
Abstract
Insulin drives growth and reproduction which trade-off against longevity. Genetically predicted insulin, i.e., insulin proxied by genetic variants, is positively associated with ischemic heart disease, but sex differences are unclear, despite different disease rates and reproductive strategies by sex. We used Mendelian randomization in 392,010 white British from the UK Biobank to assess the sex-specific role of genetically predicted insulin in myocardial infarction (MI) (14,442 cases, 77% men), angina (21,939 cases, 65% men) and heart failure (5537 cases, 71% men). Genetically predicted insulin was associated with MI (odds ratio (OR) 4.27 per pmol/L higher insulin, 95% confidence interval (CI) 1.60 to 11.3) and angina (OR 2.93, 1.27 to 6.73) in men, but not women (MI OR 0.80, 95% CI 0.23 to 2.84, angina OR 1.10, 95% CI 0.38 to 3.18). Patterns were similar for insulin resistance and heart failure. Mitigating the effects of insulin might address sexual disparities in health.
Similar content being viewed by others


Introduction
Cardiovascular disease (CVD) is the leading cause of global morbidity and mortality1, accounting for over 30% of all deaths2. This burden of disease calls for more effective prevention and treatment strategies. Notably, men have higher incident rates of ischemic CVD than women, for reasons which have not been fully explained by traditional cardiovascular risk factors, such as smoking, lipid profile and blood pressure3. Clarifying the sex disparity in CVD might provide clues to identifying new targets as well as addressing sexual disparities in health.
A novel explanation for the sex difference, from the perspective of evolutionary biology, is that longevity trades-off against growth and reproduction, with possibly sex-specific trade-offs for reproductive investment versus cardiovascular risk4,5. Genetic evidence consistent with ischemic heart disease (IHD) trading off against fertility exists6. Genetically predicted gonadotrophin releasing hormone increases IHD7. Applying this concept to disease prevention and treatment, factors that downregulate growth or reproduction might lower the burden of CVD8, differentially in men and in women, as drivers of reproduction are sex specific. Insulin is a key driver of growth and reproduction9,10, which is responsive to nutritional factors, such as a high-fat diet11, and to treatment with insulin and insulin secretagogues12.
Insulin is an invaluable life-saving treatment for type 1 diabetes13. Unexpectedly and controversially, use of insulin has long been suspected to play a role in CVD14, especially in men15. Genetically predicted insulin and insulin resistance are consistently positively associated with higher risk of IHD16,17,18, independent of adiposity18. Patients switching from metformin to an insulin secretagogue, sulphonylurea, have a higher risk of myocardial infarction (MI)19. Higher insulin doses are also associated with an unfavorable cardiovascular risk factor profile20. No large randomized controlled trials (RCTs) have assessed the role of insulin in CVD, and it may not be ethical to do so. In these circumstances, Mendelian randomization (MR) taking advantage of genetic endowment randomly allocated at conception21, can obtain unconfounded estimates. Here, we used MR to assess overall and sex-specific effects of insulin, and for completeness insulin resistance, on MI, angina, heart failure and their key risk factors (low-density lipoprotein (LDL) cholesterol, apolipoprotein B (ApoB)22, and blood pressure) using individual data in a large cohort, the UK Biobank23, or the largest available genome-wide association study (GWAS). Red blood cell attributes have long been suspected to be relevant to cardiovascular disease24, however, which trait matters is not well established. The most recent evidence from an MR study suggests the red blood cell trait, reticulocyte count, may be a causal factor for IHD25, so we similarly examined the role of insulin and insulin resistance in reticulocyte count. We also validated the findings for MI using summary statistics from a large genetic study, CARDIoGRAMplusC4D 1000 Genomes26.
Results
Genetic instruments for insulin and insulin resistance
We identified 12 single-nucleotide polymorphisms (SNPs) independently predicting insulin and 12 SNPs for BMI-adjusted insulin (extracted from Table 1 of the GWAS of Scott et al.27), as previously17,28. All reached genome-wide significance (5 × 10−8). The insulin resistance genetic score was constructed based on 10 SNPs (rs4846565, rs10195252, rs2943645, rs17036328, rs3822072, rs6822892, rs4865796, rs459193, rs2745353 and rs731839), as in the previously validated score29.
