor or humans, intentional or not, the fundamental basis for domestication originated from a reduced fear of humans, i.e., tameness2. Thereafter, humans could continue the domestication process by breeding individuals harboring favorable traits through a process termed artificial selection. Domestication, however, is not limited to artificial selection, but also includes the relaxation of natural selection pressures such as predation and starvation, and the indirect, unintentional effects on traits correlated with captivity and those artificially selected2. In addition to tameness, the domestication of animals has led to a suite of morphological, physiological, and behavioral changes common to many species. These shared traits—including tameness, changes in coat color, modified reproductive cycles, altered hormone and neurotransmitter levels, and features of neotenization—are collectively referred to as the ‘domestication syndrome’ (DS)3.
In general, two hypotheses have been proposed to govern the relationship between the development of DS and the underlying genes responsible. First, Crockford4 suggested that the regulation of thyroid hormone concentrations during development may be linked to the neotenized phenotype of DS (thyroid hormone hypothesis; THH). The thyroid hormones triiodothyronine and its precursor tetraiodothyronine are produced during embryonic and fetal development, and also play key roles in postnatal and juvenile development4,5. The THH has been supported by research in domestic chickens, for example, where a fixed mutation in the thyroid stimulating hormone receptor gene has been extensively linked to the characteristic traits of DS6.
The second hypothesis proposed by Wilkins et al.3 predicts that DS is a consequence of mild deficits in neural crest cells during embryonic development; a product of artificial selection for behavior on standing genetic variation (neural crest cell hypothesis; NCCH). In horses, for example, selected genes were enriched for functions such as associative learning, abnormal synaptic transmissions, ear shape, and neural crest cell morphology, in addition to genes transcribed in brain regions containing neurons related to movement, learning and reward7. In cats, genomic regions under selection were associated with (i) neurotransmitters, responsible for serotonergic innervation of the brain, maintaining specific neuronal connections in the brain and fear conditioning, (ii) sensory development like hearing, vision and olfaction, and (iii) and neural crest cell survival8. Comparisons between genomes of village dogs and wolves also highlighted the role of neural crest cell migration, differentiation, and development in dog domestication9. Although evidence for both hypotheses exists, they are not necessarily mutually exclusive, and the relative contribution of each may vary along a continuum5. Furthermore, despite DS being generally shared among domesticated species, a universal set of underlying genetic initiators may not exist and each case of DS may arise from independent mechanisms. Extensive examination of the genes artificially selected by humans across a variety of species and conditions will help to advance the understanding of DS.
Old World camels offer a unique opportunity for studies of domestication because they have maintained relatively high levels of genetic variation, are largely multipurpose, and lack the secondary bottlenecks associated with specific breed development often characteristic of domestic species10,11,12,13. In essence, domestic Old World camels represent features of the “initial stages” of the domestication process, which were primarily focused on the selection for tameness and docility. Of the three extant species of Old World camels, two are domesticated (single-humped dromedaries, Camelus dromedarius, and two-humped Bactrian camels Camelus bactrianus) and one remains wild (two-humped wild camels Camelus ferus). The two-humped camels, C. ferus and C. bactrianus, shared a common ancestor ~1 million years before present (ybp)14, whereas the common ancestor of all three Old World camelid species existed between 4.4 and 7.3 million ybp14,15. Domesticated camels are an essential resource, providing food, labor, commodities, and sport to millions of people. Furthermore, each species possesses a variety of adaptations to harsh desert conditions, including mechanisms to tolerate extreme temperatures, dehydration, and sandy terrain. Recent genomic studies of camels have identified patterns of selection consistent with the aforementioned adaptations15,16, in addition to quantifying genetic variation and examining demographic history15,16,17,18. However, these studies are limited to analyses from a single genome of each species, thus biasing many inferences of selection and adaptation. For example, with a small sample size and closely related species, differences between sequences may not indicate fixation events but rather unobserved segregating polymorphisms; resulting in exaggerated estimates of the Ka/Ks ratio19. Furthermore, draft genomes are susceptible to errors in the estimated number of genes—thereby distorting conclusions of adaptation based upon orthologous genes between species (e.g., Ka/Ks ratio, gene expansion–contraction tests)20.
In this study, we take a genomics approach to inferring both positive selection and demographic history of Old World camelids with an emphasis on genes potentially contributing to the DS phenotype. Considering that the direct wild ancestors of each domestic camelid (C. dromedarius and C. bactrianus) have been extinct for millennia, unlike in most other livestock, we inferred positive selection independently for each domesticated camelid using tests specific for the pattern of relationship between them and their wild counterpart (C. ferus). By re-sequencing multiple genomes from each species, we found evidence for positive selection on genes associated with both hypotheses of DS. These results, along with the extensive genomic resources made available, are an important contribution to understanding both the evolutionary history of camels and the underlying genomic features of their domestication.
Results
Whole genome re-sequencing
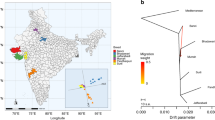
We assembled a collection of 25 Old World camel samples from throughout their range to examine the patterns and distribution of genomic variation (Supplementary Fig. 1 and Supplementary Data 1). Our collection included representatives of all three extant species (Fig. 1a), C. dromedarius (n = 9), C. bactrianus (n = 7), and C. ferus (n = 9). In both domesticated varieties (C. dromedarius and C. bactrianus), we sampled across geographical regions in an attempt to minimize effects of genetic drift8. Using the Illumina HiSeq2000 system, we generated ~36 gb of sequence for each individual (Supplementary Fig. 2 and Supplementary Data 2). An average of 76% (SD 2%) of bases uniquely aligned to the C. ferus reference genome16, resulting in mean genome coverage of 13.9×(SD 1.6×) per sample (Supplementary Data 2). When mapping reads to the C. ferus reference genome, no difference in genome coverage was observed among species (Kruskal–Wallis rank sum test, χ2 = 3.58, df = 2, p value = 0.167), but mean mapping quality varied significantly (Kruskal–Wallis rank sum test, χ2 = 11.35, df = 2, p value = 0.0034) (Table 1). To determine if the difference could be attributed to a larger divergence between C. dromedarius and the other Camelus species15, we repeated the mapping of reads from dromedaries to the C. dromedarius reference genome18 (Table 1). The intraspecific mapping of dromedary reads resulted in a similar mean coverage (12.7× and 12.5× when mapped to C. ferus and C. dromedarius references, respectively) and no difference in mapping quality (Wilcoxon rank sum test, V = 30, p value = 0.43). Mapping quality continued to vary among species (Kruskal–Wallis rank sum test, χ2 = 14.33, df = 2, p value = 0.001) when including the intraspecific dromedary alignments. These results indicated that the relative consequences of mapping our dromedary sequences to either the C. ferus or C. dromedarius reference genomes were of little concern. Previous studies of aligned camelid genomes found a high degree of synteny, including >90% coverage of C. dromedarius when aligned to either C. ferus or C. bactrianus, and concluded a majority of divergence can be attributed to single base mutations and small rearrangements15,18. As a result, subsequent analyses and comparisons across species were performed using dromedary reads mapped to the C. ferus reference, although for completeness, we report results from alignments to both species when calculating parameters within dromedaries.

