
Abstract
The role of n-3 polyunsaturated fatty acids (PUFAs) in alcoholic liver disease (ALD) has been controversial. N-3 PUFA oxidation in animal feeding stuffs was rarely concerned, likely contributing to inconsistent outcomes. Here, we report the impacts of oxidized fish oil (OFO) on ALD in C57BL/6 mice. Alcohol exposure increased plasma aminotransferase levels and hepatic inflammation. These deleterious effects were ameliorated by unoxidized FO but exacerbated by OFO. Sequencing analysis showed the accentuated intestinal dysbiosis and the increased proportion of Proteobacteria in OFO-fed mice. Intestinal sterilization by antibiotics completely abolished OFO-aggravated liver injury. Additionally, alcohol exposure leads to the greater increase in plasma endotoxin and decrease in intestinal tight junction protein expressions in OFO-fed mice. Stabilization of intestinal barrier by obeticholic acid markedly blunted OFO-aggravated liver injury in alcohol-fed mice. These results demonstrate that OFO exacerbates alcoholic liver injury through enhancing intestinal dysbiosis, barrier dysfunction, and hepatic inflammation mediated by gut-derived endotoxin.
Similar content being viewed by others


Introduction
Prolonged and excessive amount of alcohol consumption causes alcoholic liver disease (ALD), ranging from simple hepatosteatosis, hepatitis, hepatic fibrosis, and cirrhosis1,2. In 2016, alcohol abuse was responsible for ~3 million deaths and 132.6 million disability-adjusted life years worldwide3. The critical role of gut–liver axis has been well established both in clinical and experimental models of ALD4. Alcohol exposure disturbs gut microbiota homeostasis by alteration in bacteria composition, particularly the overgrowth of pathogenic bacteria. Accumulating evidence indicated that gut microbiota manipulation, such as fecal microbial transplantation and the treatment of probiotics or antibiotics, ameliorates alcoholic liver injury in humans and rodent models4,5. The overgrowth of intestinal microbiota, especially Gram-negative, increases the bacterial endotoxin or lipopolysaccharides (LPS). Alcohol exposure also impairs intestinal barrier integrity, leading to the increased intestinal permeability, and subsequent the enhanced translocation of bacteria and LPS to the portal circulation6. Clinical and experimental studies have indicated that the circulating level of LPS is indeed correlated with the severity of alcoholic liver injury7,8. The gut-derived LPS activates hepatic Kupffer cells by Toll-like receptor 4 (TLR4)-mediated nuclear factor κB (NF-κB) signaling, resulting in the overproduction of inflammatory cytokines, which, in turn, induces hepatocyte damage9.
Intestinal barrier dysfunction is a major cause of alcohol-induced bacterial translocation, and has been considered as a prerequisite in the development of ALD10,11. Although the exact molecular mechanisms underlying intestinal barrier dysfunction in the pathogenesis of ALD are not sufficiently elucidated. Acetaldehyde, a metabolite of alcohol, is correlated with the disruption of intestinal integrity by tyrosine phosphorylation of intestinal epithelial tight junction (TJ) proteins12. TJ proteins are consisted of a variety of transmembrane proteins, including intracellular plaque protein zonula occludin (ZO-1), occludin, claudin-2, and caudin-413. Accumulating evidence has shown that dysbiosis-caused intestinal inflammation also contributed to the increased intestinal permeability in ALD14,15. TNF-α, a well-characterized mediator of inflammatory response, is increased in the jejunum of mice with enteric dysbiosis, and has demonstrated to be associated with increasing intestinal permeability after alcohol feeding16.
In recent decades, long-chain n-3 polyunsaturated fatty acids (PUFAs), including eicosapentaenoic acid (EPA, 20:5 n-3) and docosahexaenoic acid (DHA, 22:6 n-3), have been well documented to exert the potentially therapeutic efficacy in nonalcoholic liver disease17, and liver injury induced by various chemicals, including carbon tetrachloride18, acetaminophen19, concanavalin A20, and D-galactosamine/lipopolysaccharide21. An increasing evidence has demonstrated that n-3 PUFAs exerted the beneficial effects on ALD9,22,23,24. However, the role of n-3 PUFAs in ALD has been still inconsistent and controversial. Several studies have shown that the supplement of n-3-rich fish oil (FO) exacerbates alcoholic liver injury and promotes the pathogenesis of ALD, which is attributed to the intensified oxidative stress and inflammation in the liver25,26,27,28. But it is well known that n-3 PUFAs with high degree of unsaturation are highly susceptible to oxidation and exert antioxidative and anti-inflammatory properties. It remains unclear whether the high levels of oxidative stress and inflammation observed in these studies with opposite results are caused by n-3 PUFAs or their oxidation products already existing in FO used2,29.
Therefore, the aim of the present study is to investigate the probable impacts of the oxidized fish oil (OFO) on the pathogenesis of ALD. Here we show that oxidation of FO exacerbates alcoholic liver injury through enhancing intestinal dysbiosis, barrier dysfunction, and hepatic inflammation mediated by gut-derived endotoxin.
Results
OFO exacerbates the features of ethanol-induced liver injury
The primary and secondary lipid-oxidation products were measured to determine oxidation status in FO and OFO. After air exposure at 65 °C for 2 weeks, OFO showed the higher degree of oxidation as evidence by the greatly elevated levels of peroxide value (POV), p-anisidine value (AV), total oxidation value (Totox), and thiobarbituric acid-reactive substances (TBARS), compared to the unoxidized FO (Table 1). As determined by gas chromatography–mass spectrometry (GC-MS), the contents of n-3 PUFAs, including EPA, DPA and DHA, in OFO were greatly decreased, compared to unoxidized FO. Approximately 50.9% EPA and 58.7% DHA in FO were oxidized during the process (Supplementary Table 1 and Supplementary Fig. 1). Liver injury induced by ethanol exposure was evaluated by biochemical and histological analysis, and oxidative stress in liver. Acute-on-chronic alcohol feeding (Supplementary Fig. 2) elevated plasma levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) by 4.5-fold and 3.0-fold, respectively, in CO-treated mice. These elevations were greatly augmented to 8.1-fold and 4.6-fold, respectively, by replacing a half of CO with OFO, but significantly decreased by supplement with FO (Fig. 1a). OFO-treated mice had a pronounced increase in plasma triglyceride (TG) and hepatic TG levels, compared to the CO-treated mice after alcohol exposure (Fig. 1b). Alcohol feeding caused oxidative stress in the liver from both CO- and OFO-treated mice, as characterized by the decreased antioxidant parameters (e.g., CAT, SOD, GSH), and increased the hepatic malonaldehyde (MDA) level, but the effects were much greater in OFO-treated mice (Fig. 1c). Lipid accumulation in the liver was increased by alcohol exposure in CO-treated mice, and markedly increased in OFO-treated group, but remarkably decreased by FO treatment, as measured by hematoxylin and eosin (H&E) (Fig. 1d) and oil red O staining (Supplementary Fig. 3a). Taken together, these results indicate that hepatic steatosis and oxidative stress induced by alcohol exposure are exacerbated by dietary OFO but reversed by FO.