Of the 12 SNPs related to insulin, we dropped 5 SNPs due to pleiotropy, specifically 2 SNPs (rs10195252 in GRB14 and rs9884482 in TET2) related to alcohol drinking (p value 9.4 × 10−7 and 2.3 × 10−6, respectively), 2 SNPs (rs1167800 in HIP1 and rs7903146 in TCF7L2) related to BMI (p value 8.7 × 10−13 and 9.8 × 10−7, respectively), and 1 SNP (rs1421085) in the FTO gene (a well-established obesity predictor), so 7 SNPs were used (Table 1 and Supplementary Table 1).
Of the 12 SNPs related to insulin adjusted for BMI, we dropped 3 pleiotropic SNPs, specifically, 2 SNPs (rs974801 in TET2 and rs10195252 in GRB14) related to alcohol drinking (p value 8.5 × 10−7 and 9.4 × 10−7, respectively) and 1 SNP (rs6912327 in C6orf107) related to BMI (p value = 1.6 × 10−14) in the UK Biobank, leaving 9 SNPs (Table 1).
In sensitivity analysis for insulin resistance in men, we further dropped 1 SNP (rs3822072 in FAM13A1) associated with BMI in men (p value = 5.5 × 10−6) but not women. Given the unclear causal role of alcohol in CVD, we kept the SNPs related to alcohol drinking in sensitivity analysis. For completeness, we also kept the SNPs related to BMI (Table 1).
Associations with MI, angina and heart failure
Genetically predicted insulin, BMI-adjusted insulin and insulin resistance score were all positively associated with MI overall (Table 2 and Supplementary Fig. 1) and also in men, but not in women (p values for sex differences were 0.02, 0.04 and 0.04 respectively) (Table 2). The overall association was validated in CARDIoGRAPMplusC4D 1000 Genomes (Table 2). Insulin and insulin resistance were also associated with higher risk of angina in men only (Table 2), with a more obvious sex difference for BMI-adjusted insulin (p value for sex difference 0.04), than insulin (p value for sex difference 0.08) (Table 2). The pattern of associations were generally robust to different analytic methods (Supplementary Table 2), and sensitivity analysis including alcohol-related SNPs (Supplementary Table 3), all potentially pleiotropic SNPs (Supplementary Table 4) and excluding rs3822072 for BMI-adjusted insulin in men (Supplementary Table 5), despite some associations including the null value due to wider confidence intervals.
The replication for MI using a different study provides additional validation, and enabled us to test causality in a cost-efficient way30. Specifically, the studies for MI with over 56,000 cases, at an approximate R2 of 0.01 (variance in insulin/BMI-adjusted insulin explained by the genetic predictors), has 0.8 power to detect an odds ratio (OR) of about 1.13 per one standard deviation increase in the exposure. The UK biobank has 0.8 power to detect an OR of about 1.27 for MI overall, of 1.31 for MI in men and 1.64 in women; an OR of 1.22 for angina overall, of 1.28 for angina in men and 1.39 in women; an OR of 1.46 for heart failure overall, of 1.57 for heart failure in men and 2.01 in women31. The larger number of cases in men than women enabled us to test a smaller effect size in men, however, there is sufficient power for both men and women when using the insulin resistance score on angina. The difference in power does not explain the sex-disparity in the magnitude of the point estimates and/or direction of associations.
Associations with CVD risk factors
Insulin and BMI-adjusted insulin were unrelated to LDL cholesterol, but the latter was associated with higher ApoB (Fig. 1). We could not test whether these associations differ by sex, because relevant genetic data is not publicly available. Insulin and BMI-adjusted insulin were positively associated with systolic blood pressure and reticulocyte count in both men and women. The associations of BMI-adjusted insulin with reticulocyte count appeared to be stronger in men than in women, although the sex difference was not statistically significance (p value for sex-difference 0.17).

Associations of genetically predicted insulin and BMI-adjusted insulin with cardiovascular disease risk factors overall and by sex. ApoB apolipoprotein, BMI body mass index, DBP diastolic blood pressure, LDL low-density lipoprotein, SBP systolic blood pressure. Beta coefficients and 95% confidence intervals (CI) for the associations of insulin and BMI-adjusted insulin with CVD risk factors have been depicted. Gray denotes the 95% CI included the null, purple denotes the 95% CI did not include the null. n = 188,577 for LDL cholesterol, n = 24,925 for ApoB, n ≤ 361,194 for blood pressure and reticulocyte count
Discussion
Our Mendelian Randomization study suggests a positive association of insulin and insulin resistance with MI overall and in men, and with angina in men but not women, with validation for MI overall in CARDIoGRAPMplusC4D 1000 Genomes. Insulin and BMI-adjusted insulin increased blood pressure and reticulocyte count and BMI-adjusted insulin increased ApoB. We found no effect of insulin on LDL cholesterol, but sex-specific analysis for LDL cholesterol and ApoB were not examined thus sex differences cannot be excluded.
