
Abstract
Multispecies host-parasite evolution is common, but how parasites evolve after speciating remains poorly understood. Shared evolutionary history and physiology may propel species along similar evolutionary trajectories whereas pursuing different strategies can reduce competition. We test these scenarios in the economically important association between honey bees and ectoparasitic mites by sequencing the genomes of the sister mite species Varroa destructor and Varroa jacobsoni. These genomes were closely related, with 99.7% sequence identity. Among the 9,628 orthologous genes, 4.8% showed signs of positive selection in at least one species. Divergent selective trajectories were discovered in conserved chemosensory gene families (IGR, SNMP), and Halloween genes (CYP) involved in moulting and reproduction. However, there was little overlap in these gene sets and associated GO terms, indicating different selective regimes operating on each of the parasites. Based on our findings, we suggest that species-specific strategies may be needed to combat evolving parasite communities.
Similar content being viewed by others


Introduction
Interactions between hosts and parasites shape biodiversity by driving coevolutionary arms races and by regulating populations over ecological timescales. Parasitism is a remarkably successful strategy, occurring in nearly half of the recognized animal phyla1,2. Parasites depend on their hosts which form their principal ecological niches and provide essential resources to survive3. Commonly, multiple parasite generations elapse per host generation. Thus parasites are prone to evolve rapidly and may circumvent any gained host advantage4. The similar selective regimes acting on phylogenetically unrelated parasites resulted in reproducible evolution such as genome size reduction, functional losses associated with reduced metabolism, and adaptive functional gains linked to parasite virulence5. Most models of parasite coevolution are limited to interactions between single focal parasites and their hosts, in part because multispecies dynamics are much more complex6,7. In nature, the reality is that parasite’s success and evolution will also depend on interactions with co-infecting conspecific and heterospecific parasites within its host niche8,9,10. Yet, coevolutionary dynamics involving more than one parasitic species remain poorly understood11,12.
Co-infecting parasites occupying the same host can exclude competitors via direct, or interference competition13,14. One strategy to reduce interspecific competition can be spatial separation within regions of the host gut as adopted by cestodes that naturally co-infect sticklebacks15. Within the same host, spatial segregation that allows parasite coexistence can ultimately lead to intrahost speciation, as found in gill parasites (Dactylogyrus) of freshwater fish (Cyprinidae)16. Niches may also be partitioned temporally, which allows two fungal sibling species (Erysiphe quercicola and E. alphitoides) to exploit the same host tree (Quercus robur) by reducing direct interference17. Alternatively, parasites may shift their strategies to exploit different aspects of the host niche. For example, fire ants (Solenopsis spp.) are parasitized by a genus of decapitating phorid fly (Pseudacteon), which are highly host-specific and have the same life cycle, but specialize on attacking ants in different contexts (e.g., while foraging, vs. at the nest)18. However, almost nothing is known about evolutionary trajectories of speciating parasites. On one hand, they share physiology and host-specific adaptations, which may predispose them to evolve along common lines, particularly in allopatry. On the other hand, selective regimes may be less predictable, or even disruptive, in the case of character displacement.
We investigated the evolution of speciating parasites by sequencing genomes of two economically important mite species that specialize on honey bees (Apis sp.): Varroa jacobsoni and Varroa destructor. The honey bee colony, which is a densely packed community of genetically similar individuals, can host many diseases and parasites. Varroa mites are obligatory ectoparasites which feed on fat bodies of developing larvae/pupae and adult honey bees (shown in V. destructor)19 and reproduce in the brood20. Obviously, host-feeding by Varroa mites injures vital organs of the honey bee, which can also lead to abnormal development21 and to reduced lifespan22. In addition, Varroa mites are known to vector several viruses23 and to transmit pathogenic bacteria24. These combined negative effects on individual honey bee decrease the fitness of the colony as a superorganism. Without acaricide treatments, managed western honey bee colonies were reported to decline and die within two to 3 years20. As its name indicates, V. destructor is considered as the main devastating force behind colony collapses and impact deeply western honey bee (A. mellifera) beekeeping25,26.
The first record of Varroa mite parasitism dates back to 1904 in Java in where female mites were described from an Indo-Malayan Apis cerana honey bee colony (ex. Apis indica)27. Not until the mid-20th century did Varroa draw attention again when they were found to infest western honey bee A. mellifera colonies introduced in far eastern Asia28. Originally from Europe, Africa, and the Middle East, the western honey bee was allopatric to the rest of the Asian Apis species, and had no indigenous brood ectoparasites29. The host-switched mites rapidly spread worldwide and caused dramatic losses of A. mellifera colonies. They were identified as Varroa jacobsoni until the 2000s when a key taxonomic revision of the genus described them as a cryptic sister species V. destructor30. The taxonomic confusion existed and persisted for a while because both mites have similar life cycles and morphology30 and both originally parasitized the widespread eastern honey bee (Apis cerana). The Varroa cryptic species complex was resolved by a combination of morphometrics and mitochondrial sequencing, which identified at least four mite species: V. destructor, V. jacobsoni, V. rindereri, and V. underwoodi30.
As western honey bees were brought into contact with the eastern honey bee in Asia and Oceania, both V. destructor repeatedly colonized this novel host, followed by V. jacobsoni in 2008 in Papua New Guinea28,31. In contrast to the population collapses of A. mellifera, the host-parasite interaction with A. cerana is a balanced relationship in which a tolerance equilibrium is maintained by a range of parasite adaptations and host counter-adaptations32,33,34,35,36,37. Specifically, the eastern honey bee has impressive host defense traits, including (i) self and collective grooming behaviors38, (ii) hygienic behavior where mites are removed from infested cell39, (iii) worker brood death that stops parasite reproduction34,35, (iv) entombing of mites in drone brood, and (v) shorter worker development cycle limiting the mites’ offspring number33. By contrast, most western honey bee populations lack these defensive mechanisms, and hives collapse quickly after Varroa infestation. By contrast, Varroa mites have evolved highly specialized strategies to find, select, feed, reproduce, and hide on their host40. While many mite adaptations have already been discovered20,40, the recent use of ‘omics approaches has quickly broadened our understanding on V. destructor hidden specialization to host37,41,42,43. For example, transcriptomic and proteomic profiling of different V. destructor life stages boosted the identification of genomic regions related to chemosensory system41,43,44, reproductive success42, and linking oogenesis initiation or failure to parasitic reduced metabolism and host defense45,46.
Here, we ask how both mite species evolved toward a tolerance equilibrium with A. cerana—did they follow similar or different evolutionary paths? On one hand, the two species naturally occur in parapatry, which is expected to result from divergent local adaptation, perhaps to the distribution of different A. cerana lineages47,48 or to the climate. In actuality, what drives the parapatric distribution is unclear and recent evolutionary history shows that massive host-associated range shifts are possible. For instance, V. destructor, which was historically absent from Southeast Asia, now occurs there on its novel host A. mellifera in sympatry with V. jacobsoni, which does not parasitize A. mellifera outside of Melanesia. Thus, it is possible that the current geographical distribution resulted from secondary contact following allopatric speciation. This view is further supported by the fact that both species have the genetic capacity to shift to A. mellifera as a novel host, suggesting a level of physiological similarity. We tested these alternative hypotheses (parallel vs. divergent evolution) by generating high-quality hybrid de novo genome assembly of both species and examining them for signatures of adaptive evolution. We also compared them to the recently published genome of a distantly related ectoparasitic mite Tropilaelaps mercedesae, to see whether the patterns appear universal at different timescales. We found here that although the two Varroa species share 99.7% sequence identity across the nuclear genome they have gone on largely dissimilar evolutionary trajectories after their split.
Results
Two new reference genomes for V. destructor and V. jacobsoni
We took advantage of the haplodiploid sex determination of both V. destructor and V. jacobsoni to generate data to reduce the ambiguity due to heterozygous sites for a high contiguous hybrid genome assembly. The sequencing strategy adopted in this study combined shorts reads (Illumina) for which input DNA can be obtained from a single mite individual, long reads of tens of kilobases (PacBio), which requires a large quantity of high-molecular-weight DNA and Hi-C to capture chromosome conformation and interactions. We generated 28.6 Gb (333 ± S.D. 56 bp) and 56.2 Gb (409 ± S.D. 61 bp) of de-duplicated, decontaminated, and merged Illumina reads for V. jacobsoni and V. destructor, corresponding to a coverage of 80× and 152×, respectively. PacBio sequencing yielded 20.3 Gb of V. destructor reads with an N50 of 13.5 kb (66× coverage), which was used to fill 84.4% of the gaps in the scaffolded HiC assembly. Final de novo assemblies were named Vdes_3.0 (GCA_002443255.1) and Vjacob_1.0 (GCA_002532875.1) and are available on NCBI.
Through the k-mer analysis, the genome size estimates ranged from 369.02 to 365.13 Mbp using k = 42 (Supplementary Fig. 1). Both Vdes_3.0 and Vjacob_1.0 final assembly size were close to the predicted estimates but V. jacobsoni genome size was slightly shorter than V. destructor with 365.59 < 368.94 Mbp, respectively (Table 1). Previous assemblies size and contiguity for V. destructor with Vdes_1.049 and Vdes_2.0 were improved as detailed in Table 1. According to the N50 statistics, more than 50% of the genome was in scaffolds of 58.5 Mbp or more for Vdes_3.0 while the Vdes_2.0 N50 was around 0.1 Mbp. When comparing to invertebrate assemblies, Vdes_3.0 genome is among the most highly contiguous with to date the best-reported scaffold N50 and best contig N50 for Acari and Mesostigmata as reported on the i5k database (i5k.github.io/arthropod_genomes_at_ncbi)50.
