
Abstract
Differentiation of osteoclasts (OCs) from hematopoietic cells requires cellular interaction with osteoblasts (OBs). Due to the difficulty of live-imaging in the bone, however, the cellular and molecular mechanisms underlying intercellular communication involved in OC differentiation are still elusive. Here, we develop a fracture healing model using the scale of trap:GFP; osterix:mCherry transgenic zebrafish to visualize the interaction between OCs and OBs. Transplantation assays followed by flow cytometric analysis reveal that most trap:GFPhigh OCs in the fractured scale are detected in the osterix:mCherry+ fraction because of uptake of OB-derived extracellular vesicles (EVs). In vivo live-imaging shows that immature OCs actively interact with osterix:mCherry+ OBs and engulf EVs prior to convergence at the fracture site. In vitro cell culture assays show that OB-derived EVs promote OC differentiation via Rankl signaling. Collectively, these data suggest that EV-mediated intercellular communication with OBs plays an important role in the differentiation of OCs in bone tissue.
Similar content being viewed by others

Introduction
Over the past few decades, the zebrafish and medaka have emerged as attractive genetic models in a variety of research fields due to their many unique advantages, including external development, large offspring numbers, transparency of embryos, short generation time, and availability of transgenic and mutant lines. In the field of bone research, some transgenic lines have been generated to visualize osteoclasts (OCs) and osteoblasts (OBs) in zebrafish and medaka1,2. The zebrafish scale is a thin membranous bone embedded in the skin that consists of OBs, OCs, and bone matrix. Although the structure of zebrafish scales is rather simple compared with mammalian bones3,4,5, various in vivo and in vitro studies have demonstrated that OCs and OBs in teleost scales respond to hormones and other substances as one would predict from observation of the mammalian bone6,7,8,9,10,11,12, indicating that fundamental cellular and molecular programs that regulate OCs and OBs are highly conserved amongst vertebrates. In addition, this “surface” bone tissue enables in vivo live-imaging of OCs and OBs using fluorescent transgenic lines.
OCs differentiate from hematopoietic stem cells and fuse to become mature multinucleated OCs, which can resorb bone via secretion of hydrochloric acid13,14. In contrast, OBs arise from mesenchymal stem cells and can produce bone matrix in response to a wide variety of growth factors15. OBs also regulate the formation and activity of OCs via signaling molecules16, highlighting the importance of cell–cell communication in osteoclastogenesis.
There are two well-documented modes of intercellular communication in OC differentiation. The first mode is through direct contact between OBs and OC precursors, allowing membrane-bound ligands and receptors to interact and initiate intercellular signaling (e.g. receptor activator of nuclear factor kappa B ligand (RANKL)-RANK signaling). The second mode is dependent on diffusible paracrine factors, including cytokines secreted by OBs and acting on OC precursors via diffusion (e.g. macrophage-colony stimulating factor (M-CSF))17,18,19. Recent studies indicate that OB-derived extracellular vesicles (EVs) can be considered a third mode of communication in OC differentiation. Two groups demonstrated that EVs shed from OBs contain RANKL proteins and can transfer signals to OC precursors through the cell-surface receptor RANK, leading to the formation of OCs20,21.The molecular composition of EVs is thought to be strictly regulated in the releasing cell by the external stimuli22,23. Multiple studies have consistently demonstrated that EVs transfer proteins, lipids, and RNAs between various cell types, thus mediating intercellular communication24,25. It is also reported that OC-derived EVs promote OB differentiation or inhibit osteoclastogenesis26,27. Due to the difficulty of live-imaging in the bone, however, it is still unknown if immature OCs actually obtain OB-derived EVs in vivo to promote their differentiation.
In the present study, we generated a double transgenic zebrafish, trap:GFP; osterix:mCherry, which labels OCs and OBs with GFP and mCherry, respectively. Combined with intubation anesthesia, it is possible to visualize and trace the dynamics of OCs and OBs during the fracture-healing process in the scale. Taking advantage of this system, we uncovered that immature OCs engulf OB-derived EVs under fracture stress, leading to the differentiation of OCs.
Results
Convergence and fusion of OCs in the fractured scale
To visualize OCs and OBs in the zebrafish scale, we generated a double-transgenic zebrafish, trap:GFP; osterix:mCherry, which expresses GFP and mCherry under the control of the OC-specific trap (tartrate-resistant acid phosphatase, also known as acid phosphatase 5a, tartrate resistant (acp5a)) and OB-specific osterix (also known as sp7 transcription factor) enhancer, respectively. Confocal imaging of extracted scales revealed that osterix:mCherry+ cells were distributed throughout the scale including the epidermis and dermis area under physiological conditions, while osterix:mCherrybright cells were limited at the edge region of the scale. In contrast, only a few, mostly small and round trap:GFP+ cells were observed (Fig. 1a). In order to induce fracture stress in the scale, the epidermis area of the scale was cut with fine scissors and confocal imaging was performed at 2 days post-fracture (dpf). We found that the fracture site was surrounded by many trap:GFPbright cells having more than 10 nuclei (Fig. 1b), indicating that multinucleated OCs are formed under fracture stress.

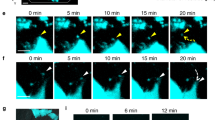
a, b Representative images of an intact a or fractured scale b in trap:GFP; osterix:mCherry double-transgenic zebrafish. Dotted lines in a show a boundary of the dermis (der) and epidermis (epd) area. The right panel in b shows a high magnification view of the white boxed area in the left panel. c Representative time-course changes of a fractured scale. The inset in the left panel shows a high magnification view of the blue boxed area. Images are orientated with the dorsal side to the top and anterior side to the left. Arrows indicate trap:GFP+ cells observed in the edge area of the fractured scale. Dotted lines in b and c show the fracture site. DIC differential interference contrast. Hoe Hoechst 33342; dpf days post-fracture; bars, 200 μm a; 10 μm (right panel in b); 100 μm (left panel in b, c). Experiments were performed twice with three biological replicates in each group a–c.
To further examine fracture healing in the scale, in vivo time-course imaging was performed using an intubation anesthesia system (Supplementary Fig. 1). As shown in Fig. 1c, some small round trap:GFP+ cells converged in the vicinity of the fracture site at 1 dpf. Interestingly, many trap:GFP+ cells were detected around the edge region near the fracture site where osterix:mCherrybright cells are abundantly observed. These trap:GFP+ cells appeared to closely interact with osterix:mCherrybright cells. At 2 dpf, trap:GFP+ cells in the edge region decreased, but a large number of trap:GFPbright cells appeared and covered the fracture site. These observations suggest that immature OCs interact with OBs mainly in the edge region of the scale prior to reaching the fracture site. After 4 dpf, the number of trap:GFP+ cells began to decrease, and the fracture site was covered with osterix:mCherry+ cells instead, leading to bone formation at the fracture site by OBs. Time-lapse imaging of the fractured scale in trap:GFP animals revealed that actively migrating trap:GFP+ cells fused to generate multinucleated OCs around the fracture site at 1 dpf. These fused cells were larger in size and brighter in GFP expression compared to unfused trap:GFP+ cells (Supplementary Movies 1 and 2). These data indicate that trap:GFP and osterix:mCherry finely label OCs and OBs, respectively, and that the fractured scale model is useful to investigate the differentiation process of OCs in vivo.