Our findings, together with previous MR studies, provide support for a potential role of insulin in IHD14,32,33. Our novel study also adds to the very limited evidence on the sex-specific effects of insulin and insulin resistance, by showing a stronger effect for men than women. Nevertheless, our study has limitations. First, MR is based on three stringent assumptions, i.e., the genetic variants are strongly related to the exposure, are not related to the exposure-outcome confounders, and the genetic variants are related to the outcomes only via influencing the exposure34,35. To satisfy the first assumption, we used genetic variants strongly associated with insulin and insulin resistance from a large GWAS27,29, as previously16,17. To satisfy the second assumption, we checked for associations with known exposure-outcome confounders, including socioeconomic position and lifestyle in the UK Biobank, where there was no association with these potential confounders. In addition, the sample for genetic variants on insulin has no overlap with the UK Biobank. Two-sample MR is usually less biased than one-sample MR36, because any relation of the genetic variants with unmeasured confounders is not expected to exist coincidently in both the sample providing genetic associations with insulin or insulin resistance and the sample providing genetic associations with the outcomes, due to the different data structures37. If bias did occur due to weak instruments, it is often towards the null, whereas in one-sample MR the bias is towards the direction of the conventional observational studies36. Population stratification might affect genetic distribution and cardiovascular risk, however, we only used participants of European ancestry, with genetic control. To test the assumption of pleiotropy, we checked for the known potential pleiotropy in three comprehensive curated databases. We also tested and corrected for pleiotropic effects using MR-PRESSO. Second, although we used the largest available source of genetic associations with heart failure, the number of cases was relatively low, which may explain the wide confidence intervals for heart failure. Third, our MR study assessing the role of endogenous insulin and insulin resistance might not be applicable to the exogenous use of insulin. MR examining a lifetime effect of an exposure may also not be comparable to exogenous treatment. However, serum insulin is responsive to exogenous supplementation, diet, and treatment12. The association for MI is also consistent with the higher cardiovascular risk when switching to sulphonylurea, an insulin secretagogue19. Fourth, our study could be affected by selection bias from selecting survivors of their genetic make-up38, and of competing risk of other specific causes of death that share risk factors. Specifically, the estimates for a potentially harmful exposure might be biased towards being less harmful if people with higher levels of exposures were already dead and not selected into the study, as in the obesity paradox39. Fifth, misclassification of the outcomes might exist. Measurement error in the outcomes might arise, but likely non-differential and so biases towards the null. Sixth, the associations in Europeans may not apply to other populations, such as Asians. However, causal effects should be consistent across settings, although their relevance may vary by population. Specifically, our findings may be particularly relevant to Asians who tend to have higher serum insulin than people of European descent40. Seventh, some of the participants may have comorbidities such as type 2 diabetes and may be taking medications for these comorbidities. Co-morbidities and their treatment may affect the cardiovascular outcomes, but should not affect the genetic predictors of exposures, so they are not confounders but their inclusion could improve the precision of the estimates. However, co-morbidities could also be consequences of insulin and insulin resistance so their consideration in the model would give the direct effects of insulin rather than the total effect sought, i.e., might create bias. As such, we did not account for co-morbidities or their treatment by adjustment or restriction, so as to obtain an unbiased, though possibly less precise, estimates. Eighth, reverse causality may occur if people with cardiovascular events change their lifestyle thereby affecting insulin or insulin resistance. However, these changes would not affect genetically predicted insulin or insulin resistance. None of the genetic variants are genome-wide significant (p < 5 × 10−8) for cardiovascular events, so it is unlikely that they predict insulin or insulin resistance by affecting cardiovascular events. Finally, the genetic variants in the sex-specific analysis were from both sexes rather than specifically for men and women. As such, the sex-specific associations are less precise may be conservative. However, the directions of the associations should be unchanged. Validation of the sex-specific associations in another cohort is warranted.