a Plasma levels of ALT and AST (n = 8–9). b Plasma TG level and hepatic TG concentration (n = 8–9). c Oxidative stress parameters in the liver, including CAT, SOD, GSH, and MDA (n = 7). d Representative H&E staining of liver sections (scale bar, 50 μm). Data were expressed as mean ± SD. Labeled means without a common letter differ within the column (p < 0.05). CO corn oil, OFO oxidized fish oil, FO fish oil.
To determine whether the absorption and metabolism of ethanol is implicated in these deteriorative effects of OFO, the circulating ethanol concentration and hepatic expression of cytochrome P450 2E1 (CYPE21) were measured. After chronic-plus-single-binge ethanol feeding, the ethanol concentrations in plasma were greatly elevated in both CO- and OFO-treated mice. However, plasma ethanol levels were comparable between CO and OFO groups after ethanol exposure (Supplementary Fig. 4a). Similar tendency was observed for hepatic expression of CYPE21 (Supplementary Fig. 4b).
OFO aggravates ethanol-induced hepatic inflammation
Alcohol exposure increased the hepatic inflammation in both CO- and OFO-treated mice, as shown by the increased pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) and the chemokine (MCP-1), and the decreased anti-inflammatory cytokine (IL-10), but the effects were much greater in OFO-treated mice. While FO supplement decreased these pro-inflammatory factors induced by ethanol feeding, but did not rescue hepatic IL-10 (Fig. 2a, b), we speculated that the inhibition of pro-inflammatory factors, but not normalization of anti-inflammatory cytokines, contribute to the protective effects of FO against alcoholic hepatic inflammation in the mouse model of chronic-plus-single-binge ethanol feeding. Consistently, the immunofluorescent staining of F4/80, a specific monocyte/macrophage marker, showed that more F4/80-positive cell number was observed in the section of liver tissue from OFO-treated mice after alcohol feeding, compared to CO group. While FO decreased the macrophage infiltration in the liver (Supplementary Fig. 3b). These data suggest that dietary OFO exacerbates hepatic inflammation upon alcohol challenge.

a Hepatic level of cytokines (TNF-α, IL-6, IL-1β, and IL-10). b chemokine (MCP-1). c Immunoblot analysis of TLR4, MyD88, phosphorylated p65, and total p65 proteins in the liver. d Immunofluorescent staining of liver sections for the phosphorylated p65 (red), nucleus was stained with DAPI (blue; scale bar, 25 μm). Data were expressed as mean ± SD (n = 6). Labeled means without a common letter differ within the column (p < 0.05). CO corn oil, OFO oxidized fish oil, FO fish oil.
Activation of Kupffer cell via TLR4/NF-κB signaling plays a vital role in hepatic inflammation induced by ethanol exposure4,30. Next, we examined the protein expressions of the genes related to TLR4/NF-κB signaling in the liver. As shown in Fig. 2c, OFO-treated mice had a remarkable increase in hepatic expressions of TLR4, and its downstream MyD88 and phosphorylated p65 (p-p65) without changing total p65 expression, compared to CO-treated mice after alcohol feeding. These increased protein expressions were reversed by supplement with FO. In addition, OFO alone mice showed slight increases in hepatic expressions of TLR4, MyD88, and p-p65, compared to CON/CO group (Fig. 2c). Consistently, the immunofluorescent staining revealed the higher hepatic expression of p-p65 in OFO-treated group after alcohol intake, compared to CO-treated mice (Fig. 2d). Collectively, our data demonstrate that TLR4/NF-κB signaling is implicated in OFO-exacerbated hepatic inflammation in alcohol-fed mice.
OFO enhances ethanol-induced intestinal barrier dysfunction
We next examined whether intestinal barrier integrity is influenced by dietary OFO. TJ proteins, the markers of intestinal integrity, play the crucial role in maintaining barrier function of intestinal epithelium31,32. Immunofluorescent and western blot analysis clearly showed the decreased expression of TJ proteins, including ZO-1, occludin, and claudin-4, in jejunum tissue from CO-treated mice after alcohol exposure, along with the increased expression of claudin-2, a TJ protein involving in the formation of paracellular water channel that is typically expressed in leaky epithelial tissues. But these changes were more severe in OFO-treated mice, while were reversed by supplement with FO (Fig. 3a, b). Importantly, OFO alone did not affect the expressions of all TJ protein investigated, compared to CO alone group (Fig. 3a, b). The impaired intestinal barrier integrity leads to the increased intestinal permeability to bacterial products, such as LPS, and the greater bacteria translocation. After chronic-plus-single-binge ethanol feeding, plasma LPS level and the hepatic expression of gram-negative bacterial 16S rRNA, a marker of bacterial translocation, were increased in both CO- and OFO-treated mice, much greater increase was observed in OFO group, but significantly decreased by FO supplement. Although OFO alone group has the slightly increased hepatic expression of 16S rRNA, compared to CO alone group, but their plasma LPS levels were comparable (Fig. 3c, d). While fecal LPS contents were increased in both CO- and OFO-treated mice, OFO alone mice showed the significantly increased LPS level in feces, compared to CO alone group (Fig. 3e). A most plausible explanation is that OFO alone cause the great increased fecal LPS, which may not be diffused into the circulation, because OFO alone cannot greatly disrupt the intestinal epithelial barrier.