Using Hi-C data, seven major scaffolds were constructed for V. destructor ranging from 39.4 to 76.9 Mbp with additional of 1,418 nuclear minor scaffolds > 0.2 Mbp. The longest genomic scaffolds correspond to the seven haploid chromosomes (2n = 14) previously detected in V. destructor karyotype51. Contrary to Vdes_3.0, which benefited from long reads and chromosome capture sequencing, V. jacobsoni Vjacob_1.0 genome was assembled from 8241 contigs into 4881 scaffolds with an N50 of 233,810 bp. Similar GC content composed 40.9% were found for both sister species Vdes_3.0 and Vjacob_1.0. The automated NCBI Eukaryote annotation pipeline predicted 10,247 protein-coding genes (out of 12,849 genes and pseudo-genes) for Vdes_3.0, and 10,739 protein-coding genes (out of 15,486 genes and pseudo-genes) for Vjacob_1.052 (based upon transcriptome data available on the database in October 2017). When single haploid male of each Varroa species reads were mapped onto the Vdes_3.0 reference genome, a total of 1,404,212 quality-filtered SNPs were detected. Relative to Vdes_3.0 genome size, sequence divergence with Vjacob_1.0 amounted to 0.38% across the nuclear genome sequence.
Identification and confirmation of Varroa mite species and mtDNA lineages
Mapping and pairwise whole mitochondrial alignment (16,505 bp) showed that Vdes_3.0 diverged of only 0.1% to the reference V. destructor mitogenome (NC004454.2)53,54. Despite presenting similar morphological phenotypes mainly distinguishable through female body size30,55 (Fig. 1), the V. jacobsoni mitochondrial genome diverged from Vdes_3.0 mtDNA at 5.1% of all nucleotides.

V. destructor and V. jacobsoni are morphologically similar sister species originally parasitizing the eastern honey bee (A. cerana). These species were recognized upon the basis of quantitative morphometric and genetic data28. Morphological differences are restricted to differences in body size and shape, as shown in a 3D surface models comparison of two fully sclerotized females. b Both mites parasitize A. cerana as their original host and can coexist in sympatry, occasionally even at the same apiary47,71, but are mainly parapatric121. c With the introduction of A. mellifera, V. destructor can be found on this novel host throughout the original V. jacobsoni range. V. jacobsoni has also extended its range into Papua New Guinea on A. cerana followed by a shift to A. mellifera, where V. destructor is currently absent31
Given that COX1 is a standard marker used to identify the Varroa mite haplogroup, this region was extracted and realigned with 26 described sequences from native and introduced populations. Divergence within the complete COX1 sequences of the V. destructor and V. jacobsoni reference genomes rose up to 5.9%. The partial COX1 sequence (426 bp) of Vdes_3.0 clustered with V. destructor sequences and was clearly distinct from Vjacob_1.0, which clustered with V. jacobsoni haplotypes (Fig. 2a, b). The concatenated COX1, ATP6, COX3, and CYTB sequence (2696 bp) from the Vdes_3.0 mitochondrial genome was identical to that of the invasive Korean K1-1/K1-2 sequence (Fig. 2c).

Inter and intraspecies mtDNA variability across the genus Varroa and host-switched lineages. Different mitochondrial markers used in the literature to discriminate the four Varroa species (and unresolved one) based on partial COX130 (a, b) and intraspecies lineages using larger COX1, ATP6, COX3 and CYTB regions69,78 (a, c). The two cryptic species V. destructor (red) and V. jacobsoni (blue) are genetically divergent as shown by the unrooted phylogenetic tree of Varroa mite partial COX1 sequences (b). Variability of V. jacobsoni is higher than that of any other Varroa species and the reference genome (blue arrow) corresponded to one of the previously detected haplotypes switching on A. mellifera in Papua New Guinea31 (b). Several haplotypes from the Korean and Japanese lineages successfully jumped on A. mellifera (bold)69,78 but only K1-1/K1-2 is quasi-cosmopolitan and can even be retrieved in the native range of the Japanese lineage as illustrated by its presence in Okinawa (red arrow) (c). Ac identified from Apis cerana, Am identified from Apis mellifera
By contrast, the V. jacobsoni (i) partial COX1 region (426 bp) was a unique new haplotype with 99% nucleotide similarity to Java (one transition point mutation G/A) and Bali (two transitions) haplotypes, and (ii) the concatenate sequence (only COX1, ATP6, and COX3) was also a distinct haplotype from the only referenced V. jacobsoni Laos 1 and 2. We named Vjacob_1.0 mtDNA haplotype as Papua New Guinea, which has been formerly reported in host switch events on A. mellifera.
Orthologue genes between Varroa and other Parasitiformes
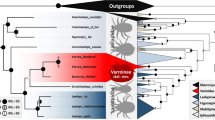
The arthropod genome database at NCBI presently contains only two other published representative Mesostigmata genomes: the brood parasitic honey bee mite Tropilaelaps mercedesae (GCA_002081605.1)56 and the non-parasitic Western predatory mite Metaseiulus (=Typhlodromus or Galendromus) occidentalis (GCA_000255335.1)57. Orthologue prediction was performed by comparing annotated Varroa genes to those of the two other Mesostigmata species gene sets and those of two additional Acari: the parasitic tick Ixodes scapularis (GCA_000208615.1) and the free-living two-spotted mite Tetranychus urticae (GCA_000239435.1). The tick presents the largest set of genes (n = 20,486) and also possessing a haploid genome more than four times larger 1.76 Gb58. Orthology prediction analysis using the six genomes clustered the genes by domain into 7502 super-orthologue groups consisting of orthologues and inparalogues59. A total of 11,123 orthologue groups were predicted and 42.8% were shared by the five parasitiformes and the outgroup spider mite Tetranychus (Supplementary Data 1). The concatenated phylogenetic tree built on the genomic sets based on 4758 genes confirmed the close relationship between the three mite species compared to other Acari (Fig. 3). The honey bee ectoparasitic mites, Varroa, and Tropilaelaps shared 813 lineage-specific orthogroups (akin to gene families) of which 65.9% were Varroidae-lineage-specific. The two sibling Varroa species shared 9628 orthologous genes including 536, which were private to Varroidae.

Positive selection and gene duplication of orthologous genes in parasitic and free-living Acari. For the honey bee mite parasites, host ranges are shown as checkboxes and individual records had to meet the following conditions: (1) Mites had to be found reproducing or observed in bee colonies in independent surveys, and (2) the identity of cryptic Tropilaelaps and Varroa species was confirmed by molecular markers (e.g., COX1 barcoding30,122). The number of orthologous genes in each lineage is circled at the nodes. The number of positively selected genes for each branch is shown in green, with the number of species-specific genes shown in purple. Gene duplications within honey bee parasitic mites are shown in boxes: white squares = shared genes, black squares = Varroa-specific genes by, red and blue squares = V. destructor and V. jacobsoni specific genes, respectively. The two Varroa mites have similar host ranges, through V. jacobsoni also occurs on A. nigrocincta, a close relative of A. cerana, possibly as a result of a recent secondary host shift30,55. Despite sharing the same ancestral host, the Varroa sister species show different patterns of adaptive molecular evolution and exhibit different gene expansion patterns, suggesting different evolutionary trajectories
Orthologous genes specific to the Parasitiformes ancestral branch (i.e., before T. urticae and other Acari species split) was enriched for molecular functions and biological processes involved in several transporters and channel activities (Supplementary Data 2). When considering the Mesostigmata ancestral branch (i.e., before M. occidentalis and the three honey bee parasites species split), GO terms related with structural constituents of the cuticle were the most represented. At the Varroidae ancestral branch level (i.e., before the two species split), orthologous genes showed GO terms mainly related to endonuclease activity.
Divergent patterns of selection among honey bee mites
A total of 234 and 225 genes were detected under positive selection for V. destructor and V. jacobsoni branches, respectively, though only 13 were shared among this list (2.8%) (Fisher’s exact test p = 0.003) (Fig. 3). Additionally, 40 genes were detected under positive selection when looking at the Varroa ancestral branch, i.e., before the two species split. On these set combined, V. destructor and V. jacobsoni presented 81.0% and 80.4% species-specific genes with positive selection signals, respectively (Supplementary Fig. 2 and Supplementary Data 3). The remaining proportion of positively selected genes shared involved the other honey bee mite T. mercedesae rather than with any other Acari. The genomic regions associated with genes under positive selection were found and distributed along all major chromosomes of V. destructor, and, assuming preservation of synteny, in V. jacobsoni as well (Fig. 4a).