Our study, together with previous evidence41,42, suggests that insulin and insulin resistance have symbiotic roles that may both ultimately play a role in CVD. Our study adds to the current evidence by showing a sex-disparity in these associations. Insulin resistance has previously been proposed as a mechanism underlying the metabolic syndrome and hence susceptibility to CVD, i.e., Reaven’s hypothesis43. However, before the advent of MR, Reaven’s hypothesis was difficult to test conclusively, and included cholesterol, when ApoB, rather than LDL cholesterol, may be driven by insulin and correspond better to the target of lipid-modifying treatment22. Insulin modulates human ApoB mRNA translation44. Verification in RCTs would be worthwhile. Insulin might operate via increasing growth or sex hormones, such as androgens45, which may increase the risk of MI46. Coagulation factors, which were not examined in this study, may also be a mechanism. Hyperinsulinemia promotes a procoagulant state47, increasing in several coagulation factors, such as thrombin generation47, and plasminogen activator inhibitor type 148, which have been identified as potential causes of IHD and MI49,50.
Our study suggests that a lifestyle, which lowers serum insulin, might lower cardiovascular risk. Conversely, less limited living conditions that enable higher levels of insulin, with corresponding effects acting on MI and angina via male reproductive factors may explain the higher rates of CVD in men than women that emerge with economic development51. Insulin causing MI may also partly explain the unexpected off-target effects of insulin raising treatments, such as sulfonylureas19. Similarly, insulin therapy has a relatively higher risk of MI than insulin sensitization therapy48. Several medications for type 2 diabetes, such as metformin, thiazolidinediones, sodium-glucose transport inhibitors, and glucagon-like peptide 1 agonists may reduce the need for insulin52. From the perspective of clinical and public health practice, our findings suggest that medications or dietary factors that operate other than by increasing insulin might prevent and treat CVD. Our findings also draw attention to the possibility that insulin may operate via red blood cell attributes, such as reticulocyte count. Clarifying these pathways, especially as regards any sex-differences in the response to insulin, would be valuable, with relevance to the re-positioning of existing drugs and new drug development. Replication in other cohorts is needed.
Methods
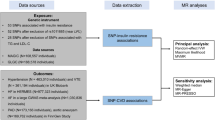
We used a two sample MR study design. Specifically, we obtained genetic predictors for insulin and insulin resistance from the largest available GWAS27, and examined their overall and sex-specific associations with MI, angina and heart failure in the UK Biobank, and with LDL cholesterol, ApoB, blood pressure, and reticulocyte count in the largest available GWAS. For MI, we conducted a validation using CARDIoGRAMplusC4D 1000 Genomes. All the data sources were shown in Fig. 2.

Flow chart of data sources
Genetic associations with insulin and insulin resistance
The exposures were genetically predicted insulin and insulin resistance. As previous MR study on insulin resistance28, we used insulin adjusted for body mass index (BMI) as an indicator of insulin resistance. We also validated the known insulin resistance findings using a validated genetic score for insulin resistance29. Genetic associations with all exposures were taken from a large meta-analysis of GWAS, conducted in adults (n = 108,557; mean age, 50.6 years; ~53% men) of European ancestry, without diabetes, adjusted for age, sex, study site and geographic covariates using an additive genetic model27. We did not use a genetic instrument for insulin resistance encompassing a lipid phenotype so as to focus more on insulin53.
To meet the three key assumptions of instrumental variable analysis, i.e., relevance, independence and exclusions-restriction34, we used genetic variants strongly and independently (r2 < 0.01) predicting the exposures. We used LD-Link (https://ldlink.nci.nih.gov/) based on Europeans to check for correlations (linkage disequilibrium) between genetic variants. We checked whether these genetic variants were independent of potential confounders from their association with Townsend index, smoking, alcohol drinking, physical activity and BMI in the UK Biobank. We dropped single nucleotide polymorphisms (SNPs) associated with any of these potential confounders at Bonferroni corrected significance (p < 0.05/2000 (number of phenotypes in the UK Biobank) = 2.5 × 10−5). We checked for known direct effects of the exposures on the outcomes (violation of the exclusion-restriction assumption) in three comprehensive curated genotype to phenotype cross-references, i.e., Ensembl (http://www.ensembl.org/index.html), the GWAS catalog (https://www.ebi.ac.uk/gwas/) and PhenoScanner (www.phenoscanner.medschl.cam.ac.uk).