a Immunofluorescent staining of intestinal TJ proteins, including ZO-1, claudin-4, occludin, and claudin-2, in jejunum tissue (scale bar, 25 μm). b Immunoblot analysis of intestinal TJ proteins. c Plasma LPS levels (n = 5–6). d The hepatic expressions of gram-negative bacteria 16S rRNA (n = 7). e Fecal LPS contents (n = 7). f Immunofluorescent staining of TNF-α (green) and F4/80 (red) in the sections of jejunum tissue (scale bar, 25 μm). Data were expressed as mean ± SD. Labeled means without a common letter differ within the column (p < 0.05). CO corn oil, OFO oxidized fish oil, FO fish oil.
An increasing evidence has demonstrated that intestinal inflammation contributed to intestinal barrier dysfunction33. To further investigate the role of OFO in intestinal inflammation in ethanol-fed mice, immunofluorescent staining of F4/80 and TNF-α in jejunum tissue was examined. As shown in Fig. 3f, the intestinal expressions of F4/80 and TNF-α were slightly increased in CO-treated group after alcohol exposure, but remarkably increased in OFO-treated mice. OFO alone group showed the higher expressions, compared to CO alone mice, even CO plus ethanol group (Fig. 3f).
OFO deteriorates alcohol-induced intestinal dysbiosis
Intestinal dysbiosis and bacterial products play the crucial role in pathogenesis of ALD5. To whether intestinal dysbiosis contributes to OFO-aggravated alcoholic liver injury, intestinal microbiome was profiled by the analysis of bacterial 16S rRNA V3-V4 region. Rank abundance curve showed the sequencing depth was enough sufficient to characterize the bacterial communities in the samples (Fig. 4a). Principal coordinate analysis (PCoA) with unweighted UniFrac distance showed the tight clusters of the samples from each group. OFO-treated mice clustered closely regardless ethanol and pair feeding, which were far apart from other groups (Fig. 4b). Furthermore, ethanol-fed group, including ETH/CO and ETH/FO mice but except ETH/OFO, had similar microbiota profiles, and clustered separately with CON/CO. These results were confirmed by unweighted pair group method with arithmetic mean (UPGMA) tree (Fig. 4c), which indicated that the diversity of intestinal microbiota was slightly changed by ethanol feeding, but strongly influenced by dietary OFO. We next identified a specific intestinal microbiota related to OFO treatment. As shown in Fig. 4d, e, Phyla analysis showed that Bacteroidetes, Proteobacteria, and Firmicutes are dominant microbiota in all groups of mice, both ETH and OFO-fed mice had the increased Bacteroidetes and Proteobacteria, and the decreased Firmicutes, compared to CON/CO group. Furthermore, the proportion of Proteobacteria was slightly higher in CON/OFO mice (22%) over CON/CO mice (9.1%), and significantly increased in ETH/OFO mice (34%), while, FO treatment exhibited the moderate decrease in Proteobacteria (16%). Cladogram analysis showed that the OFO- and ETH-fed mice mainly clustered in Proteobacteria phylum, but exhibited at different taxonomic levels. The beta-proteobacteria and delta-proteobacteria family of Proteobacteria phylum was dominant in ETH/CO mice, while grama-proteobacteria family was dominant in OFO-fed mice. Moreover, Enterobacter and Klebsiella genus was specifically clustered in ETH/OFO mice, but bacteria Desulfovibrio and Parasutterela were dominated in ETH/CO mice (Fig. 4f). These data indicated that OFO-exacerbated alcoholic liver injury might be associated with intestinal dysbiosis, especially the specific microbiota profile of Proteobacteria.

a Rank abundance curve. b Principal coordinate analysis (PCoA) (n = 6–9). c Unweighted pair group method with arithmetic mean (UPGMA) analysis for the shifts in microbial community structure (n = 8–9). d Bar chart showing the relative abundance of intestinal microbiota on phylum level. e One-way ANOVA analysis of the composition of intestinal microbiota on phylum level (n = 8–9). f Cladogram analysis showed that taxa most differently associated with mice fed with CON or ETH diet (Wilcoxon rank-sum test). Circle sizes in the cladogram plot are proportional to bacterial abundance. The circle represents from the inner circle to the outer circle: phyla, genus, class, order, and family. CO corn oil, OFO oxidized fish oil, FO fish oil.

a Fecal LPS contents (n = 8). b Plasma LPS levels (n = 8). c Hepatic expression of 16S rRNA (n = 6). d Immunofluorescent staining. e Immunoblot analysis of intestinal TJ proteins, including ZO-1, occludin, claudin-4, and claudin-2 in the sections of jejunum tissue (scale bar, 25 μm). Data were expressed as mean ± SD. Labeled means without a common letter differ within the column (p < 0.05). CO corn oil, OFO oxidized fish oil, FO fish oil.
ABx abolishes OFO-aggravated alcoholic liver injury
To reveal the role of gut bacteria in OFO-aggravated liver injury in ethanol-fed mice, nonabsorbable antibiotics (ABx), comprising of polymyxin B and neomycin, were orally administered to the mice daily during liquid-diet adaptation and subsequent alcohol-feeding period. Nonabsorbable antibiotics did not alter the intestinal absorption and metabolism of ethanol, as measured by plasma ethanol level and hepatic expression of CYPE21 (Supplementary Fig. 5a, b). Gut sterilization by ABx significantly decreased LPS levels in feces and plasma, and bacterial translocation in both ETH/CO and ETH/OFO groups, but there is no difference between ETH/CO and ETH/OFO after ABx treatment (Fig. 5a–c). Immunofluorescent (Fig. 5d) and western blot (Fig. 5e) analysis indicated that the expressions of TJ proteins, ZO-1, occludin, claudin-4, and claudin-2 in jejunum tissue were reversed by ABx treatment in ethanol-fed mice, and OFO-aggravated intestinal barrier dysfunction was completely abolished by ABx treatment. Similar tendency was observed in immunofluorescent staining of F4/80 and TNF-α in jejunum tissue (Supplementary Fig. 6a).
Consistently, nonabsorbable antibiotics remarkably inhibited the hepatic inflammation in ethanol-fed mice, as evidenced by the reduced hepatic inflammatory cytokines TNF-α, IL-6, IL-1β (Fig. 6a), and MCP-1 (Fig. 6b), the less F4/80-positive monocytes/macrophage (Fig. 6c) in the liver, and the decreased hepatic expressions of TLR4, MyD88, and p-p65 (Fig. 6d and Supplementary Fig. 6b). These results demonstrated that OFO-exacerbated hepatic inflammatory response was blunted by ABx treatment. Undoubtedly, intestinal sterilization with nonabsorbable antibiotics prevented hepatic steatosis and injury in alcohol-fed mice, as determined by plasma biochemical analysis (Fig. 7a), hepatic TG content assay (Fig. 7b), and histological staining of H&E and oil red O (Fig. 7c). These data suggest that the worse effects of OFO on alcoholic liver injury are resorted after ABx treatment.