OCs possess mCherry+ particles in the cytoplasm
To isolate and characterize OCs and OBs in the fractured scale, cells were collected from scales in trap:GFP; osterix:mCherry double-transgenic animals and analyzed by flow cytometry (FCM). We detected trap:GFP+ cells and osterix:mCherry+ cells in both intact and fractured scales at 1 dpf. Unexpectedly, however, most trap:GFP+ cells were detected in the osterix:mCherry+ fraction in the scale (Fig. 2a). The percentage of trap:GFPlow osterix:mCherry+ (“GFPlow”) and trap:GFPhigh (“GFPhigh”) cells significantly increased in fractured scales compared with intact scales (Supplementary Fig. 2a). Within the trap:GFPhigh fraction, 75.1 ± 3.8% of cells were detected in the mCherry+ fraction (n = 6, ±s.d.) (Supplementary Fig. 2b). We also examined the absolute number of trap:GFP− osterix:mCherry+ (“mCh+”), GFPlow, and GFPhigh cells in an intact or fractured scale. The number of GFPlow and GFPhigh cells was approximately 2.1 and 3.7 times higher in the fractured scale compared with the intact scale, respectively, whereas that of mCh+ cells was unchanged (Fig. 2b).

a Representative results of flow cytometric analysis of cells from intact (left panel) or fractured scales at 1 day post-fracture (dpf) (right panel). Red, orange, and green gate show trap:GFP– osterix:mCherry+ (“mCh+”), trap:GFPlow osterix:mCherry+ (“GFPlow”), and trap:GFPhigh (“GFPhigh”) cells, respectively. b Absolute number of mCh+, GFPlow, and GFPhigh cells in an intact or fractured scale at 1 dpf. Error bars, s.e.m. (n = 9 for each group); n.s., no significance; *p < 0.05; **p < 0.001 by Student’s t-test. c–e Representative fluorescent images of mCh+ c, GFPlow d, and GFPhigh cells e. Arrows indicate an osterix:mCherry+ particle observed in the cytoplasm. Numbers in bottom left of panels indicate the number of cells showing the displayed morphology over the total number of analyzed cells. DIC differential interference contrast. f Representative electron microscopic images of a GFPhigh cell. Arrowheads show vesicles, which include secondary lysosomes, early endosomes, and multi-vesicular bodies. n nucleus; m mitochondrion; g Golgi apparatus; bars, 5 μm c–e; 1 μm f. Experiments were performed twice with nine biological replicates a, b and two biological replicates c–f in each group.
Cells within the mCh+, GFPlow, and GFPhigh fraction at 1 dpf were separately sorted, plated on a fibronectin-coated glass-bottom dish, and analyzed by confocal microscopy. Cells within the mCh+ fraction contained at least three different types of OBs: small and round cells (type-1), small and spindle-shaped cells (type-2), and large, highly spread cells (type-3). Type-1 mCh+ cells were most frequently observed (~60%) (Fig. 2c). Cells within the GFPlow fraction appeared mostly small and round and were mononucleated (type-1). Some GFPlow cells did not express GFP diffusely, but contained GFP+ fragments in the cytoplasm, appearing to be phagocyte-like cells (type-2) or mCh+ OBs (type-3) (Fig. 2d). In contrast, cells within the GFPhigh fraction contained different types of OCs. Some cells were small and round with a single nucleus (type-1), while others exhibited an ameba-like morphology with many pseudopodia and one or two nuclei (type-2). A few GFPhigh cells had three to four nuclei and were relatively larger in size (type-3), suggesting that various stages of OCs may exist in the GFPhigh fraction (Fig. 2e). We also found that type-2 OCs very actively migrated in vitro in contrast to type-1 and type-3 OCs, which exhibited low motility (Supplementary Movies 3 and 4). Interestingly, most trap:GFP+ cells possessed mCherry+ particles in the cytoplasm (Fig. 2d, e). Electron microscopic analysis revealed that GFPhigh cells had small protrusions, irregular-shaped nuclei, abundant mitochondria, and compact Golgi apparatus located close to the nucleus (Fig. 2f), which are representative morphological features of OCs28. We also observed various types of vesicles, including secondary lysosomes, early endosomes, and multi-vesicular bodies, in the cytoplasm of GFPhigh cells (Fig. 2f).
Transplantation confirms uptake of OB-derived EVs in OCs
Having shown that most trap:GFP+ cells possessed mCherry+ particles, we hypothesized that immature OCs engulf OB-derived EVs to become mature OCs under fracture stress. Since OCs have been shown to originate from hematopoietic stem cells in mammals29, we performed two different hematopoietic cell transplantation assays in order to examine if donor-derived OCs can obtain OB-derived particles in the recipient scale. First, kidney marrow cells (KMCs), which contain a variety of hematopoietic cells including hematopoietic stem cells30,31, were collected from trap:GFP; osterix:mCherry double-transgenic animals and were transplanted into wild type recipients irradiated with sublethal dose of X-ray. At 20 weeks post-transplantation, fractured scales in recipients were analyzed by FCM or confocal microscopy (Fig. 3a, “Transplantation-1”). We detected only trap:GFP single-positive cells, but not osterix:mCherry-expressing cells, in the scale of recipients (Fig. 3b, c; n = 3), indicating that trap:GFP+ OCs originate from hematopoietic stem cells in zebrafish, as has been shown in mammals29. We next transplanted KMCs from trap:GFP single-transgenic animals into irradiated osterix:mCherry single-transgenic recipients (Fig. 3a, “Transplantation-2”). In this experiment, we detected GFP and mCherry double-positive cells in the fractured scale of osterix:mCherry recipients. The mean percentage of mCherry+ cells within the trap:GFPhigh fraction was approximately 48.1 ± 13.5% (n = 4, ±s.d.) in recipient scales (Fig. 3b).

a Schematic diagram of transplantation assays. Hematopoietic cells from trap:GFP; osterix:mCherry double-transgenic zebrafish kidney were transplanted into wild type recipients irradiated with sublethal dose of X-ray (Transplantation-1, n = 3). Hematopoietic cells from trap:GFP single-transgenic zebrafish kidney were transplanted into osterix:mCherry single-transgenic recipients (Transplantation-2, n = 4). After 20 or 8 weeks post-transplantation, cells in scales at 1 day post-fracture (dpf) were analyzed by flow cytometry (FCM) and/or confocal microscopy. b Representative results of FCM analysis in fractured scales from a recipient in Transplantation-1 (left) and recipient in Transplantation-2 (midle). Red, orange, and green gate show trap:GFP− osterix:mCherry+ (“mCh+”), trap:GFPlow osterix:mCherry+ (“GFPlow”), and trap:GFPhigh (“GFPhigh”) cells, respectively. GFPhigh cells in a recipient of Transplantation-2 are displayed in an osterix:mCherry vs. side scatter (SSC) dot plot (right panel). c Representative images of a fractured scale from a trap:GFP; osterix:mCherry double-transgenic animal (left) and recipient in Transplantation-1 (right). Dotted lines indicate the fracture site. Both images showed merged channels of GFP and mCherry. Bars, 100 μm. Experiments were performed twice with three or four biological replicates in each group.