Genes and pathways under positive selection in the Varroa sister species. a Red and blue bars (in 5 kb windows) represent locations of genes in V. destructor and V. jacobsoni, respectively. V. jacobsoni data are mapped to V. destructor scaffold positions for comparison. Black asterisks indicated genes under positive selection, shared by both species (n = 12) and detected in the Varroa ancestral lineage prior to the split of the two species (n = 40). b Semantic space analysis of significantly enriched GO terms (BP, CC, and MF) over-represented in genes detected under positive selection for Varroa mites Bubble color indicates the species for which GO terms were enriched (all p-value < 0.05) and size indicates the frequency of the GO term found in the GOA database. There was little overlap in analyses at both gene (2.8%) and functional levels (0.8%), suggesting different selective pressures on the two sister species since they split
Semantic distribution of the GO terms for the genes under positive selection in each Varroa species showed non-significant overlap (p = 0.385 > 0.050). The few terms that overlapped involved biological processes in essential functions such as the molting cycle—chitin-based, fatty acid, and lipid metabolism and responses to pH (Fig. 4b). More specifically, chitin is the main constituent of the mite cuticle hardening it against both bees and the environment. Its metabolism varies throughout the mite life cycle, especially during reproduction or molting42. Fatty acid metabolism could be related to the feeding on the fat body on the host, as well as oogenesis. Although few overlaps exists when GO terms list were reduced using Resnik’s measure, the REVIGO scatter plot and treemap visually suggest functionally different routes to host adaptation (Fig. 4b, Supplementary Data 4). In V. destructor, GO terms were mainly associated with (1) regulation of membrane depolarization, (2) retinal cone cell development and myofibroblast differentiation, (3) mRNA cis splicing via spliceosome whereas GO terms in V. jacobsoni related to (1) protein processing involved in targeting to mitochondria, (2) vitamin K metabolic process, (3) response to pH and other processes detailed in (Supplementary Fig. 3).
Tandem duplication was detected in 51 genes for V. destructor, 35 in V. jacobsoni and 70 in T. mercedesae (Fig. 3). Numbers of duplication events and gene clusters within a species varied from two to six units and generally involved adjacent chromosomal locations in V. destructor (Fig. 5a, Supplementary Data 5). In duplicated genes found only in V. jacobsoni, 23 had no orthologs in the V. destructor genome. However, most duplications are on the same scaffold with the exception of the ABC gene family where genes are present on two scaffolds. Manual checks of duplicated clusters with more than two genes validated all of the duplications except for the Histone 2 A family where LOC111255102 gene located on the minor scaffold BEIS01000444.1 was removed since it is 100% identical to LOC111250948 located on chromosome 1 and may be an artefact of mis-assembly.

Duplicated genes in V. destructor are found throughout the genome and are involved in different biological pathways. a Chromosomal location and similarity of duplicated genes (arrows) for V. destructor. Different genes and chromosomal regions underwent duplication in the two species. b Cluster analysis of significantly enriched GO terms for biological processes over-represented among duplicating genes in Varroa mites. Not only were most of the GO terms species-specific, but they also comprised non-overlapping categories of biological processes. Neither gene duplication nor selection analysis suggests a substantial degree of parallel evolution in these mites
Only five duplicated gene clusters were shared by all three honey bee parasites. These amounted to 9.8% and 14.3% of detected tandem gene duplications for V. destructor and V. jacobsoni, respectively (Fig. 2, Supplementary Data 6). GO terms associated with these duplicated gene clusters related to chitin, amino sugars, drug and carbohydrate derivatives. Conversely, the two Varroa sister species shared 14 gene clusters undergoing tandem duplication, representing 27.5% and 40.0% of the total size for V. destructor and V. jacobsoni, respectively. When summarizing the list of duplicated shared genes by both Varroa species, GO terms were associated with biological processes in glycerophospholipid catabolism, body development, regulation of DNA-templated transcription/elongation and amino-sugar metabolism (Supplementary Data 7). Finally, 62.7% of the duplicated genes were exclusive to V. destructor whereas 45.7% were only detected in V. jacobsoni. Genes duplicated in V. destructor were associated with GO terms related to biological processes such as mechanical/external stimulus, regulation of endoribonuclease activity and phagocytosis (Fig. 5b, Supplementary Data 7-8). In contrast, genes duplicated in V. jacobsoni were involved in biological processes of the skeletal myofibril assembly, Golgi calcium ion transport, and striated muscle contraction (Fig. 5b).
Chemosensory and cytochrome P450 genes in honey bee mites
Lipid carrier proteins: Based on the comparison of six amino acid sequences of OBPs previously described from V. destructor legs transcripts60, we found seven homologs (with conserved domain IPR036728 or OBP-like) in V. destructor annotated genome (Supplementary Fig. 4). Each OBP gene found in V. destructor presented an ortholog in V. jacobsoni coding for an identical OBP sequence (Supplementary Data 9). An analogous pattern of strong conservation was found for eight NCP2s (IPR003172) or similar with a Ganglioside GM2 activator conserved domain (IPR028996) in both sibling Varroa mites (Supplementary Fig. 5). None of the genes encoding for OBP and NCP2 showed signatures of positive selection or tandem duplication in Varroa sister species.
Membrane-bound receptors: A total of four genes per Varroa genome were found to encode for GR proteins presenting either the 7m_7 or Trehalose receptor conserved domains (IPR013604 and IPR009318, respectively). All four GR were closely related to GRs proteins found either in the honey bee parasitic mite T. mercedesae or in the parasitic tick I. scapularis (Supplementary Fig. 6). As previously reported, IGR protein profiles are the largest in V. destructor. According to the IGR phylogeny, this is also the case for V. jacobsoni with 35 identified genes (mainly presenting IPR001828, IPR019594 or IPR001320 signatures) (Fig. 6). Among these, 19 proteins found in Varroa genomes had closest homolog in T. mercedesae indicating high conservation of the gene family within honey bee parasites. Interestingly, three genes encoding for IGR or IGR-like proteins out of the 37 identified in V. destructor had no orthologous in V. jacobsoni. The opposite situation was also true but only for one gene in V. jacobsoni (LOC111272277). Additionally, divergent selective trajectories were found as a glutamate [NMDA] receptor subunit 1-like gene (LOC111270396) was under positive selection for V. jacobsoni while a metabotropic glutamate receptor 3-like gene (LOC111245193) show positive selection signal for V. destructor. Finally, despite a small number of SNMPs (with conserved domain IPR002159) detected in V. jacobsoni, we also found that one scavenger receptor class B member 1-like gene (LOC111267160) was under positive selection but none for V. destructor (Fig. S7).

The largest IGR repertoire in honey bee parasites show divergent selective trajectories for host and environment chemosensing between V. destructor and V. jacobsoni. The phylogenetic tree was constructed using 153 amino acid sequences from Acari V. destructor (Vdes = genome and Var = protein sequences83), V. jacobsoni (Vjac), T. mercedesae (Tmer), M. occidentalis (Mocc), I. scapularis (Isca), and T. urticae (Turt) which were aligned with MAFFT (see list in Supplementary Data 9). Best fit model computed for the tree is VT + R5 using IQ-TREE. Bootstrap values were estimated using an SH-like aLRT with 1000 and bootstraps over 95% are shown by a purple circle
Ecdysteroids and Halloween genes: All CYP enzymes sequences from V. destructor and V. jacobsoni annotation were extracted by filtering gene description list containing cytochrome p450 or exhibiting the associated conserved domain cl12078 or pfam00067. A total of 36 and 38 CYP (one isoform per gene) were aligned together with the seven Halloween genes reported from D. melanogaster and A. aegypti and other CYPs from Acari. Previously, only some members of this pathway, namely orthologues of spook, shade, and disembodied were found from a genomic sequence generated by 454 pyrosequencing45. Homologs proteins of CYP302A1 and CYP314A1 (~32% and ~28% sequence identity to disembodied and shade of D. melanogaster, respectively) were found for both Varroa sibling species but surprisingly no homolog spook (CYP307A1) was found for V. jacobsoni, though there was one in V. destructor (Fig. 7). Phylogenetic relationships among Varroa mites, T. urticae and M. occidentalis and D. melanogaster CYP315A1 sequences (~36% identity with the fruit fly) suggest that both V. destructor and V. jacobsoni Varroa mites possess shadow homologs. Likely, shroud and phantom are also presents in both Varroa species but not neverland. In the CYP family, we detected three genes under positive selection only in V. jacobsoni, including shadow-like (LOC111271433) and disembodied (LOC111265704) (Fig. 7 and Supplementary Data 9).

CYP repertoire in Varroa mites is broader than expected with two functionaly important Halloween genes shade and disembodied under positive selection in V. jacobsoni. The phylogenetic tree was constructed using 174 amino acid sequences from Acari V. destructor (Vdes = genome and Var = protein sequences83), V. jacobsoni (Vjac), T. mercedesae (Tmer), M. occidentalis (Mocc), I. scapularis (Isca), and T. urticae (Turt). Additionally, sequences from the fruit fly Drosophila melanogaster (Dmel), mosquito Aedes aegypti (Aaeg) and the honey bee A. mellifera (Amel) and A. cerana (Acer) were downloaded form NCBI and aligned with MAFFT. Best fit model computed for the tree is LG + R5 using IQ-TREE. Bootstrap values were estimated using a SH-like aLRT with 1000 and bootstraps over 95% are shown by a purple circle. V. destructor was previously believed to have only disembodied (CYP302a1), shade (CYP314a1), and spook (CYP307a1) homologs of the seven Halloween genes (background in black)45
Other genes possibly related to nutrition and detoxification: Some annotated genes detected under positive selection or undergoing tandem duplication in Varroa have been linked to play a role in external stress tolerance (e.g., in-hive temperature), nutrition, molting, reproduction, and metabolizing toxic xenobiotics possibly leading to acaricide resistance (detailed in Table 2). On V. destructor chromosome 4 (NW_019211457.1), two duplicated genes were also under positive selection and were involved in cuticular proteins pathway. These genes are involved in physical properties of the mite cuticle, which in addition to providing protection may constitute the first barrier to reduce the penetration of external chemical agents61. On the other hand, two duplicate clusters were also detected undergoing positive selection and annotated as part of the ABC family (ATP-Binding Cassette sub-family A) and to aminopeptidase M1-B-like, in V. jacobsoni. Finally, the most remarkable tandem duplication occurred on Vdes_3.0 chromosome 1 (NW_019211454.1) with six duplicated genes coding for heat shock 70 kDa protein. These were not detected for V. jacobsoni (Table 2). Both positive gene selection and duplication patterns in the V. destructor and V. jacobsoni genomes and related GO terms showed that the two sister species have undergone dissimilar evolutionary trajectories.