a Hepatic level of cytokines (TNF-α, IL-6, IL-1β, and MCP-1) (n = 6–7). b Chemokine (MCP-1) (n = 7). c Immunofluorescent staining of F4/80 in the liver section (scale bar, 25 μm). d Hepatic expressions of TLR4, MyD88, p65, and phosphorylated p65 proteins. Value were expressed as mean ± SD. Labeled means without a common letter differ within the column (p < 0.05). CO corn oil, OFO oxidized fish oil, FO fish oil.

a Plasma levels of ALT, AST, and TG. b Hepatic TG contents. c Representative H&E and Red Oil O staining of liver tissues (scale bar, 200 μm). Value was expressed as mean ± SD (n = 8). Labeled means without a common letter differ within the column (p < 0.05). CO corn oil, OFO oxidized fish oil, FO fish oil.
OCA blunts OFO-aggravated liver injury in alcohol-fed mice
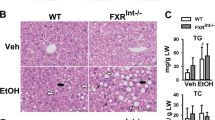
Alcohol and its metabolites disrupt intestinal barrier integrity, resulting in the increased permeability of intestinal barrier. Our data also indicated that OFO-induced intestinal inflammation may be involved in intestinal barrier dysfunction. Obeticholic acid (OCA), a potent agonist of farnesoid X receptor (FXR), is effective in the treatment of nonalcoholic steatohepatitis with liver fibrosis in animal models34 and in patients35. OCA has been also shown to ameliorate gut barrier dysfunction and bacterial translocation in cholestatic36 and in cirrhotic rats37. To further elucidate the role of intestinal barrier in OFO-aggravated alcoholic liver injury, OCA was gavaged daily (30 mg kg−1) to the mice during alcohol-feeding period. As expected, FXR agonist OCA upregulated the expressions of TJ proteins, ZO-1, occludin and claudin-4 in jejunum tissue, and suppressed the intestinal inflammation in ethanol-fed mice (Supplementary Fig. 7). OCA treatment remarkably decreased LPS levels in plasma, but not changed in feces (Fig. 8a). Consistently, OCA inhibited the hepatic inflammation in ethanol-fed mice, as manifested by the reduced hepatic inflammatory cytokines TNF-α and IL-6 (Fig. 8b), and less F4/80-positive monocytes/macrophage in the section of liver tissue (Fig. 8c). Eventually, OCA treatment prevented hepatic steatosis and injury in alcohol-fed mice, as examined by histological analysis (Fig. 8d), plasma levels of AST and ALT (Fig. 8e), and hepatic TG quantification (Fig. 8f). These results demonstrate that stabilization of intestinal barrier by the treatment of FXR agonist OCA blunts OFO-aggravated liver injury in alcohol-fed mice.

a Fecal LPS contents. b Plasma LPS levels. c Inflammatory cytokines, TNF-α, and IL-6 in the liver. c Immunofluorescent analysis of hepatic F4/80 expression. d Representative H&E (scale bar, 100 μm) and Red Oil O staining of liver tissues (scale bar, 50 μm). e Plasma levels of ALT and AST. f Hepatic TG contents. Value was expressed as mean ± SD (n = 4). Labeled means without a common letter differ within the column (p < 0.05). CO corn oil, OFO oxidized fish oil, FO fish oil.
Discussion
In recent decades, an increasing number of studies has demonstrated that n-3 PUFAs exerted the beneficial effects on hepatic steatosis and liver injury induced by excess alcohol exposure9,22,23,24. However, the impact of n-3 PUFAs in ALD have been still inconsistent or controversial. Numerous studies indicated that the combination of n-3-rich FO and ethanol exacerbates hepatic steatosis, inflammation, and liver injury, which was even used as pathological animal model for ALD25,26,27,28. The explanatory hypotheses proposed in these studies are that n-3 PUFAs in FO offer the preferential substrates for lipid peroxidation, further increasing oxidative stress and inflammation in the liver, eventually resulting in the exacerbated ALD26,27,28. While n-3 PUFAs with high degree of unsaturation, especially long-chain n-3 PUFAs, are more vulnerable to oxidation. The exogenous supplement of n-3 PUFAs might competitively reduce the potential of PUFAs in phospholipid bilayer of cell membrane to react with reactive oxygen species induced by alcohol exposure, leading to the protective effects against cell membrane damage. Interestingly, the positive results were invariably observed in the mouse-fed n-3 PUFAs with high purity29, and fat-1 transgenic mice22,38,39 in which n-3 PUFAs endogenously convert from n-6 PUFAs, thus avoiding PUFA oxidation ex vivo. It is unclear whether the exacerbated alcoholic liver injury observed in those studies with negative results is caused by n-3 PUFAs or its oxidative products already presenting in FO. Actually, the n-3 PUFA oxidation in the animal feeding stuffs was rarely concerned in the most of n-3 PUFA-related studies, likely contributing to the inconsistent outcomes2. In the present study, the probable impacts of the OFO on the pathogenesis of alcohol-induced liver injury were investigated. Our results demonstrated that alcohol-induced hepatic steatosis and inflammation are exacerbated by dietary OFO but prevented by unoxidized FO. The OFO-aggravated liver injury was associated with accentuating alcohol-induced intestinal dysbiosis and barrier dysfunction, subsequently enhancing gut-derived endotoxin-mediated inflammation in the liver.