a Summary of the phylogenetic relationships among Old World camels using the alpaca (Vicugna pacos) as an outgroup. b Results from the population clustering analysis. Each horizontal bar corresponds with the proportion of ancestry in either two or three population clusters. c The first two principal components explain 23.3% and 10.1% of the SNP variation, respectively, and easily differentiate the three camel species. d Euler diagram showing the number of segregating SNPs unique to each species and shared between them. Note that the number of C. bactrianus individuals (n = 7) was less than the number of C. ferus and C. dromedarius individuals (n = 9). Approximately 2.1 million SNPs fixed between species are not included.
Comparison of genetic variation
We identified single nucleotide polymorphisms (SNPs) after a series of recalibrating procedures that are known to improve variant detection21. The ratio of transitions to transversions (Ts/Tv) across all SNPs, a metric commonly used to assess variant quality, was 2.54—suggesting a high-quality set of variants22. Using our conservative approach, we identified ~10.8 million SNPs, of which 8.71 million (80.5%) were polymorphic within species and the remaining were fixed differences between species (monomorphic within a species, but differed between species). The illustration in Fig. 1d shows the number of polymorphic SNPs shared among species. The most segregating SNPs were observed in C. bactrianus (5.2 million), much higher than observed in C. ferus (3.9 million) and C. dromedarius (2.7 million) and despite sampling fewer individuals (n = 7 for C. bactrianus compared with n = 9 for the other two species). Of all the SNPs identified, only 107,662 (1.0%) were segregating within all three species. A large number of SNPs (~2.3 million) were shared between C. bactrianus and C. ferus, whereas much less were shared between C. dromedarius and C. ferus (48,509) than between C. dromedarius and C. bactrianus (520,658). Within C. dromedarius sequences aligned to the intraspecific reference, both Ts/Tv (2.49) and the number of SNPs (2,818,163) were comparable with the interspecific alignment (Table 1). The large number of SNPs shared between the domesticated species is consistent with known introgression events and has been observed in other genomic studies of camels23. Anthropogenic hybridization between the domesticated dromedary and Bactrian camel, especially in central Asia, is a widely practiced tradition of cross-breeding aimed at improving milk production (F1 backcrossed with dromedary), wool and meat yield, cold resistance (F1 backcrossed with Bactrian camel), or for camel wrestling24,25.
The various summary measures of genetic variation across populations are provided in Table 1, along with individual level heterozygosity in Fig. 2a. The mean population mutation rate θ (0.41 × 10−3) and nucleotide diversity π (0.52 × 10−3) were reduced in C. dromedarius relative to the other species and greatest in C. bactrianus (0.85 × 10−3 and 0.88 × 10−3, respectively). These patterns of genetic variation and heterozygosity are consistent with the relative differences observed between single genomes of the same species15,16,17,18. Interestingly, Ming et al.23 reported similar patterns of genetic variation in C. bactrianus (π = 0.95 × 10−3 − 1.1 × 10−3) and C. ferus (π = 0.88 × 10−3 compared with 0.71 × 10−3 in this study), but nearly threefold higher levels in C. dromedarius (π = 1.5 × 10−3). Although this finding by Ming et al. conflicts with our results and that of the previously mentioned studies15,16,17,18, the authors attributed this to the small sample size (n = 4) and being sampled from Iran where hybridization with C. bactrianus is commonplace.

a Boxplots of individual heterozygosity inferred from approximately 190,000 nonoverlapping 10-kb windows across the genome and colored by species. The boxplots’ elements include the median (center line), first through third quartiles (colored box), and 1.5× interquartile range (whiskers). b The demographic history of each individual camel colored by species (see legend). All results were scaled using a generation time of five years and a mutation rate of 1.1 × 10−8 (changes · site−1 · generation−1). The asterisk indicates that C. dromedarius analyses were repeated with mapping to the C. dromedarius reference.
Demographic reconstruction
We inferred historical changes in effective population size (Ne) using the pairwise sequentially Markovian coalescent model (PSMC)26 (Fig. 2b). In dromedaries, the patterns of demographic history were nearly identical when using either C. dromedarius or C. ferus as the reference genome sequence. Also, historical Ne was remarkably similar across individual dromedaries and consistent with previous analyses from single genomes15,18. It appeared that dromedaries suffered a large bottleneck beginning around 700,000 ybp that reduced Ne from nearly 40,000 to 15,000 by 200,000 ybp. The dromedary population further collapsed during and after the last glacial maximum (16,000–26,000 ybp), a finding shared with previous dromedary genomes and Northern Hemisphere mammalian megafauna15,18,27.
Both C. bactrianus and C. ferus shared the same pattern of historical Ne (~25,000) until 1 million ybp, matching the estimated divergence time (1.1 million ybp) reported between these species from mitogenomic sequences14 and further supporting differentiation between these species prior to domestication by humans. Within C. bactrianus, Ne peaked between 25,000 and 40,000 individuals ~400,000 ybp, and has since suffered a long-term decline over the last 50,000 years. The wild camel, on the other hand, experienced a large expansion between 50,000 and 20,000 ybp, but dramatically declined immediately thereafter; also consistent with potential effects of the last glacial maximum as observed in dromedaries. Although we interpreted the PSMC results assuming the demographic history represented changes in Ne of a single population, these patterns can be confounded by past changes in gene flow or population structure, such as potential hybridization between wild camels and the wild ancestor of extant domestic Bactrian camels. However, given that the population expansion and contraction timeline fits generally well with the previously estimated divergence time and the known climatic changes, the main PSMC based conclusions should be relatively robust.