Discussion
In this study, we developed high-quality de novo reference genomes and annotations for economically important honey bee parasites V. destructor and V. jacobsoni. Because Varroa mites are, to a large extent, responsible for the global honey bee crisis, and research efforts worldwide target these parasites, these genomic resources respond to a pressing need37. Improved genome assembly and annotation with transcriptomic data from different life stages produced the most comprehensive gene catalogue to date, as reflected by our discovery of previously missing Halloween genes homologs45. The supposed absence of such genes has led to speculation on control measures development or that host counter adaptation could in the future target this pathway46. Our analysis also revealed that the two Varroa species share 99.7% sequence identity across the nuclear genome. Yet, after the two species have split, they have gone on largely dissimilar evolutionary trajectories, as evidenced by different patterns of positive selection and gene duplication. In particular, signals of positive selection were found in functionally important genes involved in chemosensing, molting, and reproduction. Thus, it seems likely that the two species underwent different patterns of adaptation while continuing to exploit the same host, the eastern honey bee (A. cerana). Differences between the two species most likely result from local adaptation to host geographic subspecies, the environment or their interaction. Though not enough data exist to distinguish between these alternatives, below we summarize what is already known and outline promising avenues for investigation.
Inferring what constitutes the ancestral ecological niche of the two mites is key to understanding what selective pressures they have faced and how they responded to them. V. destructor and V. jacobsoni are very similar morphologically, and until 20 years ago they were mistakenly considered the same species30. Their host specificity overlaps as they both parasitize the eastern honey bee A. cerana throughout Asia. The only exception reported for the Indonesian A. nigrocincta infested by V. jacobsoni, though possibly as a result of a recent host switch from A. cerana30,62. The most recent common ancestor of V. destructor and V. jacobsoni might have also been a parasite of the A. cerana lineage. This hypothesis is supported by the host range of V. underwoodi, another closely related mite that also parasitizes A. cerana in addition to A. nigrocincta and A. nulensis63,64. With no known varroid fossils, no information exists about speciation timing in Varroa mites. Based on the COX1 gene, a recognized molecular clock for insects with a divergence rate of 2.3% per Myr65, the divergence between V. destructor and V. jacobsoni of 5.9% suggests that the two species split ~2-3 Myr ago. This is after the split between A. cerana and A. mellifera, which occurred 6–9 Myr ago based on COX1 and ND266,67 to 17–19 Myr using a combination of mitochondrial, ribosomal, and nuclear loci68. Thus, host ranges and molecular estimates suggest that most Varroa evolution occurred on ancestors of A. cerana.
The major ecological difference between these mites is their parapatric distribution (Fig. 1). Whether parapatry resulted from their respective biogeographic history, adaptation to different subspecies of A. cerana, or is shaped by competition is not known. In fact, even the extent of the geographical overlap between the mite distributions is uncertain, since few surveys actually used molecular markers for identification and most have focused on associations with A. mellifera30,64,69,70. Yet, parapatry is clearly not due to the inability of the mites to complete their phoretic life stage outside the hive, as host-switched V. destructor successfully expanded its range following the range extension of A. mellifera. While allopatry could result from adaptation to local hosts/conditions or competitive exclusion, both mites have been reported from the same Thai apiary on A. cerana47,71. Unfortunately, there are not enough data on A. cerana infestation under natural conditions to determine the likelihood that the two Varroa species interact and compete. One survey in southern Thailand found 98% parasitism by V. jacobsoni under natural conditions, albeit at variable levels of infestation72, suggesting that encounters between the two species are indeed likely. If the divergent evolutionary history of the two Varroa species resulted from competition, we predict that long-term coexistence within a hive of A. cerana should be unlikely, a hypothesis that we hope will be tested in the future in the zone of sympatry.
In addition, or as an alternative to interspecific competition, it is possible that the divergence between V. destructor and V. jacobsoni was driven by local adaptation to different A. cerana populations. For instance, V. destructor was found to infect the Northern, Himalayan and the Indo-Chinese A. cerana morpho-clusters identified48 whereas V. jacobsoni was reported on the Indo-Chinese and the Indo-Malayan one. These A. cerana populations differ in their nesting behaviour, such as the number of combs or even in their body size73. In China alone, A. cerana populations have been shown to be as genetically divergent for more than 300,000–500,000 years as recognized subspecies of A. mellifera74. In addition to genetic differences between the hosts, the climactic environments across Asia are highly variable and could result in local adaptation, though, as mentioned earlier, this did not prevent range expansions after switching hosts (Fig. 1). Consequently, the dissimilar selective pressures observed in the mites may have resulted from selection due to different host genotypes.
Understanding the dynamics of co-infestation by both mites will become important if the host-switched V. destructor and V. jacobsoni are ever found to co-occur on A. mellifera. Presently, host-switched V. jacobsoni is restricted to Papua New Guinea, where V. destructor is absent31. However, if its range expands, it may impose an additional parasitic burden on A. mellifera, but whether the effects of this extra parasite are cumulative or whether the parasites will simply replace each other locally is unknown. Our data suggest that if the parasites are able to tolerate each other within A. mellifera colonies, given their different suites of molecular adaptations, they may in fact impose a greater cost than either parasite alone. Clearly, this is another avenue where further experimental work is urgently needed.
Likewise, it is unknown whether chemical treatment options developed for V. destructor will also be effective against V. jacobsoni. Interestingly, both mite genomes show signs of selection on a range of genes previously found to be involved in stress and pesticide resistance (Table 2). Since V. destructor has evolved resistance to acaricides, it cannot be ruled out that positive selection resulted from recent acaricide pressures rather than coevolution with their host. By contrast, acaricides are not yet widely used on V. jacobsoni, though the number of genes under selection is similar, suggesting that most selection is ancient. In the case of V. jacobsoni, and possibly generally, these genes have the potential to become co-opted for pesticide resistance. This has been the case with pesticide tolerance in the sheep blowfly (Lucilia cuprina), where sequencing of museum specimens has shown that resistance variants pre-dated pesticide use75. Similarly, the two-spotted mite (Tetranychus urticae) appears to have co-opted existing detoxification machinery in a selective response to miticides76. However, more detailed population-level work comparing switched and unswitched populations of mites will be necessary to test this hypothesis. Museum specimens or other historical material would be particularly useful in understanding evolutionary trends77.
Despite the economic importance of brood parasitic mites in A. mellifera, it is clear that various pressing research needs still exist concerning their biology, cryptic diversity in the native range, possible geographic ranges of host-switched species, and how co-infestations may proceed. These new reference genomes will allow researchers to pursue such investigations, develop better tools for Varroa control, and to better understand the invasion and evolution of Varroa on A. mellifera. In particular, we hope that these new resources will greatly benefit future studies to track the population genetic changes and demography of V. destructor’s worldwide spread, which has been difficult due to the low polymorphism of previously available markers70. More Varroa spp. are continuously testing new host78,79 and may remain undetected. Finally, the recent boom in -omics methods for the study of honey bee parasites and diseases underlines the need to improve our knowledge of genome structure and organization to investigate the genetic basis of newly reported acaricide resistance, virulence level or even new host tolerance. Such knowledge combined with progress in novel manipulation tools, such as RNAi80,81, and more experimental tools like gene drive82, may provide ways of specifically controlling mite pests without harming honey bees or other insects.
Methods
Sequencing strategy and Varroa mite collection
Haploid males (~0.7 mm by 0.7 mm) are only found inside capped brood cells after initiation of reproduction while diploid females (~1 mm by 1.5 mm) can be found in the brood and on adult honey bees. All V. destructor samples were collected from A. mellifera colonies of Okinawa Institute of Science and Technology (OIST) experimental apiary in Japan (26.477 N 127.835 E) in August–September 2016. One mature male V. destructor was collected within a worker cell at the red-eye pupa developmental stage (day 15–16) using a brush and kept in absolute ethanol at −80 °C. Mature adult females were collected from adult honey bee workers. In order to obtain a large number of mites infecting adult workers, we modified the standard powdered sugar method55 for an entire managed colony as follows: one hive box containing three to four frames with honey bee workers and no queen was placed on top of a collecting white tray; approximately 500 g of icing sugar was then applied using sieve filter powder strainer; the sugar and honey bees were shaken in the tray for 2 min and then separated using a sieve filter, placing the sugared-workers directly within the hive. This process was repeated on adjacent colonies. Varroa mites were separated from their hosts and trapped in the icing sugar. Following a water rinse on a gauze mesh, a total of 1207 alive Varroa females were snap frozen at −80 °C until laboratory processing (inactive, sluggish or dead mites were discarded). Samples of V. jacobsoni were obtained from its first detection survey where it was found reproducing in the western honey bee31. One mature male (VJ856) collected from A. mellifera drone brood cell in Papua New Guinea (between EHP and Henganofi border, 30/05/2008) was used to prepare a whole-genome library.