Accumulating studies has demonstrated that gut microbiota dysbiosis plays a crucial role in the pathogenesis of ALD5. Alcohol exposure causes the overgrowth of pathogenic bacteria in the gut, in particular Gram-negative bacteria, leading to the overproduction of endotoxin4. It is well documented that gut microbiota manipulation, including fecal microbial transplantation and the treatment of probiotics or antibiotics, ameliorate endotoxemia, and liver injury induced by alcohol exposure in humans or rodent models4,5. In this study, sequencing of 1S ribosomal RNA indicated that both alcohol exposure and OFO induced a significant increases in the proportions of Gram‐negative bacteria Bacteroidetes and Proteobacteria, and the decrease in Firmicutes, a Gram‐positive bacteria, compared to the pair-fed group, which are consistent with the previous studies40,41. Furthermore, the proportion of Proteobacteria was dramatically increased in ETH/OFO mice, and decreased in ETH/FO group, over alcohol-alone and OFO alone groups, indicating the alternation of Proteobacteria might be related to OFO treatment. Importantly, the specific bacteria taxa of Proteobacteria were discriminated between alcohol-fed mice and OFO-fed mice. These results were in line with the previous studies in which a high abundance of Proteobacteria was observed in fecal microbial communities of alcoholics and cirrhotic patients, and the degree of its overgrowth was correlated with the severity of the liver injury42. Proteobacteria has been considered as the microbial signature of intestinal dysbiosis, and its composition alternation was also observed in patients with intestinal inflammation-related diseases and metabolic syndrome43. However, the potential reason for the bacterial overgrowth caused by OFO remains to be further investigated. In addition, PCoA analysis showed that ETH/CO and ETH/FO group exhibited close microbiota profiles and clustered separately with CON/CO (Fig. 4b), which was confirmed by UPGMA analysis (Fig. 4c). These results indicated that overall composition of intestinal microbiota was greatly disturbed by alcohol exposure regardless CO- and FO-feeding, and FO treatment failed to recover microbial community structure influenced by alcohol. However, phyla analysis showed that FO treatment exhibited the significant decrease in the specific bacteria taxa of Proteobacteria that greatly elevated by alcohol exposure. Collectively, FO treatment may inhibit alcohol-induced overgrowth of Proteobacteria, but not largely change microbial community structure, which contribute to the protective effects of FO against alcoholic liver injury.
The overgrowth of gut microbiota, especially Gram-negative, increases the bacterial products, endotoxin, or LPS. A large body of clinical and animal studies has supported that the circulating levels of gut-derived LPS is closely correlated with the development of liver injury induced by alcohol exposure7,8. Gut-derived LPS diffuses via the disrupted intestinal barrier from the intestine into the portal vein, subsequently activates hepatic Kupffer cells by TLR4 signaling pathway, resulting in inflammation in the liver9. In this study, alcohol exposure caused the increases in the LPS levels in both plasma and feces, these increases were further enhanced by dietary OFO. Consistently, OFO aggravated alcohol-induced hepatic inflammation and liver injury, these effects were completely abolished by intestinal sterilization using nonabsorbable antibiotics. Our findings suggest that OFO-exacerbated ALD is associated with intestinal dysbiosis and gut-derived LPS-mediated hepatic inflammation.
Another contributor to alcoholic endotoxemia is gut leakiness. Alcohol and its metabolite acetaldehyde disrupt the integrity of the intestinal epithelial cell layer and increase intestinal permeability, facilitating the diffusion of endotoxin and translocation of intestinal bacteria. Besides the elevated plasma LPS level, we observed the increased hepatic expression of bacteria 16S rRNA, a signature of bacterial translocation, in alcohol-fed mice. It is intriguing that OFO alone group has the comparable plasma LPS, but significantly increased level of fecal LPS, indicating that the high level of intestinal LPS induced by OFO is not allowed to influx into the portal vein. A most plausible explanation is that OFO alone could not impair intestinal integrity, which was further confirmed by immunofluorescent and western blot analysis of intestinal TJ proteins. These results suggest that the main functions of OFO and alcohol are to induce intestinal dysbiosis and intestinal barrier dysfunction in OFO-aggravated alcoholic liver injury, respectively.
TJ proteins, including ZO-1, occludins, and claudins, serve as specialized junctional complexes that offer physical intestinal barrier. Acetaldehyde induces tyrosine phosphorylation of TJ proteins, leading to intestinal hyperpermeability to macromolecules. The downregulated intestinal TJ proteins were observed in rodent models and the patients with ALD44. In this study, alcohol exposure impaired intestinal barrier function, as evidenced by the deceased expression of ZO-1, occludin, and claudin-4, and the increased expression of claudin-2, a mediator of barrier dysfunction during intestinal inflammation45, which was further aggravated by combination of OFO and alcohol. However, OFO alone did not affect the TJ protein expression, suggesting that beside the action of acetaldehyde, another factor might be implicated in intestinal barrier dysfunction. Accumulating clinical and experimental evidence has indicated that intestinal inflammation induced by dysbiosis and TNF-α receptor I signaling in epithelial cells contributes to disruption of the intestinal barrier46,47. Our data indicated that dietary OFO significantly increased intestinal expression of TNF-α and F4/80, suggesting that intestinal inflammation is implicated in OFO-deteriorated intestinal barrier dysfunction in alcohol-fed mice.
Collectively, our data clearly show that the OFO exacerbates alcoholic liver injury via enhancing intestinal dysbiosis, barrier dysfunction, and hepatic inflammation mediated by gut-derived LPS in mice, as illustrated in Fig. 9. To our knowledge, this study provides the new insight into the inconsistent outcome regarding the role of n-3 PUFAs in ALD and other diseases.

Alcohol and its metabolite, acetaldehyde, mainly disrupt intestinal integrity by down-regulation of intestinal epithelial tight junction proteins. OFO exacerbates intestinal dysbiosis, particularly the overgrowth of bacteria Proteobacteria, leading to the increased bacterial endotoxin. The impaired intestinal barrier facilitates endotoxin translocation from the intestinal lumen into the portal circulation. Subsequently, the gut-derived endotoxin activates hepatic Kupffer cells by TLR4-mediated NF-κB signaling pathway, resulting in hepatic inflammation, which induces hepatocyte damage. In summary, OFO exacerbates alcoholic liver injury via enhancing intestinal dysbiosis, barrier dysfunction and hepatic inflammation mediated by gut-derived endotoxin in mice. NF-κB nuclear factor κB, TLR4 Toll-like receptor 4, OFO Oxidized fish oil.
Methods
OFO preparation
FO (45.5% EPA, 33.5% DHA, and 13% other n-3 PUFAs) was purchased from KangDao Biotechnology Limited Company (Shenzheng, China). OFO was prepared by heating the fresh FO on a watch glass at 65 °C for 2 weeks for the adequate oxidation.