Population clustering
We employed two methods to explore the global, or genome-wide average, ancestry of Old World camels using unlinked SNPs. First, we observed the clustering of individuals using principal components analysis (Fig. 1c). Coinciding with phylogenetic predictions (Fig. 1a), the largest component of variation (23.3%) separated one-humped C. dromedarius from its two-humped congeners, and the second component (10.1%) separated C. ferus from C. bactrianus (Fig. 1c). The remaining components accounted for increasingly less variation (<6%) and tended to separate single individuals (Supplementary Fig. 4). The analysis does suggest a slight potential for introgression between species, notably between a single C. ferus individual and C. bactrianus. The second method employed a Bayesian model-based approach28 to clustering individuals and indicated high support for both two and three genetic clusters that corresponded with each species (Fig. 1b). Similar to the principal components analysis, the results indicated that a small amount (6.0%) of C. dromedarius ancestry is present in a C. bactrianus individual, and larger amount of C. bactrianus ancestry in a C. ferus individual (16.6%). These values are markedly similar to values reported by Ming et al.23, where C. dromedarius ancestry in C. bactrianus ranged between 1 and 10%, and C. bactrianus ancestry in three C. ferus individuals ranged between 7 and 15%. Introgression from the domestic species into both Mongolian and Chinese populations of the Critically Endangered wild camel has been reported elsewhere using mitochondrial DNA29, microsatellites30 and the Y chromosome31, potentially jeopardizing the wild camel’s genomic integrity and evolutionary independence (~1.1 million years)14.
Positive selection in dromedaries
We identified candidate genes under positive selection in dromedaries using a combination of tests based on the ratios of polymorphism to divergence within genes (homogeneity and Hudson–Kreitman–Aguadé tests32) and among genomic windows (low nucleotide diversity [π] and high divergence [DXY]). For the former, we were able to examine 10,297 (57.5%) genes with at least one fixed variant and 84 (0.82%) genes passed our criteria for positive selection (Supplementary Data 3 and 4). Using the latter approach, we identified 17 100-kb genomic windows containing both the lowest 0.5 percentile of π and highest 99.5 percentile of DXY. These windows contained a total of 23 protein-coding genes (Supplementary Data 5 and 6). For both sets of putative positively selected genes, we found no significant enrichment of any category of gene ontology (GO) terms or KEGG pathways after correction for multiple comparisons (Supplementary Data 4 and 6).
Among the 107 genes linked to recent, positive selection, a variety of functions and processes were represented, including numerous neurological and endocrine functions as predicted by the NCCH and THH during domestication. At least TUBGCP6, SYNE1, BPTF, KIDINS220, MYO5A, VPS13B, TBC1D24) are known to contain mutations causing various neuropathies (Supplementary Data 3 and 5). These neuropathies often manifest as features characteristic of DS such as microcephaly, facial dysmorphism, and intellectual disability (e.g., Cohen syndrome caused by a mutation in VPS13B). The genes VPS13B and BPTF, in particular, have also been reported as linked to domestication from genomic scans of chicken33 and dogs9, respectively (Fig. 3a). Four genes were previously identified as under positive selection in both C. dromedarius and C. bactrianus (CENPF, CYSLTR2, HIVEP1) or just in C. dromedarius (CCDC40)15, suggesting either a convergent consequence of domestication, or a more general role in camel adaptation. The latter is more likely, considering that CENPF, CYSLTR2, and CCDC40 are each related to ciliopathies and/or respiratory diseases such as asthma—indicating a possible adaptation to the respiratory challenge posed by dust in highly arid environments15.

The genes under positive selection in a Camelus dromedarius, b C. bactrianus, and c C. ferus were of functions that could be associated with the neural crest cell (NCC; blue) and thyroid hormone (pink) hypotheses of domestication syndrome, or of other, unknown relationship (green). Genes are shown with an icon of the corresponding domestic species where positive selection has also been reported. Arrows are directed toward a summary icon of known functions for that gene.
Several other genes (SSH2, CABIN1, NOS1, NEO1, INSC, EXOC3) were also known to have important functional and/or developmental roles in the neural system. For example, SSH2, which is also linked with domestication in dogs9, is a phosphatase critical for neurite extension34. In the mammalian brain, the gene NOS1 is an important neurotransmitter35 and EXOC3 is active in neurotransmitter release36. Both genes CABIN1 and NEO1 are critical during development for the proper migration of neural crest cells37,38—a key prediction of the NCCH3. Of special interest is the gene ATRN, whose pleiotropic effects include pigmentation phenotypes (e.g., the mahogany phenotype in mice)39 and a crucial role in the proper myelination of the central nervous system40. ATRN has also been reported as under positive selection during yak domestication41 and during long-term experimental selection for tameness in foxes42.
Among the candidate positively selected genes, several potentially associated with the THH were identified (PDPK1, PLCD3, CCNF, GFRA4). Both PDPK1 and PLCD3 are components of the thyroid hormone signaling pathway (KEGG pathway ko04919), and PDPK1 expression is known to be increased in follicular cell thyroid carcinoma in dogs43. The gene CCNF, which in dromedaries contained two, fixed non-synonymous substitutions, is known to be regulated by thyroid hormone during development44. In addition, CCNF participates in the ubiquitination and targeting of certain proteins for degradation and has been linked to neuronal degeneration disorders such as congenital amyotrophic lateral sclerosis45. The GFRA4 is not only important from a neurological perspective as it binds neurotrophic factors in the GDNF/RET signaling pathway, but also the expression and splicing of GFRA4 has been linked to endocrine cell development, including the thyroid where its expression is localized in adult humans46.