Supporting these observations, live-imaging analysis of the fractured scale in a trap:GFP; osterix:mCherry double-transgenic animal revealed that a trap:GFP+ OC actively interacted with osterix:mCherry+ OBs and obtained mCherry+ particles prior to reaching the fracture site (Supplementary Movie 5). Interestingly, we observed that a trap:GFP+ cell extended the protrusion toward mCherry+ particles and engulfed them into the cytoplasm (Fig. 4, Supplementary Movie 6). Collectively, these data suggest that trap:GFP+ OCs obtain OB-derived EVs in the fractured scale.

a Representative time-lapse imaging of the fractured scale in a trap:GFP; osterix:mCherry double-transgenic animal. Six sequences from Supplementary Movie 6 are presented, documenting the stepwise engulfment of OB-derived EVs (arrowheads) by a trap:GFP+ cell (arrows). b Single plane of a z-stack of a trap:GFP+ cell at the time point of 39 min. Images show a green (trap:GFP), red (osterix:mCherry), and merged channel. Bars, 10 μm. Experiments were performed six times with similar results.
OB-derived EVs contain abundant signaling molecules
To isolate and characterize OB-derived EVs in the scale, we next attempted to isolate OB-derived EVs in fractured scales by FCM using Hoechst 33342 (Hoe), a fluorescent dye that stains the DNA in living cells. Cells dissected from fractured scales at 1 dpf were stained with Hoe and analyzed by FCM. We detected not only an mCh+ Hoehigh fraction but also an mCh+ Hoelow fraction, in which non-nucleated osterix:mCherry+ particles with very low forward scatter (FSC) intensity were identified (Fig. 5a). Ultrastructure of isolated mCh+ Hoelow particles showed that a part of particles, especially large particles, contained some cell compartments, including mitochondria and vesicles, while small particles showed vesicular nature without cellular organelles (Fig. 5b). Negative staining followed by electron microscopic analysis revealed that most mCh+ Hoelow particles were 0.6–1.5 μm in diameter, whereas a minority of particles were more than 2 μm (Fig. 5c, d). We found that the number of mCh+ Hoelow particles was approximately three times higher in the fractured scale than the intact scale, suggesting that OB-derived EVs are released in response to fracture stress (Fig. 5e).

a Representative flow cytometric analysis of cells in fractured scales at 1 day post-fracture (dpf) from an osterix:mCherry single-transgenic zebrafish. Gated regions in the left panel indicate the osterix:mCherry+ Hoecht 33342high (mCh+ Hoehigh) cell fraction and mCh+ Hoelow EV fraction. mCh+ Hoehigh cells and mCh+ Hoelow EVs are displayed in a forward scatter (FSC) vs. side scatter (SSC) dot plot (middle and right panels, respectively). b Representative electron microscopic images of isolated large and small EVs. c Representative image of isolated EVs negatively stained. Arrows indicate an EV. d Percent size distribution of EVs (n = 223). e Absolute number of mCh+ Hoelow EVs in an intact and fractured scale at 1 dpf. **p < 0.01 (n = 9 for each group). Bars, 1 μm b; 2 μm c. Experiments were performed twice with two biological replicates a–d and nine biological replicates e in each group.
To further characterize OCs, OBs, and OB-derived EVs, we performed RNA-seq analysis on four different populations in the fractured scale at 1 dpf: GFP– mCh+ Hoehigh (“mCh+” fraction), GFPlow mCh+ Hoehigh (“GFPlow” fraction), GFPhigh Hoehigh (“GFPhigh” fraction), and mCh+ Hoelow (“EV” fraction) (Fig. 6a). Principal component analysis (PCA) showed that GFPlow and GFPhigh were very closely associated, whereas mCh+ and EV were far from each other (Fig. 6b). We found that OC-related genes, such as nfatc1 (nuclear factor of activated T cells 1), ctsk (cathepsin K), mmp9 (matrix metallopeptidase 9), csk (C-terminal Src kinase), itgb3b (integrin beta 3b), and atp6v (ATPase, H + transporting V) family genes, were enriched in the GFPhigh fraction (Fig. 6c). In contrast, OB-related genes, such as alpl (alkaline phosphatase, biomineralization associated), col1a1a (collagen, type I, alpha 1a), runx2b (RUNX family transcription factor 2b), wnt10b (wingless-type MMTV integration site family, member 10b), efnb2a (ephrin-B2a), csf1a (colony stimulating factor 1a), and tgfb1a (transforming growth factor, beta 1a), were highly expressed in the mCh+ fraction (Fig. 6d).

a Representative flow cytometric analysis of cells in scales at 1 day post-fracture (dpf) from a trap:GFP; osterix:mCherry double-transgenic animal. Gated regions in the left panel indicate the Hoechst 33342high (Hoehigh) cell fraction and osterix:mCherry+ Hoelow fraction (“EV”). Cells in the Hoehigh fraction are displayed in the right panel to further divide into three populations, trap:GFP− osterix:mCherry+ (“mCh+”), trap:GFPlow osterix:mCherry+ (“GFPlow”), and trap:GFPhigh (“GFPhigh”). b Principal component analysis (PCA) based on the read per million (RPM) of each sample. c, d Hierarchical clustering of selected OC-related c and OB-related genes d in the mCh+, GFPlow, GFPhigh, and EV fraction in the fractured scale. e Gene ontology enrichment analysis of highly expressed genes in the mCh+, GFPlow, GFPhigh, and EV fraction. Experiments were performed once with two biological replicates.
To further determine the characteristics of each fraction, gene ontology enrichment analysis was performed using significantly up-regulated genes in each fraction. Genes involved in “proton transmembrane transport” and “ATP metabolic process” were predominantly expressed in the GFPhigh fraction, highlighting that cells in the GFPhigh fraction possess the representative molecular signature of OCs. In contrast, genes involved in “leukocyte activation” and “immune system process” were up-regulated in the GFPlow fraction (Fig. 6e). Based on morphological analysis and expression data, cells within the GFPlow fraction appear to contain mainly monocytes/macrophages that are induced by fracture stress. In contrast, genes involved in “extracellular matrix organization” and “response to wounding” were highly expressed in the mCh+ fraction. Interestingly, genes involved in “vesicle-mediated transport”, “intracellular signal transduction”, and “phagocytosis” were enriched in the EV fraction (Fig. 6e), suggesting that OB-derived EVs contain abundant signaling molecules.