Measurements of lipid oxidation and fatty acid profile
POV assay was performed by visual titration of iodine method to evaluate the primary lipid-oxidation products as described previously48. The POV was expressed as milliequivalents peroxide per kilogram (meq kg−1). Measurement of p-anisidine value (AV) was determined using a colorimetric method49. The results of AV were calculated according to the formula “AV = 10 × [1.2 × (A2 − A1)]/SW, where SW is the sample weight (g). The POV and AV were obtained to evaluate the total oxidation value (Totox) according to the formula “Totox = POV × 2 + AV.” In order to measure the second oxidation products in the oil samples, the TBARS assay was conducted with minor modifications as described previously50. The malonaldehyde (MDA) was used as the standard. TBARS were expressed as mg MDA equivalents/kg sample. In addition, the fatty acid profiles in FO and OFO were measured by GC-MS, as previously described51.
Animal model and drug administration
Male C57BL/6 mice (10–12 weeks old) were housed in specific pathogen-free room at Experimental Animal Center, Guangdong Pharmaceutical University (Guangzhou, China). Alcoholic liver injury was induced by a NIAAA model of chronic-plus-binge alcohol feeding as described previously52. Briefly, after 1 week of acclimatization with liquid control diet, all mice were fed the modified Lieber-DeCarli liquid diets containing ethanol (ETH) or isocaloric maltose dextrin as the CON diet (TROPHIC Animal Feed High-Tech Co., Ltd Nantong, Jiangsu, China), supplementing with CO or OFO for 10 days. CO was used as fat source in the modified Lieber-DeCarli liquid control diets to eliminate the possible effects of different energy intake and other type of fats. An additional group of mice received ethanol liquid-diet containing unoxidized FO was used to compare the effects of OFO. Diet compositions were shown in Supplementary Table 2. On day 11, ethanol-fed mice and control mice were received with a single dose of ethanol (31.5%, v/v, 5 g kg−1) and an isocaloric maltose dextrin, respectively. After 9-h fasting, all mice were euthanized, and plasma, liver, intestine, and fresh feces were harvested for further analysis. Animal feeding process was illustrated in Supplementary Fig. 2a. Animal practices were approved by the Animal Ethics Committee, Institute of Chinese Medical Sciences, University of Macau.
The second group of C57BL/6 male mice was fed the Lieber-DeCarli alcohol liquid diets supplemented with CO or OFO, nonabsorbable antibiotics (ABx; Polymyxin B, 150 mg kg−1 BW and Neomycin, 200 mg kg−1 BW) or an equal volume of vehicle were orally administered to the mice daily during liquid-diet adaptation and subsequent alcohol-feeding period (Supplementary Fig. 2b)16.
The third group of C57BL/6 male mice was fed the Lieber-DeCarli alcohol liquid diets supplemented with CO or OFO, the mice were gavaged with OCA (30 mg kg−1) or equal volume of vehicle during the period of alcohol feeding (Supplementary Fig. 2c).
Biochemical and histological analysis
Plasma activities of ALT and AST were determined using the corresponding commercial reagent kits (Nanjing Jianchen Bioengineering Institute, Nanjing, China). TG level and ethanol concentration in plasma were analyzed by commercially available quantification kits (BHKT Clinical Reagent Co., Ltd, Beijing, China) and an Ethanol Assay Kit following the manufacturers’ instructions (BioVision, Milpitas, CA, USA), respectively. LPS levels in plasma and feces were determined using a commercially available ELISA kit (Cloud-Clone Corp., Business Co., Ltd, Wuhan, China). Histopathological changes of liver were observed by H&E staining as standard protocol elsewhere.
Measurement of hepatic steatosis
To evaluate hepatic lipid accumulation, liver samples were determined by Oil Red O staining and triacylglycerol quantification. After fixing in 4% phosphate-buffered paraformaldehyde, frozen sections (8 μm) were stained with Oil Red O (Sigma-Aldrich, St Louis, USA) for 10 min and subsequently counterstained with hematoxylin according to a standard protocol. For the quantification of hepatic TG level, 50 mg of liver sample was homogenized and mixed with chloroform/methanol (2:1, v/v) overnight. After centrifugation, the low lipid phase was dissolved in chloroform, and was collected to determine lipid level using the TG quantification kit (Beijing BHKT clinical reagent Co., Ltd, Beijing, China). Hepatic TG content was normalized to correspondingly liver weight and expressed as mg kg−1.
Measurements of oxidative stress parameters and cytokines
Liver homogenates were prepared in the cold radioimmunoprecipitation assay (RIPA) buffer (Beyotime Institute of Biotechnology, Nanjing, China) following the manufacturer’s instructions. The enzymatic activities of CAT and SOD, and levels of hepatic GSH and MDA by commercially available kits according to the manufacturer’s instruction (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). For measurement of the hepatic inflammatory cytokines and chemokines, the liver tissues were homogenized and collected to determine the levels of TNF-α, IL-1β, IL-10, and MCP-1 using Mouse ELISA MAX™ Standard kits according to the manufacturer’s instruction (BioLegend Inc., San Diego, CA, USA), respectively. All values were normalized to hepatic total protein.
Immunofluorescent staining
Immunofluorescence analysis for the expression of F4/80 and p65 in liver tissue, and TJ proteins in jejunum tissue were performed as described9. In brief, the cryosections of liver or intestine tissues (8 μm) were fixed in cold acetone for 15 min, washed with phosphate-buffered saline (PBS) for three times, and blocked nonspecific binding with 5% normal goat serum in PBS. For p65 staining, permeabilization is prior to block the endogenous peroxidase. After incubation with primary antibodies (1:100) overnight at 4 °C, the sections were washed with PBS three times, subsequently incubated with Alexa Fluor® 568 or Alexa Fluor® 488 goat anti-rabbit IgG (1:200, Life Technologies) at room temperature for 2 h in dark. Nuclei were stained with DAPI, followed by visualization using a Leica TCS SP8 confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany).
Immunoblot assay
Total proteins of partial liver and jejunum tissues were extracted with RIPA lysis buffer. Equivalent amount of protein samples in each group were separated on 8–10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After electrophoresis, proteins were transferred into polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA). After blocking with 5% non-fat dry milk, the membranes were incubated with specific primary antibodies (Table S3) independently overnight at 4 °C, and then incubated with corresponding horseradish-peroxidase-conjugated secondary antibodies.