Positive selection in domestic Bactrian camels
In C. bactrianus, it was possible to perform the homogeneity test above in only 90 genes because the close relationship with C. ferus resulted in very few fixed differences within genes thus reducing the power of the test. Of these 90 genes, none showed evidence of positive selection in C. bactrianus. To mitigate this issue, we subsequently tested for excessive allele frequency divergence in C. bactrianus relative to C. ferus and using C. dromedarius as an outgroup. This test, known as the population branch statistic (PBS)47, produced 39 windows that passed our criteria for positive selection and overlapped ten protein-coding genes (Supplementary Data 7). In addition, as performed for dromedaries, we identified two 100-kb windows with excess DXY and a dearth of π. No protein-coding genes were found in these regions. Although the ten putative positively selected genes were not enriched for any specific GO functions (Supplementary Data 8) or KEGG pathways, several genes were promising candidates for associations with camel domestication. For example, the histone demethylase KDM1A regulates global DNA methylation and the expression of many genes via chromatin remodeling48. Like several of the genes found in dromedaries, defects in KDM1A cause craniofacial disorders and psychomotor retardation49—again, signature features of DS. Furthermore, KDM1A is required for pituitary organogenesis50, and stress hormone activity regulated by the hypothalamic–pituitary–adrenal axis is also tightly correlated with both thyroid hormone activity51 and tameness in foxes52. The genes LUZP1 and NLK both function in neural development. Mouse knockouts of LUZP1 develop neural tube closure defects in the embryonic brain53, and NLK, also selected in domestic chickens33 (Fig. 3b), acts as part of the noncanonical Wnt/Ca2+ pathway to inhibit canonical Wnt/ß-catenin and control the migration of neural crest cells54,55.
Relaxed selection relative to wild two-humped camels
In wild camels we undertook a different approach to identify genomic regions that signify relaxed selection in the domestic species. These windows had an excessive π log-ratio \(\left( {\ln \pi _{domestic} - \ln \pi _{wild}} \right)\) in both domestic species (99.5 percentile) and a substantially negative measure of Tajima’s D (D ≤ −2) in C. ferus. Three 100-kb windows passed these conservative criteria and one of the windows contained two protein-coding genes, SPR and EXOC6B (Fig. 4; Supplementary Data 9). A region containing both these genes has also been described as differentiating between tame and aggressive foxes42 (Fig. 3c). In humans, deficiencies in SPR cause dystonia—uncontrollable muscular contractions—, psychomotor retardation, and progressive neurologic deterioration56. The gene EXOC6B is part of the exocyst complex, which is critical for cellular trafficking, and mutations in EXOC6B have been associated with intellectual disability, language delay, hyperactivity, ear malformations, and craniofacial abnormalities in humans57.

The three 100-kb windows in Camelus ferus (a–c) found to contain evidence of positive selection. The location of variants within each species are shown as vertical lines, and overlapping genes as black bars.
Discussion
Old World camels represent an interesting example in understanding the genetic impacts of domestication. Camel breeders have aimed to retain a high degree of phenotypic diversity in their herds and generally avoided selection at the level of individual animals, with the exception of traits for tameness and tolerance of humans10. Without the secondary bottlenecks associated with specific breed formation, camels thus represent an initial stage in the domestication process. Our genomic scans for selection in two domesticated camel species identified candidate genes whose functions were consistent with many features of DS (e.g., neotenization, intellectual disability, neuropathies). More specifically, we found evidence of selection in genes that were associated with both the NCCH and THH, and shown to be under selection during the domestication of other species (i.e., chicken, yak, dog, fox, rabbit). These results prompted two important conclusions. First, the results supported that the NCCH and THH need not be mutually exclusive—the pathways are not completely independent and their relative contributions can vary across domestication events in space and time5. Second, the results supported that a shared set of domestication genes between camels does not exist, even across independent domestication processes of two evolutionary close species (divergence time ~4.4–7.3 mya14,15). It is possible that because the direct wild ancestors of each domestic camel are extinct, our tests for selection may recover positive selection occurring prior to domestication. Although we cannot completely exclude this possibility, we chose tests for selection that detect more recent events based upon comparisons of polymorphism to divergence, and we did identify genes associated with domestication in camels that overlapped those described in other domestic species. Future studies targeting the analysis of insertion/deletion (indel) polymorphisms may also be useful for identifying additional targets of selection. Indel polymorphisms were omitted from our analyses because their accurate genotyping remains quite challenging, especially without high coverage (≥60×58) and in non-model species with incomplete, or draft genome assemblies. With regards to domestication selection, a specific, universal set of domestication genes may not exist, but we do speculate that there may be a ‘core’ set of genes shared across multiple domestication processes. This core set of genes, however, is small compared with the set of ‘pan’-domestication genes (the sum of all genes selected across all domestication events). Nonetheless, the wealth of genomic data across domesticated animals suggests that future meta-analyses are warranted to determine the components of this core and pan-domestication genome.
Patterns of demographic history across all three species demonstrated widespread population declines during the late Pleistocene. Although the exact reason for these declines is unclear, it is consistent with declines and extinctions in other megafauna as a result of either climatic changes or human persecution. Dromedaries, in particular, have experienced a long-term, exceptional decline (possibly to as few as six maternal lineages13) reducing their nucleotide diversity to nearly half that of the other species. Wild camels, despite having more genetic variation than dromedaries, remain one of the most critically endangered of all mammals and are at substantial risk of extinction resulting from their continued population decline. Our study contributes a large set of genomic resources for Old World camels. These resources, combined with the existing and ongoing development of other resources (e.g., chromosome-level assemblies, SNP chip), will aid in the prudent application of breeding and selection schemes to conserve and manage the genomic diversity of camels. In turn, these efforts will preserve the evolutionary potential of the wild species in addition to the promise and sustainability of domestic camels as a valuable livestock in arid environments.
Methods
Sample collection and sequencing
We collected EDTA-preserved blood from 25 Old World camels including nine dromedaries (C. dromedarius), seven domestic Bactrian camels (C. bactrianus), and nine wild camels (C. ferus) (Supplementary Data 1 and Supplementary Fig. 1; see Ethics statement below). We included domesticated camels that represent a variety of geographic locations and/or ‘breeds’ according to information supplied from the camels’ owners, although microsatellite evidence for both dromedaries and domestic Bactrian camels suggest little genetic differentiation among breeds11,12,13. In dromedaries, moderate differentiation exists between camels from northwest Africa (e.g., Canary Islands and Algeria), the Horn of Africa (e.g., Ethiopia, Somalia, Kenya), and their remaining range including northeast Africa, the Middle East, and Pakistan11,13. We extracted DNA using the Master PureTM DNA purification kit for blood (Epicentre version III) and generated a 500 bp paired-end library for each sample. We sequenced each library with a single lane of an Illumina HiSeq (Illumina, USA) according to standard protocols. Due to the sampling procedure, which included a wild endangered species, it was not possible to retrieve tissue samples that would facilitate expression studies and/or functional analyses. A follow-up project will consider this next analytical step.