Quantitative PCR analysis also showed that trap, nfatc1, and ctsk were highly expressed in the GFPhigh fraction, whereas osterix, alpl, col1a1a, and osteocalcin (also known as bone gamma-carboxyglutamate protein (bglap)) were enriched in the mCh+ fraction (Supplementary Fig. 3). It is known in mammals that OC precursors express a cell-surface RANK, while OBs express its ligand RANKL, on the surface of their plasma membrane. This RANKL–RANK signaling activates a variety of downstream signaling pathways required for OC differentiation32. The expression of both rank and rankl was predominantly detected in both the GFPlow and EV fractions in the fractured scale (Supplementary Fig. 3). These data raised the possibility that OB-derived EVs can potentially induce the differentiation of OCs.
OB-derived EVs promote OC differentiation via Rankl signals
In general, EVs are classified into three types according to their sizes and origins: exosomes, microvesicles (MVs), and apoptotic bodies (ABs)33. Exosomes and MVs are shed from a variety of cell types and have been implicated in cell–cell communication34. ABs are formed during the apoptotic process and are then engulfed by macrophages and other immune cells35,36. Since exosomes are recognized as very tiny vesicles (<100 nm)37,38, it is likely that the OB-derived EVs isolated by FCM and visualized by confocal microscopy are mainly MVs and/or ABs rather than exosomes. To distinguish MVs and ABs, we further performed cell staining with Sytox Red and Annexin-V, which distinguish MVs and ABs as well as live, pre-apoptotic, and apoptotic cells39. FCM analysis showed that mCh+ Hoehigh cells, which mainly contain OBs, were subdivided into three fractions, Sytox Redlow Annexin Vlow (live cell fraction), Sytox Redlow Annexin Vhigh (pre-apoptotic cell fraction), and Sytox Redhigh Annexin Vhigh (apoptotic cell fraction). We found that 79.2 ± 2.4% of mCh+ Hoehigh cells were detected in the live cell fraction, and 7.1 ± 1.3% and 11.2 ± 1.0% were in the pre-apoptotic and apoptotic cell fraction, respectively (n = 4, ±s.d.). Interestingly, 65.4 ± 5.0% of mCh+ Hoelow EVs were detected in the Sytox Redlow Annexin-Vlow “MV” fraction, while 32.2 ± 5.9% were in Sytox Redlow Annexin-Vhigh “AB” fraction (n = 4, ±s.d.) (Fig. 7a, b), indicating that mCh+ Hoelow EVs contain both MVs and ABs.

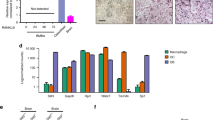
a Representative flow cytometric analysis of cells in scales at 1 day post-fracture (dpf) from a osterix:mCherry single-transgenic animal. Gated regions indicate the mCh+ Hoehigh cell fraction and mCh+ Hoelow EV fraction. b mCh+ Hoehigh cells and mCh+ Hoelow EVs were displayed in an Annexin-V-FITC vs. Sytox Red dot plot. mCh+ Hoehigh cells were subdivided into three fractions, “live”, “pre-apoptotic”, and “apoptotic”, whereas mCh+ Hoelow EVs were divided into two fractions, “microvesicle” (MV) and “apoptotic body” (AB). c Schematic diagram of in vitro cell culture assays. Kidney marrow cells (KMCs) from trap:GFP single-transgenic zebrafish were co-cultured with OBs, MVs, or ABs from fractured scales of osterix:mCherry single-transgenic zebrafish in a fibronectin-coated plate. At 2 days of co-culture, the number of trap:GFP+ cells was counted in each well. Non-co-cultured KMCs were used as a control. d The average number of trap:GFP+ cells in each type of wells. Error bars, s.d. (n = 4 for each group). e Representative images of trap:GFP+ cells co-cultured with OBs (left panel), MVs (middle panel), or ABs (right panel). trap:GFP+ cells contained OB-derived EVs in the cytoplasm (arrows). Bars, 5 μm. f Schematic diagram of zebrafish rankl locus. gRNA target sites and primer recognition sites are shown in red bars and blue arrows, respectively. g qPCR analysis of rankl in the adult fin of wild type control and rankl gRNA-injected animals. Results from seven individual gRNA-injected animals are shown. Data are mean ± s.d. from three independent experiments. h The average number of trap:GFP+ cells in non-co-cultured KMCs (control), or KMCs co-cultured with OBs or EVs from wild type or rankl gRNA-injected animals. Error bars, s.d. (n = 4 for each group); n.s., no significance; *p < 0.05; **p < 0.01; ***p < 0.001 by one-way ANOVA followed by Dunnett’s test. Experiments were performed twice with four biological replicates in each group d, h.
To determine if OB-derived EVs are involved in the differentiation of OCs, 60,000 KMCs from trap:GFP animals were co-cultured with 2000 mCh+ Hoehigh Sytox Redlow Annexin-Vlow “OBs”, mCh+ Hoelow Sytox Redlow Annexin-Vlow “MVs”, or mCh+ Hoelow Sytox Redlow Annexin-Vhigh “ABs” on fibronectin-coated plates. After 2 days of culture, the number of trap:GFP+ cells was counted in each well (Fig. 7c). We found that the number of GFP+ cells significantly increased in KMCs co-cultured with MVs or OBs when compared to non-co-cultured controls. Surprisingly, the number of GFP+ cells also significantly increased in KMCs co-cultured with ABs (Fig. 7d). Importantly, most trap:GFP+ cells co-cultured with OBs, MVs, or ABs contained mCherry+ EVs in the cytoplasm (Fig. 7e), similar to OCs observed in the fractured scale of trap:GFP; osterix:mCherry double-transgenic animals (shown in Fig. 2e).
To clarify the role of OB-derived EVs, KMCs were further cultured with or without OB-derived EVs (mCh+ Hoelow). Although the number of GFP+ cells increased, the total number of KMCs was unchanged by treatment with OB-derived EVs (Supplementary Fig. 4a, b), suggesting that treatment with EVs does not affect hematopoietic cell proliferation, but does affect OC differentiation from hematopoietic cells. In addition, the percentage of multinucleated GFP+ cells significantly increased by treatment with EVs, but the percentage of mononucleated GFP+ cells decreased. We also observed a few GFP+ cells with more than three nuclei in KMCs treated with OB-derived EVs (Supplementary Fig. 4c). Taken together, these data suggest that treatment with OB-derived EVs promotes differentiation and fusion of OCs in vitro.