One sclerotized mature female V. destructor (VDOKI-01) collected from OIST apiary in the reproductive phase and one sclerotized mature female V. jacobsoni (VJ347-11) collected from A. cerana originating in Java (Cililin central Java, 13/06/1998 by D. Anderson) were processed for X-ray microtomography (micro-CT) using a Zeiss Xradia 510 Versa scanner. 3D surface models were reconstructed from micro-CT outputs using Amira software to visualize the slight morphological differences when focusing on idiosoma ventral and dorsal views and whole body in profile (Fig. 1a).
DNA extraction, genome sequencing, assembly, and annotation
Genomic DNA was extracted from each single haploid male V. jacobsoni and V. destructor using QIAamp DNA Micro Kit (Qiagen) following manufacturer’s instructions. The total amount of dsDNA was measured using Quant-iT™ PicoGreen™ dsDNA Assay Kit (Invitrogen). Short-insert of 250 bp paired-end libraries were prepared for both individuals using NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (Biolabs) and 10 PCR cycles. Size-selection and cleanup was applied using SPRI beads and library was subsequently checked using a Bioanalyzer High Sensitivity DNA kit (Agilent). Both paired-end libraries were each sequenced on two lanes of Illumina HiSeq 2500 at the OIST sequencing center in paired-end 250 cycle mode.
Prior to assembly paired-end reads were de-duplicated using ParDRe83, and host and microbial contamination was filtered using DeConSeq (parameters: -i 97 -c 95)84, screening against genomes of Apis cerana, Apis mellifera, as well as bacterial genomes and genomes of bee diseases and parasites. The cleaned reads were then merged using PEAR (parameters:–min-overlap 10–memory 80 G–threads 10 -n 200 -m 600 -p 0.0001)85 and assembled using Newbler (v. 2.6) (parameters: -large -m -cpu 24 -mi 98 -siom 390 -l 1000 -a 500 -urt -novs -a 1000 -minlen 45 -ml 100)86. The contigs were then scaffolded using publicly available RNA-seq data assembled using Trinity87,88. For V. destructor further scaffolding was carried out using Hi-C, performed by Phase Genomics. Hi-C read pairs were aligned to the initial assembly using BWA-MEM with the -5 option89, keeping only primary properly paired alignments (samtools with the -F 2316 filter90). Scaffolding was then performed using Phase Genomics' Proximo Hi-C scaffolding pipeline91, which builds on the LACHESIS method92.
To estimate genome characteristics, K-mers from cleaned reads were counted using jellyfish version 2.2.793 for the following values of K: 19, 25, 31, and 42 and coverage cutoff of 1000. Frequencies were only computed on HiSeq 2500 reads from one individual haploid male of each species. Computed and selected K-mer histograms for k = 42 were analyzed with GenomeScope to estimate genome size, kcoverage, number of duplicates94. We used the bioinformatically derived genome sizes for genome coverage estimation.
Small batches of adult females V. destructor (1,207 mites total) were pooled and crushed by pestle in 1.5 mL Eppendorf tubes. The powder was then lysed using proteinase K and lysis buffer at 56°C for 20 min. Sample lysate was thus transferred to a Maxwell Cartridge. The remaining step was fully performed on the Maxwell 16 automated purification system (Promega) with the LEV Blood DNA mode. Library preparation for PacBio RSII sequencing was performed using SMRTbell Template Prep Kit 1.0 (Pacific Biosciences) following the manufacturer's recommendations. SMRTbell libraries were prepared and sequenced at the OIST sequencing center on 48 SMRT cells. These reads were trimmed using proovread95 and then used to gap-fill the Hi-C scaffolded V. destructor assembly using PBJelly (parameters: -minMatch 8 -minPctIdentity 70 -bestn 1 -nCandidates 20 -maxScore -500 -nproc 24 -noSplitSubreads) (v. 15.8.24)96.
Annotation for both genomes Vdes_3.0 and Vjacob_1.0 was carried out by NCBI using NCBI’s automated genome annotation pipeline. This pipeline takes advantage of species-specific RNA-seq data, as well as extensive protein homology data stored in GenBank52.
Identification of Varroa mite strains using mtDNA sequences
Most previous taxonomic work relied on mitochondrial fragments. We used mitochondrial analysis to determine and compare the lineages of Varroa species used for genome sequencing to the previously described haplotypes. The whole mitogenome of V. destructor (accession NC_004454.253,54) was available from the NCBI database and used as a reference for read mapping. Raw Illumina® reads for both V. destructor and V. jacobsoni males were mapped onto the entire reference sequence (16,476 bp) using NextGenMap 0.5.097. Mapped reads were sorted and PCR duplicates were removed using Samtools 1.3.190. Maximum coverage of 500x was subsampled on all mapped reads using VariantBam 1.4.398. Subsequently, variant calling was performed using FreeBayes v1.1.0-4699, with the following parameters others than the default: minimum mapping quality of 20 and minimum base quality of 5. Consensus mitochondrial sequence was generated for each individual using vcf2fasta from Vcflib library100. To check the divergence level across the nuclear genome, we also mapped haploid male data from each species to the Vdes_3.0 genome using the same mapping and variant calling parameters.
One standard method to identify Varroa species and haplogroup is to sequence a partial region of the COX1 gene (426 bp) from the mitochondrial DNA55. The same region was extracted from both Vdes_3.0 and Vjacob_1.0 generated mitochondrial sequence and aligned together with 26 Varroa spp. haplotypes previously described30,69,79,101. Given its worldwide bee pest status, V. destructor haplogroups were better described than V. jacobsoni and additional larger COX1, COX3, ATP6, and CYTB mitochondrial sequences were available on NCBI. Therefore, a more resolutive comparison within V. destructor was done by aligning concatenated sequences of the four genes with 22 haplotypes described previously69,78. Sequences were aligned using ClustalW, checked by hand and a consensus maximum likelihood tree was constructed after 5000 bootstraps in Mega 7.0.18102.
A distribution map of the Varroa mites infecting A. cerana was built from reviewing literature (Fig. 1b). Conservatively, only the presence of Varroa spp. confirmed by mtDNA barcoding after the taxonomic revision was plotted30,47,69,79,101,103,104,105 onto the potential native and introduced range of the Asian honey bee host48,73 (Fig. 1c). The map was produced using QGIS 2.16.1 (Open Source Geospatial Foundation Project qgis.osgeo.org) and the public domain natural 10 m earth raster NE1_HR_LC github.com/nvkelso/natural-earth-raster). An additional interactive map showing the reported distribution of the 26 mtDNA haplotypes is available at github.com/MikheyevLab/varroa-denovo-genomes with layers control options.
Gene orthology, selection, and duplication in Varroa mites
The Orthonome pipeline (www.orthonome.com)59 was applied to predict orthologues (pairwise and 1:1 orthogroups), inparalogues and lineage-specific gene expansions from the two parasitic Varroa mites’ genomes sequenced and annotated here and four Acari annotated reference genomes available from NCBI (www.ncbi.nlm.nih.gov/genome/browse/#!/overview/Acari). The Orthonome pipeline uses phylogenetic information and microsynteny to identify gene copy number changes and tandem duplications between pairs of species using the MSOAR2.0 pipeline106. We included four outgroups which had fully sequenced genomes, including the eastern honey bee ectoparasitic mite Tropilaelaps mercedesae56, the parasitic tick Ixodes scapularis58; the free-living Western predatory mite Metaseiulus occidentalis107 and the free-living two-spotted mites Tetranychus urticae108. Total and pairwise/lineage intersection of orthologs gene sets were represented using Upset plots109 using R. The 1:1 orthogroups with both Varroa species represented and >3 species were used for positive selection hypothesis tested on phylogeny using HyPhy to calculate the mean dN/dS and test using aBSREL (adaptive Branch-Site Random Effects Likelihood)110. This test allowed estimation of selection along specific branches of the tree, including ancestral branches. Only branches with a significant p-value (<0.05) after FDR corrections were considered as positively selected in each orthogroup. Additionally, tandem duplications in the three parasitic mites were identified from inparalogues in immediate vicinity of the orthologues. All orthologous genes in Varroa mites under positive selection and/or duplicated in tandem were checked by hand by using BLAST. Duplication events with more than two genes were systematically checked by aligning the annotated coding sequence (CDS) with ClustalW alignment implemented in Geneious R8 (Biomatters, Ltd., Auckland, New Zealand) and construct a tree with IQ-TREE111.