Read processing and alignments
We trimmed the 3′ end of sequence reads to a minimum phred-scaled base quality score of 20 (probability of error <1.0%) and excluded trimmed reads <50 bp in length using POPOOLATION v1.2.259. We aligned all processed reads to the C. ferus CB1 reference genome (Genbank accession: GCA_000311805.2) using BWA v0.6.260 with parameters ‘-n 0.01 -o 1 -e 12 -d 12 -l 32’. We removed duplicate reads and filtered alignments to only include reads that are properly paired and unambiguously mapped with a mapping quality score >20. We realigned reads around insertions/deletions and performed a base quality score recalibration using the Genome Analysis Toolkit (GATK) v3.1-1 following guidelines presented by Van der Auwera et al.21. As input into the base quality score recalibration step we generated a stringently filtered set of SNPs using the overlap of three different variant-calling algorithms (SAMTOOLS v1.161; GATK HAPLOTYPECALLER v3.1-121; ANGSD v0.56362). The overlapping SNPs were filtered to exclude those with a quality score (Q) < 20, depth of coverage (DP) > 750 (~30×/individual), quality by depth (QD) < 2.0, strand bias (FS) > 60.0, mapping quality (MQ) < 40.0, inbreeding coefficient < −0.8, mapping quality rank sum test (MQRankSum) < −12.5, and read position bias (ReadPosRankSum) < −8.0. Furthermore, we excluded SNPs if three or more were found within a 20-bp window, were within 10 bp of an insertion/deletion, or were found in an annotated repetitive region.
Identification of sex chromosome-linked scaffolds
We identified scaffolds from the reference genome that can putatively be assigned to the sex chromosomes (the reference genome was male) (Supplementary Fig. 3). This was a necessary step in order to remove variants from downstream analyses that require accurate estimates of allele frequencies assuming diploid samples (our samples consisted of both the homogametic and heterogametic sexes). We first aligned all scaffolds to the cattle X and Y assemblies (UMD3.1 and Btau4.6.1 assemblies, respectively) using LASTZ v1.02.0063 with parameters ‘--step=1 --gapped --chain --inner=2000 --ydrop=3400 --gappedthresh=6000 --hspthresh=2200 --seed=12of19 --notransition’. For each scaffold with high-scoring alignments, we calculated the ratio of the scaffold coverage to the genome-wide mean coverage in each individual. To assign scaffolds to the X chromosome, we identified scaffolds whose coverage ratio in males was significantly less than the ratio in females using a Wilcoxon Rank Sum test (p < 0.05) and whose total alignment length was ≥20% of the total scaffold length. For the Y chromosome, we identified scaffolds whose coverage ratio did not differ significantly from 0.5 in males and was significantly less than 0.5 in females using a Wilcoxon Rank Sum test (p < 0.05) and whose total alignment length was ≥20% of the total scaffold length.
Variant identification
We generated another set of SNPs from the realigned and recalibrated alignment files using the GATK HAPLOTYPECALLER and filtering criteria as described above. We further excluded SNPs on scaffolds putatively assigned to the X and Y chromosome (see ‘Identification of Sex Chromosomes’ below), with a minimum allele count <2, missing a genotype in more than five individuals, with 4 > DP > 30 per genotype, and deviating from Hardy-Weinberg equilibrium (p < 0.0001) in VCFTOOLS v0.1.12b64. We used this set of SNPs as a training set to perform variant quality score recalibration in GATK, assigning a probability of error to the training set of 0.1. This recalibration develops a Gaussian mixture model across the various annotations in the high-quality training dataset then applies the model to all variants in the initial dataset. After variant recalibration, we excluded all SNPs with VQSLOD score outside the range containing 95% of the SNPs in the training set. We hard-filtered any remaining variants missing genotypes in more than five individuals. Variants on the X and Y putative scaffolds were excluded from all analyses except for gene-based analyses described below (See Homogeneity and HKA tests below).
We assessed the quality of the final set of SNPs by calculating the ratio of transitions to transversions (Ti/Tv ratio) in VCFTOOLS. The Ti/Tv ratio is often used as a diagnostic parameter to examine the quality of SNP identification22. When substitution is random Ti/Tv = 0.5 because there are twice as many transversions possible as transitions. However, in humans the genome-wide Ti/Tv is ~2.0–2.2, so values much less than this are indicative of an excess of false-positive SNPs22. Estimates of genetic variation (i.e. π, θ, and heterozygosity) and Tajima’s D were averaged across nonoverlapping 10-kb windows using VCFTOOLS and excluding SNPs on the putative X and Y scaffolds.
Population clustering
To infer population clustering, we used SNPRELATE 1.10.165 to calculate linkage disequilibrium between pairs on SNPs within a 1-Mb sliding window and randomly removed one locus from each pair with a correlation coefficient (r2) >0.5. The resulting dataset contained 90,918 unlinked SNPs. We used the unlinked SNPs to examine the global ancestry of Old World camels using the principal components analysis method in SNPRELATE and a Bayesian model-based approach implemented in ADMIXTURE v1.2328. We restricted the Bayesian analysis to the maximum likelihood estimation of individual ancestry proportions (Q values) in three ancestral populations. Likelihood searches were terminated for each point estimate when the log likelihood increased by less than 0.0001 between iterations (parameters: -C 0.0001 -c 0.0001).
Demographic inference
We used PSMC (v0.6.426) to examine the demographic history of the three camelid species. The PSMC model infers the historical effective population size (Ne) from a single diploid genome by examining the distribution of coalescent rates across the genome. Because the coalescent rates across the genome are dependent upon the density of polymorphic sites, we employed a strict set of conditions as described previously by our group18. Briefly, for each individual we first constructed a genome sequence by applying the individual-specific alleles from our final set of SNPs to the C. ferus reference genome. Furthermore, we masked all repetitive regions and putative X and Y contigs from the analysis. We ran PSMC for a total of 25 iterations using the parameters ‘-t15 -r5 -p “4 + 25 × 2 + 4 + 6”’ and verified that ~10 recombination events occurred in the final set of intervals spanned by each parameter26. We ran 100 bootstrap replicates to assess the variance in the final inference of Ne. For C. dromedarius, we repeated the PSMC analysis as described above using the intraspecific reference. We scaled the final results using a generation time of five years and mutation rate of 1.1 × 10−8.