Since the high level of rankl mRNA was detected in OB-derived EVs, we next questioned if OB-derived EVs mediate Rankl signals to promote OC differentiation. We utilized a gene knockdown method based on the CRISPR/Cas9 system, in which injection of multiple guide RNAs (gRNAs) redundantly targeting a single gene leads to a high proportion of null phenotypes at the G0 generation40. Four different gRNAs targeting either exon 1 or 4 of rankl gene were co-injected with Cas9 protein into one-cell stage embryos from trap:GFP and/or osterix:mCherry zebrafish (Fig. 7f). Embryos injected with rankl gRNA could partially survive into adulthood. However, most adult animals showed severe body curvature (Supplementary Fig. 5a). Efficiency of rankl knockdown was examined in the adult fin by qPCR using two different sets of primers that recognize gRNA target sites of rankl gene. We detected ~60–90% reduction of rankl expression by each primer set in all seven individual animals tested (Fig. 7f, g), suggesting that wild type rankl expression largely decreased in rankl gRNA-injected adult animals. The scale formation was, however, nearly unaffected in rankl gRNA-injected animals in terms of the size and morphology (Supplementary Fig. 5b). In mice, deficiency of Rankl results in the reduction of multinucleated OCs in bone tissue41. Similar to Rankl-deficient mice, rankl gRNA-injected zebrafish showed reduction in both the percentage and absolute number of trap:GFPhigh OCs compared with wild type zebrafish. In contrast, the number of OBs (GFP– mCh+ Hoehigh) and OB-derived EVs (mCh+ Hoelow) was unaffected in rankl-gRNA-injected zebrafish (Supplementary Fig. 5c–e), suggesting that knockdown of rankl does not affect secretion of EVs in OBs. We co-cultured KMCs from wild type trap:GFP animals with OBs (mCh+ Hoehigh) or EVs (mCh+ Hoelow) from wild type or rankl gRNA-injected osterix:mCherry animals. We found that the number of GFP+ cells significantly decreased in KMCs co-cultured with EVs from rankl gRNA-injected animals compared with those from wild type animals, whereas it was nearly unchanged between KMCs co-cultured with OBs from wild type and ranklgRNA-injected animals (Fig. 7h). Taken together, these data suggest that the uptake of OB-derived EVs promotes the differentiation of OCs in a Rankl signaling-dependent manner.
Discussion
In the present study, we have developed a fracture healing model using double-transgenic zebrafish scales to visualize and isolate OCs and OBs. Our data showed that immature OCs engulf OB-derived EVs prior to convergence at the fracture site. Moreover, co-culture of hematopoietic cells with OB-derived EVs promoted the differentiation of OCs via Rankl signaling. These findings provide insights into the fundamental regulatory mechanisms of OC differentiation by OBs in bone tissue.
Many metabolic or genetic bone diseases are associated with the disruption of the intercellular communication between OCs and OBs17. Regulation of OC differentiation by OBs has been shown by many in vitro studies in mice. Monocyte/macrophage precursors can give rise to OCs in the presence of M-CSF and RANKL. M-CSF is secreted in part by OBs and binds to its receptor, c-Fms, expressed on OCs to regulate the survival, migration, and bone resorption activity in OCs19. RANKL–RANK signaling activates its downstream targets, including the TNF receptor-associated factor (TRAF) family, which regulates OC formation, survival, and activation via multiple signaling pathways42. In vitro studies have thus elucidated roles of various signaling molecules that regulate OC differentiation and function. It is still challenging, however, to investigate OC–OB interactions in vivo, and hence the cellular and molecular mechanisms involved in cell–cell communication between these two types of cells remain largely elusive. Ishii’s group applied intravital two-photon imaging, which allows to visualize the behavior of living OCs in mouse bone, and detected dynamic communication between OCs and OBs. They described two subsets of functional OCs in terms of their motility and function, ‘static—bone resorptive’ and ‘moving—non-resorptive’, which can be shifted by a direct contact with mature OBs43,44. In the medaka fin model, it has also been shown that there are two types of OCs in the amputated fin, early-induced and late-induced OCs. The former was relatively small with low TRAP-activity and resorbed bone fragments, while the late-induced OCs were large with high TRAP activity and remodeled the callus45. These imaging analyses provided insight into the functional divergence of OCs, highlighting the importance of in vivo live-imaging analysis to better understand OC and OB functions. We could also distinguish at least three different types of OCs and OBs based on morphology in the fractured scale. While further work is needed to precisely describe the maturation process of these cell types, these morphological features may reflect different stages of OBs and OCs. Our model system using the transgenic zebrafish scale enables visualization of the whole tissue at a single cell level throughout the process of bone resorption and formation. Indeed, we have successfully captured the moment of cell–cell fusion between two OCs as well as the convergence of OCs at the fracture site, as has been shown in mammalian OCs46,47. In addition, the recent advent of gene knockout/knockdown methods in zebrafish based on the CRISPR/Cas9 system40,48,49 will extend our scale model to rapid and scalable mutagenesis strategies to further probe the function of OCs and OBs. Our imaging strategy in the zebrafish scale will thus open new avenues to elucidate molecular cues needed to regulate OC–OB communication.
The availability of double-transgenic zebrafish, trap:GFP; osterix:mCherry, also allowed us to investigate the role of EVs in bone tissue. Different subtypes of EVs have been increasingly recognized as potent vehicles of intercellular communication in the past decade. Studies in mammals revealed that EVs released by healthy and apoptotic cells transport biologically active molecules, such as lipids, proteins, mRNAs, and microRNAs to target cells; however, our current knowledge of EVs in the diversity, internalization, and cargo delivery is still very limited34. Recently, it was reported that a zebrafish transgenic line that expresses human CD63 fused with pHluorin under the control of tissue-specific promoter/enhancer can be used to visualize the release and uptake of EVs in vivo. A role for EVs in the formation of metastatic niches in vivo could also be determined by tracking EVs shed from melanoma cells in zebrafish embryos, demonstrating that zebrafish is an excellent model for the study of EVs50. In the present study, our transplantation assays clearly demonstrated that mCherry+ particles observed in trap:GFP+ cells are derived from EVs shed from OBs. Our FCM analysis revealed that ~75% of trap:GFPhigh cells were detected in the mCherry+ fraction at 1 dpf, suggesting that the majority of mature OCs experience uptake of OB-derived EVs during their differentiation process. Interestingly, our cell culture assays showed that uptake of ABs also promotes OC differentiation in KMCs, supporting the hypothesis that accumulation of apoptotic OBs/osteocytes induces the recruitment of OCs to initiate bone resorption51. Since OCs arise from monocyte/macrophage precursors, it is likely that phagocytosis of apoptotic OBs also triggers the differentiation of OCs. In mammals, EVs derived from OCs also play an important role in osteoclastogenesis as well as osteoblastogenesis. EVs from OC precursors promote OC formation in whole bone marrow cultures, whereas EVs from OC-enriched cultures inhibit osteoclastogenesis26. Moreover, EVs released by mature OCs contain a high level of RANK and promote bone formation by triggering RANKL reverse signaling in OBs27. These observations strongly suggest that cell–cell communication in osteoclastogenesis and osteoblastogenesis is largely dependent on the release and uptake of EVs. Such EV-mediated intercellular communication represents a previously unrecognized cellular mechanism in the bone. Further analysis of EVs in bone tissue will elucidate the molecular mechanisms that regulate the balance between bone resorption and formation.
Methods
Zebrafish husbandry and fracture stimulation
Zebrafish were raised in a circulating aquarium system (AQUA) at 28.5 °C in a 14/10 h light/dark cycle and maintained in accordance with guidelines of the Committee on Animal Experimentation of Kanazawa University. For fracture stimulation, adult zebrafish were anesthetized in system water containing 0.01% tricaine (Sigma), and the epidermis area of a scale was cut ~400 μm in length with fine scissors under a fluorescent stereo microscope (Axiozoom V16, Zeiss).