Gene ontology (GO) enrichment analysis of the positively selected genes in honey bee parasitic mites were carried out using the GOstats R package112. The same process was applied for gene duplication in Varroa mites. Biological processes associated with these GO terms were summarized and visualized using REVIGO113 (http://revigo.irb.hr). The semantic similarity was measured using the Resnik’s measure (SimRel)114 and the threshold used was C = 0.5 (i.e., small). The results were then used to produce a scatter plot and treemap plot using the ggplot2 package. The same package was used to plot the localization of the genes under positive selection and/or duplicated onto V. destructor chromosomal scaffolds. Significance of overlaps between gene sets and associated GO terms were tested using Fisher’s exact test with GeneOverlap R package. Code and raw plots for exploratory analysis of positively selected genes, duplication in tandem and GO term association are available online (https://github.com/MikheyevLab/varroa-denovo-genomes).
Chemosensory and ecdysteroid pathway-related gene families
Varroa mites are blind and rely on their chemosensory system to find a suitable host within a densely packed honey bee colony40. Chemical cues are transported to the nervous system by binding to soluble carrier proteins and membrane proteins in insects. Several homologous chemosensory protein families involved in these pathways have been identified in Acari and Varroa mites41,43,44,60. Here we used the available catalogue of protein sequences reported from V. destructor forelegs transcripts44,60 containing six Odorant Binding Proteins (OBP), six Niemann‐Pick disease protein, type C2 (NPC2), seven Sensory Neuron Membrane Proteins (SNMPs), three Glutamate Receptors (GRs), and 17 Ionotropic Glutamate Receptor (IGRs). We identified these proteins in both V. destructor and V. jacobsoni annotated genomes using BLASTP and compiled predicted protein sequences (keeping one isoform if several exist for the same gene). We then aligned each family of protein sequences with other sequences from other Acari and insects using MAFFT version 7115 (parameters: E-INS-i, the BLOSUM62 matrix, and MAFFT-homolog option with 100 homologs, E threshold = 0.1). We used IQ-TREE116 to find the best substitution model for each family alignment, with state frequency estimated by maximum likelihood, 1000 ultra-fast bootstraps, 1000 replicate for SH-aLRT branch test. All generated phylogenetic trees were visualized and exported using iTOL v4117.
We ran the same process for the cytochrome P450 enzyme (CYP) genes (including the Halloween genes), known to be involved in the steroid molting hormone ecdysone biosynthesis pathway in Arthropods. Ecdysteroids derivatives are also important as they are involved in the vitellogenin production and oogenesis initiation in V. destructor after feeding on its host45,118. To assess which CYP genes and in particular the Halloween genes were present in both V. destructor and V. jacobsoni genomes, we used sequences from the fruit fly Drosophila melanogaster, the mosquito Aedes aegypti, both honey bee hosts A. mellifera and A. cerana, as well as other Acari (same name code as in a previous study on M. occidentalis119).
Statistics and reproducibility
GO terms enrichment was conducted with the R package GOstats using the hypergeometric test for association of categories and genes. The test parameters for each species and each ontology (BP, CC, and MF) using gene ID from NCBI were as follow: p-value cutoff = 0.05, not conditional and with detection of over-represented GO terms (testDirection = over). Regarding the test conducted for the significance in gene overlap, a Fisher’s exact two-tailed test with GeneOverlap R package and can be reproduced directly from the online supplementary data.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data generated or analysed during this study are included in this published article (and its supplementary information files). V. destructor (Vdes_3.0) and V. jacobsoni (Vjacob_1.0) assembled reference genomes and annotation are available on NCBI database respectively with the accession number GCF_002443255 and GCF_002532875. These data are also available for search and annotation at the i5k Workspace@NAL (https://i5k.nal.usda.gov/)130. Raw Illumina and PacBio reads are also available in the Sequence Read Archive (SRA) under the Bioprojects PRJDB6279 and PRJNA391052. Raw data of both Varroa mites micro-CT scans are available upon request.
Code availability
Online supplemental codes and map are available with an R markdown hosted from on github.com/MikheyevLab/varroa-denovo-genomes120.
References
de Meeûs, T. & Renaud, F. Parasites within the new phylogeny of eukaryotes. Trends Parasitol. 18, 247–251 (2002).
Weinstein, S. B. & Kuris, A. M. Independent origins of parasitism in Animalia. Biol. Lett. 12, 20160324 (2016).
Poulin, R., Krasnov, B. R., Mouillot, D. & Thieltges, D. W. The comparative ecology and biogeography of parasites. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 2379–2390 (2011).
Kochin, B. F., Bull, J. J. & Antia, R. Parasite evolution and life history theory. PLoS Biol. 8, e1000524 (2010).
Poulin, R. & Randhawa, H. S. Evolution of parasitism along convergent lines: from ecology to genomics. Parasitology 142(Suppl 1), S6–S15 (2015).
Rigaud, T., Perrot-Minnot, M.-J. & Brown, M. J. F. Parasite and host assemblages: embracing the reality will improve our knowledge of parasite transmission and virulence. Proc. Biol. Sci. 277, 3693–3702 (2010).
Hempel, P. S. Evolutionary parasitology: the integrated study of infections, immunology, ecology, and genetics. (Oxford University Press, 2011).
Petney, T. N. & Andrews, R. H. Multiparasite communities in animals and humans: frequency, structure and pathogenic significance. Int. J. Parasitol. 28, 377–393 (1998).
Vaumourin, E., Vourc’h, G., Gasqui, P. & Vayssier-Taussat, M. The importance of multiparasitism: examining the consequences of co-infections for human and animal health. Parasit. Vectors 8, 545 (2015).
Pouyaud, L., Desmarais, E., Deveney, M. & Pariselle, A. Phylogenetic relationships among monogenean gill parasites (Dactylogyridea, Ancyrocephalidae) infesting tilapiine hosts (Cichlidae): systematic and evolutionary implications. Mol. Phylogenet. Evol. 38, 241–249 (2006).
Rabajante, J. F. et al. Red Queen dynamics in multi-host and multi-parasite interaction system. Sci. Rep. 5, 10004 (2015).
Betts, A., Gray, C., Zelek, M., MacLean, R. C. & King, K. C. High parasite diversity accelerates host adaptation and diversification. Science 360, 907–911 (2018).
Miller, R. S. Pattern and Process in Competition. in Advances in Ecological Research (ed. Cragg, J. B.) 4, 1– (Academic Press, 1967).
Bashey, F. Within-host competitive interactions as a mechanism for the maintenance of parasite diversity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20140301 (2015).
Chappell, L. H. Competitive exclusion between two intestinal parasites of the three-spined stickleback, gasterosteus aculeatus L. J. Parasitol. 55, 775–778 (1969).
Simková, A., Morand, S., Jobet, E., Gelnar, M. & Verneau, O. Molecular phylogeny of congeneric monogenean parasites (Dactylogyrus): a case of intrahost speciation. Evolution 58, 1001–1018 (2004).
Hamelin, F. M., Bisson, A., Desprez-Loustau, M.-L., Fabre, F. & Mailleret, L. Temporal niche differentiation of parasites sharing the same plant host: oak powdery mildew as a case study. Ecosphere 7, e01517 (2016).
Orr, M., Seike, S. & Gilbert, L. Foraging ecology and patterns of diversification in dipteran parasitoids of fire ants in south Brazil. Ecol. Entomol. 22, 305–314 (1997).
Ramsey, S. D. et al. Varroa destructor feeds primarily on honey bee fat body tissue and not hemolymph. Proc. Natl Acad. Sci. USA 116, 1792–1801 (2019).
Rosenkranz, P., Aumeier, P. & Ziegelmann, B. Biology and control of Varroa destructor. J. Invertebr. Pathol. 103(Suppl 1), S96–S119 (2010).
Le Conte, Y., Ellis, M. & Ritter, W. Varroa mites and honey bee health: can Varroa explain part of the colony losses? Apidologie 41, 353–363 (2010).
Annoscia, D., Del Piccolo, F., Covre, F. & Nazzi, F. Mite infestation during development alters the in-hive behaviour of adult honeybees. Apidologie 46, 306–314 (2015).
Carreck, N. L., Ball, B. V. & Martin, S. J. Honey bee colony collapse and changes in viral prevalence associated with Varroa destructor. J. Apic. Res. 49, 93–94 (2010).
Gliński, Z. & Jarosz, J. Varroa jacobsoni as a carrier of bacterial infections to a recipient bee host. Apidologie 23, 25–31 (1992).
Neumann, P. & Carreck, N. L. Honey bee colony losses. J. Apic. Res. 49, 1–6 (2010).
Steinhauer, N. et al. Drivers of colony losses. Curr. Opin. Insect Sci. 26, 142–148 (2018).
Oudemans, A. C. Note VIII. On a new genus and species of parasitic Acari. Notes from the Leyden Museum 24, 216–222 (1904).
Crane, E. The varroa mite. Bee World 59, 12 (1978).
Eickwort, G. C. in Mites: Ecological and Evolutionary Analyses of Life-History Patterns (ed. Houck, M. A.) 218–251 (Springer US, 1994).
Anderson, D. L. & Trueman, J. W. Varroa jacobsoni (Acari: Varroidae) is more than one species. Exp. Appl. Acarol. 24, 165–189 (2000).
Roberts, J. M. K., Anderson, D. L. & Tay, W. T. Multiple host shifts by the emerging honeybee parasite, Varroa jacobsoni. Mol. Ecol. 24, 2379–2391 (2015).
Oldroyd, B. P. Coevolution while you wait: Varroa jacobsoni, a new parasite of western honeybees. Trends Ecol. Evol. 14, 312–315 (1999).