Signals of positive selection
We employed multiple approaches to identify candidate genes under positive selection in domesticated camel species. First, we used a gene-based approach that combined the homogeneity test and the HKA test32. The homogeneity test examines the intraspecific (polymorphism) and interspecific (divergence) genetic diversity, which are expected to be correlated under neutral evolution. Under positive selection, the amount of polymorphism is expected to be reduced along one branch. To perform the test in dromedaries, we calculated four values for each of the 17,912 protein-coding genes (longest isoform per gene) annotated in the camel genome:
-
(A)
Number of polymorphic sites in the dromedary samples.
-
(B)
Number of polymorphic sites in the wild camel samples.
-
(C)
Number of fixed differences between dromedaries and both wild camels and the alpaca genome sequence.
-
(D)
Number of fixed differences between wild camels and both dromedaries and the alpaca genome sequence.
We then tested the null hypothesis that \(\frac{A}{C} = \frac{B}{D} \equiv \frac{A}{B} = \frac{C}{D}\) using a Fisher exact test for a 2 × 2 contingency table. We omitted any genes with either A or C < 1. Alpaca alleles were identified by mapping all short-insert, paired-end sequencing reads from the alpaca genome assembly15 (BioProject accession PRJNA233565) to the camel reference genome as described above. Then, for each camel SNP location, we selected the most common allele (minimum depth of two) from the aligned alpaca reads. If multiple bases occurred at equal frequency, one was randomly selected. Next, we performed the HKA test by comparing ratio \(\frac{A}{C}\) for each gene to the ratio \(\frac{A}{C}\) summed across all genes analyzed \(\left( {\frac{A}{C}\left[ {gene} \right] = \frac{A}{C}\left[ {genome} \right]} \right)\) using a Fisher exact test. For the final set of putative positively selected genes, we retained those with a homogeneity test P < 0.05 and with a significant HKA test score (P < 0.05) only in the dromedary population. As suggested by Liu et al.32 the P values obtained from these tests can be misinterpreted since accurate P values can be only be obtained from simulations, but can be informative when combined with other ranking criteria. In conjunction with the recommendation by Liu et al. we emphasize that these genes are in ranked order of priority, or evidence, rather than of statistically significant effect. The above procedure was repeated for domestic Bactrian camels using the wild camels again for comparison.
The second approach to identify putative positively selected genes utilized a window-based approach. We calculated nucleotide diversity (π) within and divergence (DXY) between camelid species across 100-kb sliding windows with a step size of 50 kb using the popgenWindows.py script (https://github.com/simonhmartin/genomics_general). Only windows with at least ten polymorphic sites were included. Next, we defined candidate positively selected regions in dromedaries and domestic Bactrian camels as windows in both the lowest 0.5 percentile in π within species and the highest 99.5 percentile in DXY relative to the wild camel population. Within domestic Bactrian camels only, we calculated the PBS47. The PBS is a powerful method to detect both complete and incomplete selective sweeps over relatively short divergence times assuming two populations and an outgroup47—making this test applicable for domestic Bactrian camels. Using the windows defined above, we calculated Reynold’s FST for the three population pairs and converted them to divergence times scaled by NE using the Cavalli-Sforza transformation \(T = - \log \left( {1 - F_{ST}} \right)\) 47. The PBS was subsequently obtained from \(PBS = \frac{{T_1 + T_2 - T_3}}{2}\), where \(T_1\) is from the domestic Bactrian vs wild camel comparison, \(T_2\) is from the domestic Bactrian vs dromedary comparison, and \(T_3\) is from the wild camel vs. dromedary comparison. Windows in the top 99.5 percentile of PBS values were retained as positively selected. In C. ferus, we calculated the π log-ratio with both domestic species \(\left( {\ln \pi _{domestic} - \ln \pi _{wild}} \right)\)41 to identity windows with a relatively low level of polymorphism. As above, windows were 100 kb in length with a step size of 50 kb, and only windows with at least ten polymorphic sites across all species were retained. If a window contained no heterozygous sites within a species (π = 0), then a value less than the minimum across polymorphic windows was used (π = 10−5) to avoid logarithmic errors. In addition, in C. ferus we calculated Tajima’s D in each window. As a conservative estimate of regions undergoing positive selection in C. ferus and/or relaxed selection in the domestic species, we retained a final set of windows with an excessive π log-ratio in both domestic species (99.5 percentile) and a substantially negative measure of Tajima’s D (D ≤ −2) in C. ferus. For all window-based analyses, protein-coding genes that overlap these windows were identified.
Functional enrichment
We assigned GO terms to all annotated protein-coding genes in the C. ferus reference genome using BLAST2GO v3.0.866. BLASTP v2.2.30 (http://ncbi.nlm.nih.gov/blast) searches were conducted against metazoan protein sequences from the ‘nr’ database with an e value cut-off of 10−3 and retaining only the top 20 hits. We tested for functional enrichment of GO terms using topGO v2.28.067 with a classic Fisher exact test and minimum annotation count of five for each GO term across the full annotation set. We also tested each set of putative positively selected genes for overrepresentation of KEGG pathways using WEBGESTALT68. In WEBGESTALT, we used the Bos taurus KEGG annotations of protein-coding genes as a reference set and otherwise default parameters. In all analyses we corrected for multiple testing using a false discovery rate <0.05. Additional functional information for each protein-coding can be found in Supplementary Data 10.
Statistics and reproducibility
Summary statistics and tests were calculated using R v3.6.2. The tests included the nonparametric Wilcoxon rank sum and Kruskal–Wallis rank sum tests for comparing mapping results within and between C. dromedarius (n = 9), C. bactrianus (n = 7), and C. ferus (n = 9). The full test results are reported in the Results section. Contingency table (2 × 2) testing (homogeneity and HKA tests) was also performed in R using the Fisher exact test, and although are reported in Supplementary Data 3, P values were only used for relative prioritization rather than assessing statistical significance.