Generation of transgenic lines
Zebrafish transgenic lines of Tg(trap:GFP-CAAX)ou2031Tg and Tg(osterix:Lifeact-mCherry)ou2032Tg were generated as previously described1,2. Briefly, a 6-kb upstream regulatory region of the zebrafish trap (acp5a) gene and 4.1 kb upstream regulatory region of the medaka osterix (sp7) gene were amplified using the primers listed in Supplementary Table 1 and ligated into pT2AL200R150G containing GFP-CAAX and Lifeact-mCherry, respectively. TPase mRNA was synthesized with the pCS2+ vector by mMESSAGE mMACHINE SP6 Transcription Kit (Thermo Fisher Scientific). Transgenic zebrafish were generated by injection of the plasmid construct with TPase mRNA into 1-cell stage embryos. A stable transgenic line was obtained by screening of GFP or mCherry expression in F1 generation.
Knockdown of rankl by CRISPR/Cas9 system
Previously designed guide RNA (gRNA) sequences targeting the rankl exon 1 or 4 were utilized40. gRNAs were synthesized as previously described40. Briefly, a single-strand DNA oligo containing the gRNA target and T7 promoter sequence was annealed with a gRNA scaffold primer. Double-strand DNAs were then synthesized with MightyAmp DNA polymerase (Thermo Fisher Scientific), followed by in vitro transcription using MEGAshortscript T7 Transcription Kit (Thermo Fisher Scientific). gRNAs were purified with mirVana miRNA Isolation Kit (Thermo Fisher Scientific) according to the manufacture’s protocol. Embryos from trap:GFP and/or osterix:mCherry animals were injected with a mixture of Cas9 protein (400 ng/µL) (IDT) and gRNAs (100 ng/µL each) at one-cell stage. gRNA sequences and gRNA scaffold primer are listed in Supplementary Table 1.
Cell preparation and flow cytometry
Scales were extracted from anesthetized zebrafish under a stereo microscope, and were treated with Liberase TM (Roche) in PBS for 1 h at 37 °C. Cells were then filtered through 40 μm stainless mesh and washed with ice-cold 2% fetal bovine serum (FBS) in Hanks’ balanced salt solution (HBSS, Wako) by centrifugation (300×g). KMCs were prepared as previously described52 with some modifications. Briefly, cells were obtained by pipetting of a dissected kidney in 1 mL of ice-cold 2% FBS in HBSS. After centrifugation, the pellet was gently mixed with 1 mL of distilled water by pipetting to lyse erythrocytes by osmotic shock. Subsequently, 1 mL of 2X HBSS was added. Cells were then filtered through a 40 μm stainless mesh and washed with 2% FBS in HBSS by centrifugation. For staining with Hoechst 33342 (Hoe), cells were resuspended at a density of 106 cells/mL in 2% FBS in HBSS and were stained with 5 μg/mL Hoe (Thermo Fisher Scientific) for 90 min at 25 °C in dark with gentle agitation. For staining with Annexin-V, cells were washed with Cell Staining Buffer (BioLegend) by centrifugation, resuspended in Annexin-V-binding buffer containing Annexin-V-FITC (BioLegend), and stained for 15 min at room temperature in dark. Just before flow cytometric (FCM) analysis, the Sytox Red (Thermo Fisher Scientific) was added at a concentration of 5 nM to exclude dead cells or to detect apoptotic cells. FCM acquisition and cell sorting were performed on a FACS Aria III (BD Biosciences). Data analysis was performed using the Kaluza software (ver. 1.3, Beckman Coulter). Since the detectable size of cells/particles by FACS Aria III is more than 0.5 μm, it is not possible to separate very small EVs (<0.4 μm), such as exosomes, in this analysis. EVs were therefore isolated according to the same procedure as cells to minimize contamination with EVs of unknown origin. The absolute number of cells was calculated by flow cytometry based on acquisition events, maximum acquisition times, and the percentage of each cell fraction.
Cell culture
KMCs from trap:GFP animals were resuspended in medium containing 40% Leiboviz’s L-15 medium (Wako), 32% Dulbecco’s modified Eagle’s medium (Wako), 12% Ham’s F12 medium (Wako), 8% FBS, 2 mM l-glutamine (Wako), 15 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Sigma), 100 U penicillin (Wako), and 100 μg/mL streptomycin (Wako). Approximately 6 × 104 of KMCs were plated on a 96-well plate coated with fibronectin (Corning) and incubated for 3 h at 30 °C, 5% CO2. Subsequently, ~2000 of sorted cells or EVs were plated in the wells (four wells for each fraction) and incubated for additional 2 days at the same condition described above. The number of GFP+ cells was counted using an EVOS FL Cell Imaging System (Thermo Fisher Scientific). For counting the number of nuclei, Hoe was directly added into the medium at a concentration of 5 μg/mL. For counting the total number of cells in each well, cells were treated with 0.25% trypsin–1 mM EDTA and harvested by pipetting. The total number of cells was counted using a hemocytometer (Funakoshi).
RNA-seq and qPCR
For sorted cells and EVs, whole-transcript amplification and double-strand cDNA synthesis was performed according to the Quartz-Seq method as previously described53,54. Cells were directly sorted in a lysis buffer containing 1 μg/mL of polyinosinic–polycytidylic acid, and total RNA was extracted using RNeasy Mini Kit. Reverse transcription (RT) was performed using Super Script III (Thermo Fisher Scientific) and an RT primer, which contains oligo-dT, T7 promoter, and PCR target region sequences. After digestion of remaining RT primers by exonuclease I (Takara), a poly-A tail was added to the 3′ ends of the first-strand cDNAs using terminal transferase (Sigma). The second-strand DNA was then synthesized using MightyAmp DNA polymerase (Thermo Fisher Scientific) and a tagging primer. PCR amplification was performed using a suppression primer. The amplified double-strand cDNA was purified using QIAquick PCR Purification Kit (Qiagen). Library preparation for RNA-seq was performed using Nextera XT DNA Library Preparation Kit (illumina). Next generation sequencing of cDNA libraries was performed by GENEWIZ using the Illumina NextSeq500 (illumina), and base-calling was performed using the Illumina RTA software (ver. 2.4.11). Sequence reads were mapped to the zebrafish reference genome (GRCz11) using HiSAT2 (version 2.1.0). Reads per million (RPM) were calculated using Subread (ver. 1.6.4). Up-regulated genes of each fraction were selected with a p-value cutoff of 0.05 based on one-way ANOVA and more than two-fold change. PCA and hierarchical clustering of each subset was performed in R (ver. 3.5.0) with the Bioconductor gplots package. The data have been deposited in Gene Expression Omnibus (GEO) (National Center for Biotechnology Information) and are accessible through the GEO database (series accession number, GSE134330). Total RNAs from the adult fin were extracted with RNeasy Mini Kit (QIAGEN) and cDNAs were synthesized with ReverTra Ace qPCR RT Master Mix (Toyobo). Quantitative real-time PCR (qPCR) assays were performed using TB Green Premix Ex Taq II (TaKaRa) on a ViiA 7 Real-Time PCR System according to manufacturer’s instructions (Thermo Fisher Scientific). Primers used for whole-transcript amplification and qPCR are listed in Supplementary Table 1.