Rath, W. Co-adaptation of Apis cerana Fabr. and Varroa jacobsoni Oud. Apidologie 30, 97–110 (1999).
Page, P. et al. Social apoptosis in honey bee superorganisms. Sci. Rep. 6, 27210 (2016).
Lin, Z. et al. Reproduction of parasitic mites Varroa destructor in original and new honeybee hosts. Ecol. Evol. 8, 2135–2145 (2018).
Zhang, Y. & Han, R. A saliva protein of Varroa mites contributes to the toxicity toward Apis cerana and the DWV elevation in A. mellifera. Sci. Rep. 8, 3387 (2018).
Evans, J. D. & Cook, S. C. Genetics and physiology of Varroa mites. Curr. Opin. Insect Sci. 26, 130–135 (2018).
Peng, Y.-S., Fang, Y., Xu, S. & Ge, L. The resistance mechanism of the Asian honey bee, Apis cerana Fabr., to an ectoparasitic mite, Varroa jacobsoni Oudemans. J. Invertebr. Pathol. 49, 54–60 (1987).
Lin, Z. et al. Go East for Better Honey Bee Health: Apis cerana Is Faster at Hygienic Behavior than A. mellifera. PLoS ONE 11, e0162647 (2016).
Nazzi, F. & Le Conte, Y. Ecology of Varroa destructor, the Major Ectoparasite of the Western Honey Bee, Apis mellifera. Annu. Rev. Entomol. 61, 417–432 (2016).
McAfee, A., Chan, Q. W. T., Evans, J. & Foster, L. J. A Varroa destructor protein atlas reveals molecular underpinnings of developmental transitions and sexual differentiation. Mol. Cell. Proteom. 16, 2125–2137 (2017).
Mondet, F. et al. Transcriptome profiling of the honeybee parasite Varroa destructor provides new biological insights into the mite adult life cycle. BMC Genomics 19, 328 (2018).
Iovinella, I. et al. Proteomic analysis of chemosensory organs in the honey bee parasite Varroa destructor: a comprehensive examination of the potential carriers for semiochemicals. bioRxiv 260539 https://doi.org/10.1101/260539 (2018).
Eliash, N. et al. Chemosensing of honeybee parasite, Varroa destructor: transcriptomic analysis. Sci. Rep. 7, 13091 (2017).
Cabrera, A. R. et al. Three Halloween genes from the Varroa mite, Varroa destructor (Anderson & Trueman) and their expression during reproduction. Insect Mol. Biol. 24, 277–292 (2015).
Conlon, B. H. et al. A gene for resistance to the Varroa mite (Acari) in honey bee (Apis mellifera) pupae. Mol. Ecol. 28, 2958–2966 (2019).
Warrit, N., Smith, D. R. & Lekprayoon, C. Genetic subpopulations of Varroa mites and their Apis cerana hosts in Thailand. Apidologie 37, 19–30 (2006).
Radloff, S. E. et al. Population structure and classification of Apis cerana. Apidologie 41, 589–601 (2010).
Cornman, S. R. et al. Genomic survey of the ectoparasitic mite Varroa destructor, a major pest of the honey bee Apis mellifera. BMC Genomics 11, 602 (2010).
Thomas, G. W. C. et al. The Genomic Basis of Arthropod Diversity. bioRxiv 382945 https://doi.org/10.1101/382945 (2018).
De Ruijter, A. & Pappas, N. Varroa jacobsoni Oud. affecting honey bees: present status and needs. In Proceedings of a meeting of the EC Experts' Group/Wageningen (ed. R. Cavalloro) 41–47 (Balkema, Rotterdam, 1983).
Thibaud-Nissen, F., Souvorov, A., Murphy, T., DiCuccio, M. & Kitts, P. Eukaryotic Genome Annotation Pipeline. (National Center for Biotechnology Information (US), 2013).
Navajas, M., Le Conte, Y., Solignac, M., Cros-Arteil, S. & Cornuet, J.-M. The complete sequence of the mitochondrial genome of the honeybee ectoparasite mite Varroa destructor (Acari: Mesostigmata). Mol. Biol. Evol. 19, 2313–2317 (2002).
Evans, J. D. & Lopez, D. L. Complete mitochondrial DNA sequence of the important honey bee pest, Varroa destructor (Acari: Varroidae). Exp. Appl. Acarol. 27, 69–78 (2002).
Dietemann, V. et al. Standard methods for varroa research. J. Apic. Res. 52, 1–54 (2013).
Dong, X. et al. Draft genome of the honey bee ectoparasitic mite, Tropilaelaps mercedesae, is shaped by the parasitic life history. Gigascience 6, 1–17 (2017).
Jeyaprakash, A. & Hoy, M. A. The nuclear genome of the phytoseiid Metaseiulus occidentalis (Acari: Phytoseiidae) is among the smallest known in arthropods. Exp. Appl. Acarol. 47, 263–273 (2009).
Gulia-Nuss, M. et al. Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nat. Commun. 7, 10507 (2016).
Rane, R. V., Oakeshott, J. G., Nguyen, T., Hoffmann, A. A. & Lee, S. F. Orthonome—a new pipeline for predicting high quality orthologue gene sets applicable to complete and draft genomes. BMC Genomics 18, 673 (2017).
Eliash, N. et al. Varroa chemosensory proteins: some are conserved across Arthropoda but others are arachnid specific. Insect Mol. Biol. 28, 321–341 (2019).
Martin, S. J. Acaricide (pyrethroid) resistance in Varroa destructor. Bee World 85, 67–69 (2015).
de Guzman, L. I. & Rinderer, T. E. Identification and comparison of Varroa species infesting honey bees. Apidologie 30, 85–95 (1999).
Delfinado-Baker, M. & Aggarwal, K. A new Varroa (Acari: Varroidae) from the nest of Apis cerana (Apidae). Int. J. Acarol. 13, 233–237 (1987).
Warrit, N. & Lekprayoon, C. in Honeybees of Asia (eds. Hepburn, H. R. & Radloff, S. E.) 347–368 (Springer Berlin Heidelberg, 2011).
Papadopoulou, A., Anastasiou, I. & Vogler, A. P. Revisiting the insect mitochondrial molecular clock: the mid-Aegean trench calibration. Mol. Biol. Evol. 27, 1659–1672 (2010).
Garnery, L., Vautrin, D., Cornuet, J. M. & Solignac, M. Phylogenetic relationships in the genus Apis inferred from mitochondrial DNA sequence data. Apidologie 22, 87–92 (1991).
Arias, M. C. & Sheppard, W. S. Molecular phylogenetics of honey bee subspecies (Apis mellifera L.) inferred from mitochondrial DNA sequence. Mol. Phylogenet. Evol. 5, 557–566 (1996).
Ramírez, S. R. et al. A molecular phylogeny of the stingless bee genus Melipona (Hymenoptera: Apidae). Mol. Phylogenet. Evol. 56, 519–525 (2010).
Navajas, M. et al. New Asian types of Varroa destructor: a potential new threat for world apiculture. Apidologie 41, 181–193 (2010).
Solignac, M. et al. Characterization of microsatellite markers for the apicultural pest Varroa destructor (Acari: Varroidae) and its relatives. Mol. Ecol. Notes 3, 556–559 (2003).
Rueppell, O., Hayes, A. M., Warrit, N. & Smith, D. R. Population structure of Apis cerana in Thailand reflects biogeography and current gene flow rather than Varroa mite association. Insectes Soc. 58, 445–452 (2011).
Rath, W. & Drescher, W. Response of Apis cerana Fabr towards brood infested with Varroa jacobsoni Oud and infestation rate of colonies in Thailand. Apidologie 21, 311–321 (1990).
Koetz, A. H. Ecology, behaviour and control of Apis cerana with a focus on relevance to the Australian incursion. Insects 4, 558–592 (2013).
Chen, C. et al. Population genomics provide insights into the evolution and adaptation of the eastern honey bee (Apis cerana). Mol. Biol. Evol. https://doi.org/10.1093/molbev/msy130 (2018).
Hartley, C. J. et al. Amplification of DNA from preserved specimens shows blowflies were preadapted for the rapid evolution of insecticide resistance. Proc. Natl Acad. Sci. USA 103, 8757–8762 (2006).
Dermauw, W. et al. A link between host plant adaptation and pesticide resistance in the polyphagous spider mite Tetranychus urticae. Proc. Natl Acad. Sci. USA 110, E113–E122 (2013).
Yeates, D. K., Zwick, A. & Mikheyev, A. S. Museums are biobanks: unlocking the genetic potential of the three billion specimens in the world’s biological collections. Curr. Opin. Insect Sci. 18, 83–88 (2016).
Gajic, B. et al. Variability of the honey bee mite Varroa destructor in Serbia, based on mtDNA analysis. Exp. Appl. Acarol. 61, 97–105 (2013).
Beaurepaire, A. L. et al. Host specificity in the honeybee parasitic mite, Varroa spp. in Apis mellifera and Apis cerana. PLoS ONE 10, e0135103 (2015).
Campbell, E. M., Budge, G. E. & Bowman, A. S. Gene-knockdown in the honey bee mite Varroa destructor by a non-invasive approach: studies on a glutathione S-transferase. Parasit. Vectors 3, 73 (2010).
Huang, Z. Y., Bian, G., Xi, Z. & Xie, X. Genes important for survival or reproduction in Varroa destructor identified by RNAi. Insect Sci. 26, 68–75 (2019).