Quantitative PCR analysis
Total RNA was extracted from homogenized liver samples using commercial Trizol reagent (Takara Bio., Tokyo, Japan) according to the manufacturer’s protocol. Quantitative PCR were conducted using SYBR green (Applied Biosystems) in a Stratagene Mx3005P multiple quantitative PCR system (Agilent Technologies, Santa Clara, CA, USA). The sequences for the primer pair of 16S rRNA were shown as follows (Forward: 5′-AGAGTTTGATCCTGGC TCAG-3′, Reverse: 5′-TGCTGCCTCCCGTAGGAGT-3′), and 18S rRNA (Forward: 5′-ACGG ACCAGAGCGAAAGCAT-3′, Reverse: 5′-TGTCAATCCTGTCCGTGTCC-3′). The gene expression level of 16S rRNA was normalized to 18S RNA.
Microbial analysis
Fresh fecal pellets from individual mice were collected from each mice immediately upon defecation, frozen subsequently stored at −80 °C. Bacterial genomic DNA was extracted from fecal samples using the Power Fecal™ DNA Isolation kit (MO BIO Laboratories, Carlsbad, CA). DNA samples were sequenced and amplified using the hypervariable regions of V3-V4 of bacterial 16S ribosomal DNA (PCR1F_460: 5′-CTTTCCCTACACGACGCTCTTCCGATCTACGG RAGGCAGCAG-3′, PCR1R_460: 5′-GGAGTTCAGACGTGTGCTCTTCCGATCTTACCAGGGT ATCTAATCCT-3′). The amplicons of PCR products were extracted on agarose gels and purified using an Axygene DNA gel extraction kit (Coring, NY, USA). The purified amplicons were sequenced on Illumina Miseq 2500 sequencing platform (Major BioPharm Technology, Shanghai, China). The raw sequences from original DNA fragments were assembled using Trimmomatic and FLASH programs, and then filtered the default parameters using the quantitative insights into microbial ecology (QIIME v1.9.0)31. The non-chimeric sequences were demultiplexed clustered into species-level (97% similarity) operational taxonomic units (OTUs) using a de novo OTU picking protocol. Representative sequence for corresponding OTU was identified to annotate taxonomic assignment using the RDP classifier (version 2.2)53. Analysis of Simpson’s diversity index, Shannon–Wiener index, and rarefaction estimates were calculated using QIIME. Bacterial alpha diversity was performed using Shannon Index. Analysis of the prevalence of OTUs <5% were discarded. Data process using R software v2.14.1 restricted the same taxonomic assignment in the merged OTUs. Results are represented as the mean ± SEM. Beta diversity was performed using unweighted unifrac distances visualizing by PCoA and UPGMA. Cladograms generated from linear discriminant analysis coupled with effect size were performed to identify the most differentially abundant taxa between groups at the different taxonomy levels.
Statistics and reproducibility
Statistical analyses were conducted using GraphPad Prism 6.0 package (San Diego, CA, USA). All data, except in microbial analysis, are presented as mean ± standard deviation. One-way analysis of variance with the Student’s t test or Tukey’s post hoc test was performed to evaluate the difference between groups. The value of p < 0.05 was considered statistically significant. All experiments, except microbial analysis, were conducted in at least three independent replicates. The sample size is indicated for each experiment in the corresponding figure legend.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data supporting the findings of this work are available within the paper and its Supplementary files or available from the corresponding author upon reasonable request. Raw data underlying plots in figures are available in Supplementary Data 1. The unprocessed blot images with size markers are provided in Supplementary Information.
References
Masarone, M. et al. Epidemiology and natural history of alcoholic liver disease. Rev. Recent Clin. Trials 11, 167–174 (2016).
Wang, M. et al. n-3 Polyunsaturated fatty acids for the management of alcoholic liver disease: a critical review. Crit. Rev. Food Sci. Nutr. 59, S116–S129 (2019).
Organization, W. H. Global Status Report on Alcohol and Health. Organization W. H., Geneva (2018).
Szabo, G. Gut–liver axis in alcoholic liver disease. Gastroenterology 148, 30–36 (2015).
Bajaj, J. S. Alcohol, liver disease and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 16, 235–246 (2019).
Yan, A. W. et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 53, 96–105 (2011).
Wang, M. et al. Saponins isolated from the leaves of Panax notoginseng protect against alcoholic liver injury via inhibiting ethanol-induced oxidative stress and gut-derived endotoxin-mediated inflammation. J. Funct. Foods 19, 214–224 (2015).
Louvet, A. & Mathurin, P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 12, 231–242 (2015).
Wang, M. et al. Preventive effect of α-linolenic acid-rich flaxseed oil against ethanol-induced liver injury is associated with ameliorating gut-derived endotoxin-mediated inflammation in mice. J. Funct. Foods 23, 532–541 (2016).
Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 50, 638–644 (2009).
Keshavarzian, A. et al. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 50, 538–547 (2009).
Atkinson, K. & Rao, R. Role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 280, G1280–G1288 (2001).
Suzuki, T., Seth, A. & Rao, R. Role of phospholipase Cγ-induced activation of protein kinase Cϵ (PKCϵ) and PKCβI in epidermal growth factor-mediated protection of tight junctions from acetaldehyde in Caco-2 cell monolayers. J. Biol. Chem. 283, 3574–3583 (2008).
Cao, M. et al. Amelioration of IFN-γ and TNF-α-induced intestinal epithelial barrier dysfunction by berberine via suppression of MLCK-MLC phosphorylation signaling pathway. PloS ONE 8, e61944 (2013).
Su, C. et al. Neohesperidin dihydrochalcone versus CCl4-induced hepatic injury through different mechanisms: the implication of free radical scavenging and Nrf2 activation. J. Agric. Food Chem. 63, 5468–5475 (2015).
Chen, P. et al. Dysbiosis‐induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 61, 883–894 (2015).
de Castro, G. S. & Calder, P. C. Non-alcoholic fatty liver disease and its treatment with n-3 polyunsaturated fatty acids. Clin. Nutr. 37, 37–55 (2018).
Feng, R. et al. Endogenous n-3 fatty acids alleviate carbon-tetrachloride-induced acute liver injury in fat-1 transgenic mice. Oxid. Med. Cell. Longev. 2016, 7962948 (2016).
Feng, R. et al. Acetaminophen-induced liver injury is attenuated in transgenic fat-1 mice endogenously synthesizing long-chain n-3 fatty acids. Biochem. Pharmacol. 154, 75–88 (2018).
Li, Y. et al. Endogenous n-3 polyunsaturated fatty acids attenuate T cell-mediated hepatitis via autophagy activation. Front. Immunol. 7, 350 (2016).