Electron microscopy
The ultrastructure of cells and EVs was observed by transmission electron microscopy as previously described8 with some modifications. Briefly, sorted cells and EVs were fixed with 2.5% glutaraldehyde (Nacalai Tesque), 2% paraformaldehyde (Wako) in 0.1 M phosphate buffer (pH 7.4) at 4 °C overnight. Cells were then dehydrated and embedded in Epon 812 (TAAB Laboratories). Ultrathin sections were obtained from the Epon blocks and stained with uranyl acetate and lead citrate. For negative staining of EVs, a grid with a supporting film was placed on a drop of the EV suspension for 15 min and excess solution was removed with a filter paper. The grid was then placed in 2% uranium acetate aqueous solution for 2 min, followed by air-drying. Sections and stained EVs were observed under an electron microscope (H-7650, Hitachi).
X-ray irradiation and transplantation
Three to six zebrafish were placed in a 90 mm Petri dish in system water, and animals were sublethally irradiated with X-ray on a Faxitron RX-650 (Faxitron, 130 kVp, 1.15 Gy/min) for 20 min (~23 Gy). At 2 days post-irradiation, animals were transplanted with KMCs using a retro-orbital injection method31.
Intubation anesthesia and imaging
Intubation anesthesia was performed as previously described55 with some modifications. A flask containing 0.04% of 2-phenoxyethanol (Wako) in system water was kept in a water bath to maintain a constant temperature of 28 °C, and delivered to a glass-bottom chamber (Eppendorf) using a peristatic pump (ATTO). An adult zebrafish mounted in the chamber was orally perfused with the anesthetic water using an Intramedic polyethylene tube (BD). Excess water in the chamber was removed and returned to the flask using another peristatic pump. For imaging of extracted scales, scales were stained with 5 μg/mL of Hoe for 20 min at room temperature and mounted in a glass bottom dish (Matsunami) filled with 0.6% low-gelling agarose (Sigma). For imaging of sorted cells, cells were plated on a fibronectin-coated glass-bottom dish with culture medium described above. For imaging of cultured cells, cells were plated on a cover slip coated with fibronectin (Cosmo Bio) and were cultured as described above. Cell were then fixed with 4% paraformaldehyde and mounted with ProLong Gold Antifade Mountant with DAPI (Thermo Fisher Scientific). Fluorescent images were captured using an FV10i confocal microscope and Fluoview FV10i-SW software (ver. 2.1.1) (Olympus). For time-lapse imaging, images were captured every 3 or 5 min for scales and 30 s for cells, and movies were generated using iMovie software (ver. 10.1.12) or Fluoview FV10i-SW software. We defined GFP “bright” as having four-fold higher than the average intensity of GFP+ cells in the intact scale and mCherry “bright” as having 10-fold higher intensity than the average mCherry intensity of the whole intact scale. Visible light imaging of adult animals were captured using an Olympus Pen E-PL8 distal camera (Olympus).
Statistics and reproducibility
Statistical differences between groups were determined by unpaired two-tailed Student’s t-test or one-way ANOVA with Dunnett’s test. A value of p < 0.05 was considered to be statistically significant. All experiments were repeated at least twice with similar results.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The raw data of RNA-seq can be obtained from the GEO database (series accession number, GSE134330). The source data for Figs. 2, 5, 7, Supplementary Figs. 2–5 are provided as Supplementary Data 1. All data and materials produced by this study are available from the corresponding author upon request.
References
Chatani, M., Takano, Y. & Kudo, A. Osteoclasts in bone modeling, as revealed by in vivo imaging, are essential for organogenesis in fish. Dev. Biol. 360, 96–109 (2011).
Renn, J., Winkler, C., Schartl, M., Fischer, R. & Goerlich, R. Zebralish and medaka as models for bone research including implications regarding space-related issues. Protoplasma 229, 209–214 (2006).
Sire, J.-Y., Huysseune, A. & Meunier, F. J. Osteoclasts in teleost fish: light-and electron-microscopical observations. Cell Tissue Res. 260, 10 (1990).
de Vrieze, E., Sharif, F., Metz, J. R., Flik, G. & Richardson, M. K. Matrix metalloproteinases in osteoclasts of ontogenetic and regenerating zebrafish scales. Bone 48, 704–712 (2011).
Pasqualetti, S., Banfi, G. & Mariotti, M. The zebrafish scale as model to study the bone mineralization process. J. Mol. Histol. 43, 589–595 (2012).
Suzuki, N., Suzuki, T. & Kurokawa, T. Suppression of osteoclastic activities by calcitonin in the scales of goldfish (freshwater teleost) and nibbler fish (seawater teleost). Peptides 21, 115–124 (2000).
Suzuki, N. & Hattori, A. Melatonin suppresses osteoclastic and osteoblastic activities in the scales of goldfish. J. Pineal Res. 33, 253–258 (2002).
Suzuki, N. et al. Parathyroid hormone 1 (1-34) acts on the scales and involves calcium metabolism in goldfish. Bone 48, 1186–1193 (2011).
de Vrieze, E., Zethof, J., Schulte-Merker, S., Flik, G. & Metz, J. R. Identification of novel osteogenic compounds by an ex-vivo sp7:luciferase zebrafish scale assay. Bone 74, 106–113 (2015).
Carnovali, M., Luzi, L., Banfi, G. & Mariotti, M. Chronic hyperglycemia affects bone metabolism in adult zebrafish scale model. Endocrine 54, 808–817 (2016).
Park, C. M. et al. Ginsenoside Re Inhibits Osteoclast Differentiation In Mouse Bone Marrow-derived Macrophages and zebrafish scale model. Mol. Cells 39, 855–861 (2016).
Witten, P. E., Harris, M. P., Huysseune, A. & Winkler, C. Small teleost fish provide new insights into human skeletal diseases. Methods Cell Biol. 138, 321–346 (2017).
Raggatt, L. J. & Partridge, N. C. Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 285, 25103–25108 (2010).
Crockett, J. C., Rogers, M. J., Coxon, F. P., Hocking, L. J. & Helfrich, M. H. Bone remodelling at a glance. J. Cell Sci. 124, 991–998 (2011).
Heino, T. J. & Hentunen, T. A. Differentiation of osteoblasts and osteocytes from mesenchymal stem cells. Curr. Stem Cell Res. Ther. 3, 131–145 (2008).
Sims, N. A. & Martin, T. J. Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. Bonekey Rep. 3, 481 (2014).
Matsuo, K. & Irie, N. Osteoclast-osteoblast communication. Arch. Biochem. Biophys. 473, 201–209 (2008).
Han, Y., You, X., Xing, W., Zhang, Z. & Zou, W. Paracrine and endocrine actions of bone-the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. 6, 16 (2018).