McFarlane, G. R., Whitelaw, C. B. A. & Lillico, S. G. CRISPR-based gene drives for pest control. Trends Biotechnol. 36, 130–133 (2018).
González-Domínguez, J. & Schmidt, B. ParDRe: faster parallel duplicated reads removal tool for sequencing studies. Bioinformatics 32, 1562–1564 (2016).
Schmieder, R. & Edwards, R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE 6, e17288 (2011).
Zhang, J., Kobert, K., Flouri, T. & Stamatakis, A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620 (2014).
Margulies, M. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005).
Xue, W. et al. L_RNA_scaffolder: scaffolding genomes with transcripts. BMC Genomics 14, 604 (2013).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Bickhart, D. M. et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 49, 643–650 (2017).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Hackl, T., Hedrich, R., Schultz, J. & Förster, F. proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30, 3004–3011 (2014).
Worley, K. C. et al. in International Plant and Animal Genome Conference Xxii 10–15 (San Diego, CA, 2014).
Sedlazeck, F. J., Rescheneder, P. & von Haeseler, A. NextGenMap: fast and accurate read mapping in highly polymorphic genomes. Bioinformatics 29, 2790–2791 (2013).
Wala, J., Zhang, C.-Z., Meyerson, M. & Beroukhim, R. VariantBam: filtering and profiling of next-generational sequencing data using region-specific rules. Bioinformatics 32, 2029–2031 (2016).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv https://arxiv.org/abs/1207.3907 (2012).
Garrison, E. Vcflib: A C++ library for parsing and manipulating VCF files. GitHub https://github.com/ekg/vcflib (2012).
Wang, S. et al. Ectoparasitic Mites Varroa underwoodi (Acarina: Varroidae) in Eastern Honeybees, but not in Western Honeybees. J. Econ. Entomol. 112, 25–32 (2019).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Anderson, D. L. & Fuchs, S. Two genetically distinct populations of Varroa jacobsoni with contrasting reproductive abilities on Apis mellifera. J. Apic. Res. 37, 69–78 (1998).
Zhou, T. et al. Identification of Varroa mites (Acari: Varroidae) infesting Apis cerana and Apis mellifera in China. Apidologie 35, 645–654 (2004).
Solignac, M. et al. The invasive Korea and Japan types of Varroa destructor, ectoparasitic mites of the Western honeybee (Apis mellifera), are two partly isolated clones. Proc. Biol. Sci. 272, 411–419 (2005).
Shi, G., Zhang, L. & Jiang, T. MSOAR 2.0: Incorporating tandem duplications into ortholog assignment based on genome rearrangement. BMC Bioinforma. 11, 10 (2010).
Hoy, M. A. et al. Genome sequencing of the phytoseiid predatory mite metaseiulus occidentalis reveals completely atomized hox genes and superdynamic intron evolution. Genome Biol. Evol. 8, 1762–1775 (2016).
Grbić, M. et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature 479, 487–492 (2011).
Lex, A., Gehlenborg, N., Strobelt, H., Vuillemot, R. & Pfister, H. UpSet: Visualization of Intersecting Sets. IEEE Trans. Vis. Comput. Graph. 20, 1983–1992 (2014).
Pond, S. L. K. & Muse, S. V. in Statistical Methods in Molecular Evolution (ed. Nielsen, R.) 125–181 (Springer New York, 2005).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Falcon, S. & Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 23, 257–258 (2007).
Supek, F., Bošnjak, M., Škunca, N. & Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6, e21800 (2011).
Resnik, P. Semantic Similarity in a Taxonomy: an information-Based Measure and its Application to Problems of Ambiguity in Natural Language. arXiv https://arxiv.org/abs/1105.5444 (2011).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. https://doi.org/10.1093/bib/bbx108 (2017).
Trifinopoulos, J., Nguyen, L.-T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Frey, E., Odemer, R., Blum, T. & Rosenkranz, P. Activation and interruption of the reproduction of Varroa destructor is triggered by host signals (Apis mellifera). J. Invertebr. Pathol. 113, 56–62 (2013).
Wu, K. & Hoy, M. A. The glutathione-S-transferase, cytochrome P450 and carboxyl/cholinesterase gene superfamilies in predatory mite metaseiulus occidentalis. PLoS ONE 11, e0160009 (2016).
Techer, M. A., et al. MaevaTecher/varroa-denovo-genomes: Online material for "Divergent evolutionary trajectories following speciation in two ectoparasitic honey bee mites", Zenodo, https://doi.org/10.5281/zenodo.3372750 (2019).
Chantawannakul, P., de Guzman, L. I., Li, J. & Williams, G. R. Parasites, pathogens, and pests of honeybees in Asia. Apidologie 47, 301–324 (2016).
Anderson, D. L. & Morgan, M. J. Genetic and morphological variation of bee-parasitic Tropilaelaps mites (Acari: Laelapidae): new and re-defined species. Exp. Appl. Acarol. 43, 1–24 (2007).
Garrido, P. M. et al. Heat shock proteins in Varroa destructor exposed to heat stress and in-hive acaricides. Exp. Appl. Acarol. 76, 421–433 (2018).
Posada-Florez, F. et al. Insights into the metabolism and behaviour of Varroa destructor mites from analysis of their waste excretions. Parasitology 146, 527–532 (2019).
González-Cabrera, J. et al. Novel mutations in the voltage-gated sodium channel of pyrethroid-resistant Varroa destructor populations from the Southeastern USA. PLoS ONE 11, e0155332 (2016).
Sammataro, D., Untalan, P., Guerrero, F. & Finley, J. The resistance of varroa mites (Acari: Varroidae) to acaricides and the presence of esterase. Int. J. Acarol. 31, 67–74 (2005).
Ménard, C. et al. Multiple combinations of RDL subunits diversify the repertoire of GABA receptors in the honey bee parasite Varroa destructor. J. Biol. Chem. 293, 19012–19024 (2018).
Dermauw, W. & Van Leeuwen, T. The ABC gene family in arthropods: comparative genomics and role in insecticide transport and resistance. Insect Biochem. Mol. Biol. 45, 89–110 (2014).
Dmitryjuk, M., Zalewski, K., Raczkowski, M. & Żółtowska, K. Composition of fatty acids in the Varroa destructor mites and their hosts, Apis mellifera drone-prepupae. Ann. Parasitol. 61, 21–26 (2015).
Zalewski, K., Zaobidna, E. & Żóltowska, K. Fatty acid composition of the parasitic mite Varroa destructor and its host the worker prepupae of Apis mellifera : fatty acids of V. destructor and its host. Physiol. Entomol. 41, 31–37 (2016).
Acknowledgements
We would like to acknowledge Yoann Portugal (Ecology and Evolution OIST) for helping with the fieldwork collection of V. destructor mites, Jo Tan (Ecology and Evolution OIST) for processing in laboratory Varroa males and Hiroki Goto (OIST DNA sequencing section – Onna, Okinawa) for leading and processing the PacBio RSII sequencing. We also thank Francisco Hita Garcia (Biodiversity and Biocomplexity OIST) for his support in preparation and scanning of specimens using the micro-CT. We thank Steven Aird (OIST technical editor) for carefully checking and helping to correct our manuscript. This work was supported by the Okinawa Institute of Science and Technology Graduate University, as well as grants from the Japan Society for the Promotion of Science (Kakenhi grants 16KK0175, 16H06209 and 18H02216) and the Australian Research Council (Future Fellowship Scheme FT160100178). Genome assembly was partially supported by Driscoll’s, Inc.
Author information
Authors and Affiliations
Contributions
M.A.T. collected samples for sequencing, carried out analyses, interpreted the data, and wrote the manuscript. R.V.R. carried out analyses and wrote the manuscript. J.M.K.R. collected samples for sequencing. M.L.G. carried out the assembly and quality analyses. S.T.S. carried out the computational analyses for Hi-C genome assembly. I.L. carried out the experimental protocols for Hi-C genome assembly. A.K.C. conceived and designed experiments. J.D.E. conceived and designed experiments. A.S.M. conceived and designed experiments, carried out the assembly and analyses, interpreted the data, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
I.L. and S.T.S. declare the following competing interests: I.L. and S.T.S. are employees and shareholders of Phase Genomics, a company commercializing proximity-ligation technology. M.A.T., R.V.R., J.M.K.R., M.L.G., A.K.C., J.D.E., and A.S.M. declare no financial or non-financial competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Techer, M.A., Rane, R.V., Grau, M.L. et al. Divergent evolutionary trajectories following speciation in two ectoparasitic honey bee mites. Commun Biol 2, 357 (2019). https://doi.org/10.1038/s42003-019-0606-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-019-0606-0
This article is cited by
-
Molecular phylogeny reveals Varroa mites are not a separate family but a subfamily of Laelapidae
Scientific Reports (2024)
-
LAMP (Loop-mediated isothermal amplification) assay for rapid identification of Varroa mites
Scientific Reports (2023)
-
Genome-wide SNPs show hybridization of Varroa mites from different Apis hosts in Vietnam and Taiwan
Apidologie (2023)
-
Embryogenesis and apoptosis appears as key molecular pathways involved in Varroa destructor reproduction
Apidologie (2023)
-
Vector-virus interaction affects viral loads and co-occurrence
BMC Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