Schmocker, C. et al. Omega-3 fatty acids alleviate chemically induced acute hepatitis by suppression of cytokines. Hepatology 45, 864–869 (2007).
Wang, M. et al. Omega-3 polyunsaturated fatty acids ameliorate ethanol-induced adipose hyperlipolysis: a mechanism for hepatoprotective effect against alcoholic liver disease. Biochim. Biophys. Acta 1863, 3190–3201 (2017).
Wang, M. et al. Dietary alpha-linolenic acid-rich flaxseed oil prevents against alcoholic hepatic steatosis via ameliorating lipid homeostasis at adipose tissue-liver axis in mice. Sci. Rep. 6, 26826 (2016).
Wada, S. et al. Fish oil fed prior to ethanol administration prevents acute ethanol-induced fatty liver in mice. J. Hepatol. 49, 441–450 (2008).
Sachan, D. S., Yatim, A. M. & Daily, J. W. Comparative effects of dietary corn oil, safflower oil, fish oil and palm oil on metabolism of ethanol and carnitine in the rat. J. Am. Coll. Nutr. 21, 233–238 (2002).
Nanji, A. A. et al. Markedly enhanced cytochrome P450 2E1 induction and lipid peroxidation is associated with severe liver injury in fish oil-ethanol-fed rats. Alcohol. Clin. Exp. Res. 18, 1280–1285 (1994).
Donohue, T. M. et al. Lysosomal leakage and lack of adaptation of hepatoprotective enzyme contribute to enhanced susceptibility to ethanol-induced liver injury in female rats. Alcohol. Clin. Exp. Res. 31, 1944–1952 (2007).
Nanji, A. A. et al. Dietary saturated fatty acids: a novel treatment for alcoholic liver disease. Gastroenterology 109, 547–554 (1995).
Huang, L. L. et al. Suppression of acute ethanol-induced hepatic steatosis by docosahexaenoic acid is associated with downregulation of stearoyl-CoA desaturase 1 and inflammatory cytokines. Prostaglandins Leukot. Essent. Fat. Acids 88, 347–353 (2013).
Feng, R. et al. A combination of Pueraria lobata and Silybum marianum protects against alcoholic liver disease in mice. Phytomedicine 58, 152824 (2019).
Ferrere, G. et al. Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J. Hepatol. 66, 806–815 (2017).
Shao T. et al. Intestinal HIF-1α deletion exacerbates alcoholic liver disease by inducing intestinal dysbiosis and barrier dysfunction. J. Hepatol. 69, 886–895 (2018).
Llopis, M. et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 65, 830–839 (2016).
Jouihan, H. et al. Superior reductions in hepatic steatosis and fibrosis with co-administration of a glucagon-like peptide-1 receptor agonist and obeticholic acid in mice. Mol. Metab. 6, 1360–1370 (2017).
Mudaliar, S. et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145, 574–582.e1 (2013).
Verbeke, L. et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am. J. Pathol. 185, 409–419 (2015).
Ubeda, M. et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J. Hepatol. 64, 1049–1057 (2016).
Huang, W. et al. Endogenously elevated n-3 polyunsaturated fatty acids alleviate acute ethanol-induced liver steatosis. Biofactors 41, 453–462 (2015).
Warner, D. R. et al. Decreased omega-6:omega-3 PUFA ratio attenuates ethanol-induced alterations in intestinal homeostasis, microbiota, and liver injury. J. Lipid Res. 60, 2034–2049 (2019).
Bjorkhaug, S. T. et al. Characterization of gut microbiota composition and functions in patients with chronic alcohol overconsumption. Gut Microbes 10, 663–675 (2019).
Meroni, M., Longo, M. & Dongiovanni, P. Alcohol or gut microbiota: who is the guilty? Int. J. Mol. Sci. 20, 4568 (2019).
Hauge, T., Persson, J. & Danielsson, D. Mucosal bacterial growth in the upper gastrointestinal tract in alcoholics (heavy drinkers). Digestion 58, 591–595 (1997).
Litvak, Y. et al. Dysbiotic proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 39, 1–6 (2017).
Basuroy, S. et al. Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: protection by EGF and L-glutamine. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G367–G375 (2005).
Luettig, J. et al. Claudin-2 as a mediator of leaky gut barrier during intestinal inflammation. Tissue Barriers 3, e977176 (2015).
Mathurin, P. et al. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology 32, 1008–1017 (2000).
Yoseph, B. P. et al. Chronic alcohol ingestion increases mortality and organ injury in a murine model of septic peritonitis. PLoS ONE 8, e62792 (2013).
Yu, H. et al. Antioxidant activities of aqueous extract from Stevia rebaudiana stem waste to inhibit fish oil oxidation and identification of its phenolic compounds. Food Chem. 232, 379–386 (2017).
Albert, B. B. et al. Fish oil supplements in New Zealand are highly oxidised and do not meet label content of n-3 PUFA. Sci. Rep. 5, 7928 (2015).
Maqsood, S. & Benjakul, S. Comparative studies of four different phenolic compounds on in vitro antioxidative activity and the preventive effect on lipid oxidation of fish oil emulsion and fish mince. Food Chem. 119, 123–132 (2010).
Zhang, X. J. et al. Characterizing plasma phospholipid fatty acid profiles of polycystic ovary syndrome patients with and without insulin resistance using GC-MS and chemometrics approach. J. Pharm. Biomed. Anal. 95, 85–92 (2014).
Bertola, A. et al. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 8, 627–637 (2013).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Acknowledgements
This work was financially supported by the grants from the Research Committee of the University of Macau (MYRG2017-00035-ICMS) and the Science and Technology Development Fund, Macau SAR (File no. 065/2018/A2 and 0034/2019/A1).
Author information
Authors and Affiliations
Contributions
J.-B.W. conceived and designed the experiments, and finalized the manuscript; R.F., L.M., M.W., C.L., and R.Y. performed experiments and acquired data; R.F. wrote the initial manuscript; and H.S. and Y.Y. contributed to statistical analysis. All authors have read, commented on, and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Feng, R., Ma, LJ., Wang, M. et al. Oxidation of fish oil exacerbates alcoholic liver disease by enhancing intestinal dysbiosis in mice. Commun Biol 3, 481 (2020). https://doi.org/10.1038/s42003-020-01213-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-020-01213-8
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