Ethics statement
The blood samples for each camelid species were retrieved during routine veterinary procedure, micro-chipping, or radio-collaring of Mongolian wild camels. All domestic and wild Bactrian camel samples were collected within the framework of the legal requirements of both Austria and Mongolia. Micro-chipping of wild camels from the breeding center of the Wild Camel Protection Foundation was performed with the request and consent of the foundation (John Hare, personal communication). Capture and collaring of wild camels within the Great Gobi Strictly Protected Area “A” was conducted within a cooperation agreement between the International Takhi Group and the Mongolian Ministry of Nature, Environment and Tourism signed on 15.02.2001 and renewed on 27.01.2011.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Raw and mapped sequencing data for the 25 genomes in the study are available in GenBank (BioProject accession PRJNA276064), and additional data have been deposited in Dryad (https://doi.org/10.5061/dryad.prr4xgxj2).
Code availability
Computer code and scripts for the various analyses are available at GitHub (https://github.com/rfitak/Camel_Genomics).
References
Vigne, J.-D. The origins of animal domestication and husbandry: a major change in the history of humanity and the biosphere. C. R. Biol. 334, 171–181 (2011).
Jensen, P. Behavior genetics and the domestication of animals. Annu. Rev. Anim. Biosci. 2, 85–104 (2014).
Wilkins, A. S., Wrangham, R. W. & Fitch, W. T. The “Domestication syndrome” in mammals: a unified explanation based on neural crest cell behavior and genetics. Genetics 197, 795–808 (2014).
Crockford, S. J. in Human evolution through developmental change (eds. Minugh-Purvis N. & McNamara K. J.) 122–153 (Johns Hopkins University Press, 2002).
Wilkins, A. S. Revisiting two hypotheses on the “domestication syndrome” in light of genomic data. Vavilov J. Genet. Breed. 21, 435–442 (2017).
Karlsson, A.-C. et al. A domestication related mutation in the thyroid stimulating hormone receptor gene (TSHR) modulates photoperiodic response and reproduction in chickens. Gen. Comp. Endocrinol. 228, 69–78 (2016).
Librado, P. et al. Ancient genomic changes associated with domestication of the horse. Science 356, 442–445 (2017).
Montague, M. J. et al. Comparative analysis of the domestic cat genome reveals genetic signatures underlying feline biology and domestication. Proc. Natl Acad. Sci. USA 111, 17230–17235 (2014).
Pendleton, A. L. et al. Comparison of village dog and wolf genomes highlights the role of the neural crest in dog domestication. BMC Biol. 16, 64 (2018).
Abdussamad, A. M., Charruau, R., Kalla, D. J. U. & Burger, P. A. Validating local knowledge on camels: colour phenotypes and genetic variation of dromedaries in the Nigeria-Niger corridor. Livest. Sci. 181, 131–136 (2015).
Mburu, D. N. et al. Genetic diversity and relationships of indigenous Kenyan camel (Camelus dromedarius) populations: implications for their classification. Anim. Genet. 34, 26–32 (2003).
Chuluunbat, B., Charruau, P., Silbermayr, K., Khorloojav, T. & Burger, P. A. Genetic diversity and population structure of Mongolian domestic Bactrian camels (Camelus bactrianus). Anim. Genet. 45, 550–558 (2014).
Almathen, F. et al. Ancient and modern DNA reveal dynamics of domestication and cross-continental dispersal of the dromedary. Proc. Natl Acad. Sci. USA 113, 6707–6712 (2016).
Mohandesan, E. et al. Mitogenome sequencing in the genus Camelus reveals evidence for purifying selection and long-term divergence between wild and domestic Bactrian camels. Sci. Rep. 7, 9970 (2017).
Wu, H. et al. Camelid genomes reveal evolution and adaptation to desert environments. Nat. Commun. 5, 5188 (2014).
Jirimutu et al. Genome sequences of wild and domestic bactrian camels. Nat. Commun. 3, 1202 (2012).
Burger, P. A. & Palmieri, N. Estimating the population mutation rate from a de novo assembled Bactrian camel genome and cross-species comparison with dromedary ESTs. J. Hered. 105, 839–846 (2014).
Fitak, R. R., Mohandesan, E., Corander, J. & Burger, P. A. The de novo genome assembly and annotation of a female domestic dromedary of North African origin. Mol. Ecol. Res 16, 314–324 (2016).
Kryazhimskiy, S. & Plotkin, J. B. The population genetics of dN/dS. PLoS Genet. 4, e1000304 (2008).
Denton, J. F. et al. Extensive error in the number of genes inferred from draft genome assemblies. PLoS Comput. Biol. 10, e1003998 (2014).
Van der Auwera, G. A. et al. From FastQ data to high-confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 11.10.11–33 (2013).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Ming, L. et al. Whole-genome sequencing of 128 camels across Asia reveals origin and migration of domestic Bactrian camels. Commun. Biol. 3, 1 (2020).
Faye, B & Konuspayeva, G. In Camels in Asia and North Africa: Interdisciplinary perspectives on their past and present significance (eds. Knoll, E. & Burger, P. A.) 27–33 (Austrian Academy of Sciences Press, 2012).
Çalişkan, V. Examining cultural tourism attractions for foreign visitors: the case of camel wrestling in Selçuk (Ephesus). Turizam 14, 22–40 (2010).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011).
Lorenzen, E. D. et al. Species-specific responses of Late Quaternary megafauna to climate and humans. Nature 479, 359–364 (2011).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Silbermayr, K. et al. High mitochondrial differentiation levels between wild and domestic Bactrian camels: a basis for rapid detection of maternal hybridization. Anim. Genet 41, 315–318 (2010).
Silbermayr, K. & Burger, P. A. in Camels in Asia and North Africa: Interdisciplinary perspectives on their past and present significance (eds Knoll E. & Burger P. A.) 69–76. (Austrian Academy of Sciences Press, 2012).
Felkel, S. et al. A first Y-chromosomal haplotype network to investigate male-driven population dynamics in domestic and wild Bactrian camels. Front. Genet 10, 423 (2019).
Liu, S. et al. Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 157, 785–794 (2014).
Rubin, C. J. et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464, 587–591 (2010).
Endo, M., Ohashi, K. & Mizuno, K. LIM kinase and slingshot are critical for neurite extension. J. Biol. Chem. 282, 13692–13702 (2007).
Kano, T., Shimizu-Sasamata, M., Huang, P. L., Moskowitz, M. A. & Lo, E. H. Effects of nitric oxide synthase gene knockout on neurotransmitter release in vivo. Neuroscience 86, 695–699 (1998).