Chen, X. et al. Osteoblast–osteoclast interactions. Connect Tissue Res. 59, 99–107 (2018).
Deng, L. et al. Osteoblast-derived microvesicles: a novel mechanism for communication between osteoblasts and osteoclasts. Bone 79, 37–42 (2015).
Cappariello, A. et al. Osteoblast-derived extracellular vesicles are biological tools for the delivery of active molecules to bone. J. Bone Min. Res. 33, 517–533 (2018).
EL Andaloussi, S., Mäger, I., Breakefield, X. O. & Wood, M. J. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 12, 347–357 (2013).
Turchinovich, A., Drapkina, O. & Tonevitsky, A. Transcriptome of extracellular vesicles: state-of-the-art. Front. Immunol. 10, 202 (2019).
Gibbings, D. J., Ciaudo, C., Erhardt, M. & Voinnet, O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol. 11, 1143–1149 (2009).
Tao, S. C. & Guo, S. C. Extracellular vesicles in bone: “dogrobbers” in the “eternal battle field”. Cell Commun. Signal. 17, 6 (2019).
Huynh, N. et al. Characterization of regulatory extracellular vesicles from osteoclasts. J. Dent. Res. 95, 673–679 (2016).
Ikebuchi, Y. et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature 561, 195–200 (2018).
Baron, R., Neff, L., Tran Van, P., Nefussi, J. R. & Vignery, A. Kinetic and cytochemical identification of osteoclast precursors and their differentiation into multinucleated osteoclasts. Am. J. Pathol. 122, 363–378 (1986).
Udagawa, N. et al. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl Acad. Sci. USA 87, 7260–7264 (1990).
Traver, D. et al. Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat. Immunol. 4, 1238–1246 (2003).
de Jong, J. L. et al. Characterization of immune-matched hematopoietic transplantation in zebrafish. Blood 117, 4234–4242 (2011).
Boyce, B. F. Advances in the regulation of osteoclasts and osteoclast functions. J. Dent. Res. 92, 860–867 (2013).
Kalra, H., Drummen, G. P. & Mathivanan, S. Focus on extracellular vesicles: introducing the next small big thing. Int. J. Mol. Sci. 17, 170 (2016).
Tkach, M. & Théry, C. Communication by extracellular vesicles: where we are and where we need to go. Cell 164, 1226–1232 (2016).
Caruso, S. & Poon, I. K. H. Apoptotic cell-derived extracellular vesicles: more than just debris. Front. Immunol. 9, 1486 (2018).
Budai, Z. et al. Macrophages engulf apoptotic and primary necrotic thymocytes through similar phosphatidylserine-dependent mechanisms. FEBS Open Bio 9, 446–456 (2019).
van Niel, G., D’Angelo, G. & Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19, 213–228 (2018).
Mathieu, M., Martin-Jaular, L., Lavieu, G. & Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 21, 9–17 (2019).
Wlodkowic, D., Skommer, J. & Darzynkiewicz, Z. Flow cytometry-based apoptosis detection. Methods Mol. Biol. 559, 19–32 (2009).
Wu, R. S. et al. A rapid method for directed gene knockout for screening in G0 zebrafish. Dev. Cell 46, 112–125.e114 (2018).
Kong, Y. Y. et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397, 315–323 (1999).
Boyle, W. J., Simonet, W. S. & Lacey, D. L. Osteoclast differentiation and activation. Nature 423, 337–342 (2003).
Kikuta, J. et al. Dynamic visualization of RANKL and Th17-mediated osteoclast function. J. Clin. Invest. 123, 866–873 (2013).
Furuya, M. et al. Direct cell-cell contact between mature osteoblasts and osteoclasts dynamically controls their functions in vivo. Nat. Commun. 9, 300 (2018).
Takeyama, K., Chatani, M., Takano, Y. & Kudo, A. In-vivo imaging of the fracture healing in medaka revealed two types of osteoclasts before and after the callus formation by osteoblasts. Dev. Biol. 394, 292–304 (2014).
Søe, K., Hobolt-Pedersen, A. S. & Delaisse, J. M. The elementary fusion modalities of osteoclasts. Bone 73, 181–189 (2015).
Schell, H. et al. Osteoclastic activity begins early and increases over the course of bone healing. Bone 38, 547–554 (2006).
Shah, A. N., Davey, C. F., Whitebirch, A. C., Miller, A. C. & Moens, C. B. Rapid reverse genetic screening using CRISPR in zebrafish. Nat. Methods 12, 535–540 (2015).
Varshney, G. K. et al. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 25, 1030–1042 (2015).
Verweij, F. J., Hyenne, V., Van Niel, G. & Goetz, J. G. Extracellular vesicles: catching the light in zebrafish. Trends Cell Biol. 29, 770–776 (2019).
Plotkin, L. I. Apoptotic osteocytes and the control of targeted bone resorption. Curr. Osteoporos. Rep. 12, 121–126 (2014).
Kobayashi, I. et al. Jam1a-Jam2a interactions regulate haematopoietic stem cell fate through Notch signalling. Nature 512, 319–323 (2014).
Sasagawa, Y. et al. Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 14, R31 (2013).
Kobayashi, I. et al. Enrichment of hematopoietic stem/progenitor cells in the zebrafish kidney. Sci. Rep. 9, 14205 (2019).
Noishiki, C. et al. Live imaging of angiogenesis during cutaneous wound healing in adult zebrafish. Angiogenesis 22, 341–354 (2019).
Acknowledgements
The authors thank Dr. S. Yuge for supporting intubation anesthesia, Dr. M. Hazawa for supporting cell culture assays, Dr. Y. Furusawa for supporting RNA-seq analysis, and Dr. K. Lewis for critical evaluation of the manuscript. This work was supported in part by Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (18K06331).
Author information
Authors and Affiliations
Contributions
J.K.-S., M.K., S.Y., J.K., and M.I. performed experiments; J.K.-S., N.S., K.K., A.H., and I.K. discussed results; N.S., K.K., A.H., and M.Y. edited the manuscript; J.K.-S. and I.K. designed the research, analyzed data, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kobayashi-Sun, J., Yamamori, S., Kondo, M. et al. Uptake of osteoblast-derived extracellular vesicles promotes the differentiation of osteoclasts in the zebrafish scale. Commun Biol 3, 190 (2020). https://doi.org/10.1038/s42003-020-0925-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-020-0925-1
This article is cited by
-
Extracellular Vesicles and Exosomes in the Control of the Musculoskeletal Health
Current Osteoporosis Reports (2024)
-
Bioengineering extracellular vesicles: smart nanomaterials for bone regeneration
Journal of Nanobiotechnology (2023)
-
Isolation and characterization of the morphology, size and particle number of rainbow trout (Oncorhynchus mykiss) and zebrafish (Danio rerio) cell line derived large and small extracellular vesicles
Fish Physiology and Biochemistry (2023)
-
Regenerating zebrafish scales express a subset of evolutionary conserved genes involved in human skeletal disease
BMC Biology (2022)
-
The advances in nanomedicine for bone and cartilage repair
Journal of Nanobiotechnology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