Hsu, S. C. et al. The mammalian brain rsec6/8 complex. Neuron 17, 1209–1219 (1996).
Villanueva, A. A. et al. The Netrin-4/Neogenin-1 axis promotes neuroblastoma cell survival and migration. Oncotarget 8, 9767–9782 (2017).
Weinberger, D. R. Reverse genetic analysis of zebrafish development: requirements for CABIN1 in the nervous system and neural crest Doctor of Philosophy thesis. (University of Wisconsin-Milwaukee, 2012).
Nan, H., Kraft, P., Hunter, D. J. & Han, J. Genetic variants in pigmentation genes, pigmentary phenotypes, and risk of skin cancer in Caucasians. Int. J. Cancer 125, 909–917 (2009).
Kuramoto, T. et al. Attractin/mahogany/zitter plays a critical role in myelination of the central nervous system. Proc. Natl Acad. Sci. USA 98, 559–564 (2001).
Qiu, Q. et al. Yak whole-genome resequencing reveals domestication signatures and prehistoric population expansions. Nat. Commun. 6, 10283 (2015).
Kukekova, A. V. et al. Red fox genome assembly identifies genomic regions associated with tame and aggressive behaviours. Nat. Ecol. Evol. 2, 1479–1491 (2018).
Campos, M. et al. Upregulation of the PI3K/Akt pathway in the tumorigenesis of canine thyroid carcinoma. J. Vet. Itern. Med. 28, 1814–1823 (2014).
Das, B., Heimeier, R. A., Buchholz, D. R. & Shi, Y. B. Identification of direct thyroid hormone response genes reveals the earliest gene regulation programs during frog metamorphosis. J. Biol. Chem. 284, 34167–34178 (2009).
Galper, J. et al. Cyclin F: a component of an E3 ubiquitin ligase complex with roles in neurodegeneration and cancer. Int. J. Biochem. Cell Biol. 89, 216–220 (2017).
Lindahl, M., Timmusk, T., Rossi, J., Saarma, M. & Airaksinen, M. S. Expression and alternative splicing of mouse Gfra4 suggest roles in endocrine cell development. Mol. Cell. Neurosci. 15, 522–533 (2000).
Yi, X. et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78 (2010).
Wang, J. et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 41, 125–129 (2009).
Pilotto, S. et al. LSD1/KDM1A mutations associated to a newly described form of intellectual disability impair demethylase activity and binding to transcription factors. Hum. Mol. Genet. 25, 2578–2587 (2016).
Wang, J. et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446, 882–887 (2007).
Helmreich, D. L., Parfitt, D. B., Lu, X. Y., Akil, H. & Watson, S. J. Relation between the hypothalamic-pituitary-thyroid (HPT) axis and the hypothalamic-pituitary-adrenal (HPA) axis during repeated stress. Neuroendocrinology 81, 183–192 (2005).
Hekman, J. P. et al. Anterior pituitary transcriptome suggests differences in ACTH release in tame and aggressive foxes. G3 8, 859 (2018).
Hsu, C. Y. et al. LUZP deficiency affects neural tube closure during brain development. Biochem. Biophys. Res. Commun. 376, 466–471 (2008).
De Calisto, J., Araya, C., Marchant, L., Riaz, C. F. & Mayor, R. Essential role of non-canonical Wnt signalling in neural crest migration. Development 132, 2587–2597 (2005).
Ishitani, T. et al. The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol. Cell. Biol. 23, 131–139 (2003).
Bonafé, L., Thöny, B., Penzien, J. M., Czarnecki, B. & Blau, N. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Am. J. Hum. Genet. 69, 269–277 (2001).
Wen, J. et al. Phenotypic and functional consequences of haploinsufficiency of genes from exocyst and retinoic acid pathway due to a recurrent microdeletion of 2p13.2. Orphanet J. Rare Dis. 8, 100 (2013).
Fang, H. et al. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med 6, 89–89 (2014).
Kofler, R. et al. PoPoolation: a toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS ONE 6, e15925 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: analysis of next generation sequencing data. BMC Bioinforma. 15, 356 (2014).
Harris, R. S. Improved pairwise alignment of genomic DNA Ph.D thesis. (The Pennsylvania State University, 2007).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Alexa, A. & Rahnenfuhrer, J. topGO: Enrichment analysis for gene ontology. R package version 2.28.0 (2016).
Liao, Y., Wang, J., Jaehnig, E. J., Shi, Z. & Zhang, B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 47, W199–W205 (2019).
Acknowledgements
We thank all camel owners for dedicating aliquots of commensally collected samples for research purpose, especially J. Hare and K. Rae from the Wild Camel Protection Foundation, G. Gassner (Austria), J. Burgsteiner (Austria), A. and J. Perret (Kenya) and R. Saleh (Syria) for facilitating sample collection and for their continuous support. We thank the CSC—IT Center for Science, Finland, for generous computational resources. PAB acknowledges support by the Austrian Science Fund (FWF) project grants P24706-B25 and P29623-B25.
Author information
Authors and Affiliations
Contributions
R.R.F wrote the paper and performed bioinformatic analyses. E.M. extracted DNA, performed bioinformatic analyses, and revised the paper. J.C. provided the extensive computational resources necessary for the completion of the project and revised the paper. A.Y, B.C., O.A., A.R., P.N., C.W., and B.F. facilitated commensal sample collection and revised the paper. P.A.B. managed the project, carried out initial raw data analysis and wrote parts of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fitak, R.R., Mohandesan, E., Corander, J. et al. Genomic signatures of domestication in Old World camels. Commun Biol 3, 316 (2020). https://doi.org/10.1038/s42003-020-1039-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-020-1039-5
This article is cited by
-
Phenotype transition from wild mouflon to domestic sheep
Genetics Selection Evolution (2024)
-
Worldwide Late Pleistocene and Early Holocene population declines in extant megafauna are associated with Homo sapiens expansion rather than climate change
Nature Communications (2023)
-
Characterization and genetic diversity of MHC class II DRB genes in the Arabian camel (Camelus dromedarius)
Immunogenetics (2023)
-
Behavioral responses of wild animals to anthropogenic change: insights from domestication
Behavioral Ecology and Sociobiology (2022)
-
Nucleotide diversity of functionally different groups of immune response genes in Old World camels based on newly annotated and reference-guided assemblies
BMC Genomics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