Genetic associations with MI, angina and heart failure
Genetic associations with MI, angina and heart failure were obtained using individual-level data in the UK Biobank (under application #42468), with validation for MI using summary statistics from CARDIoGRAPMplusC4D 1000 Genomes26.
The UK Biobank is an ongoing large prospective cohort study23. The UK Biobank recruited 502,713 people aged 40–69 years, mean age 56.5 years, from Great Britain between 2006 and 2010, with 94% self-reported European ancestry, 45.6% men and median follow-up time currently 11.1 years. Disease outcomes were obtained from a nurse-led interview at recruitment, with ongoing follow-up via record linkage to all health service encounters and deaths54. Genotyping was assessed using two very similar arrays, i.e., the UK BiLEVE array and UK Biobank Axiom array. To control for population stratification, we restricted our analysis to participants with self-reported and genetically validated white British ancestry. For quality control, we also excluded participants with (1) excess relatedness (more than 10 putative third-degree relatives) or (2) mismatched information on sex between genotyping and self-report, or (3) sex-chromosomes not XX or XY, or (4) poor-quality genotyping based on heterozygosity and missing rates >1.5%. After quality control, we identified 392,010 white British in the UK Biobank, with 14,442 cases of MI (77% men), 21,939 cases of angina (65% men), and 5,537 cases of heart failure (71% men). Genetic associations with MI, angina and heart failure were obtained using logistic regression controlling for age, assay array and 10 principal components in sex-specific analysis and additionally adjusted for sex in the overall analysis, as the adjustment in our previous MR study in the UK Biobank46.
Data on coronary artery disease/MI have been contributed by CARDIoGRAMplusC4D investigators and have been downloaded from www.cardiogramplusc4d.org. CARDIoGRAMplusC4D 1000 Genomes is a large genetic study (IHD cases n = 60,801, others = 123,504), with ~70% of the cases MI. The participants are mainly of European descent (77%) with phenotyping based on medical records, clinical diagnosis, as well as medications, or indicative symptoms or procedures, such as revascularization, and/or angiographic evidence of stenosis26.
Genetic association with CVD risk factors
Genetic association with LDL cholesterol and ApoB
Genetic associations with LDL cholesterol (inverse normal transformed effect sizes), adjusted for age, age2 and sex, were obtained from the Global Lipids Genetics Consortium Results summary statistics including 188,577 participants of European descent and 7,898 participants of non-European descent, mean age 55.2 years55. Genetic associations with ApoB (inverse normal transformed effect sizes), adjusted for age, sex, time from last meal, if applicable, and first ten principal components, were obtained from a meta-analysis GWAS of metabolomics in 24,925 Europeans (45% men)56. In both GWAS, genomic control was applied to each sample and the meta-analysis results, to correct for inflated test statistics due to potential population stratification.
Genetic associations with blood pressure and reticulocyte count
We obtained overall and sex-specific genetic associations with blood pressure and reticulocyte count using summary statistics from the UK Biobank, provided by Neale Lab (http://www.nealelab.is/uk-biobank/), in 361,194 white British (167,020, 46% men). The study adjusted for age, age2, and 20 principal components in sex-specific analysis and additionally adjusted for sex and interactions of sex with age and age2 in the overall analysis.
Statistics and reproducibility
We obtained MR estimates for the associations of genetically predicted insulin with MI, angina, heart failure and cardiovascular risk factors from two-sample instrumental variable analysis. Specifically, we obtained SNP-specific Wald estimates (quotient of genetic association on outcome and genetic association on insulin) and then meta-analyzed them using inverse variance weighting (IVW) with multiplicative random effects. We validated the findings for MI using summary statistics from a large genetic study, CARDIoGRAMplusC4D 1000 Genomes (IHD cases n = 60,801, others = 123,504), with ~70% of the cases MI26. A consistent direction in both studies gives more confidence. As sex-specific summary statistics were not available in CARDIoGRAMplusC4D 1000 Genomes, we only used overall statistics to replicate overall associations with MI. For insulin resistance genetic score, logistic regression was used to obtain the associations of the genetic score with MI, angina and heart failure controlling for age, assay array and 10 principal components in sex-specific analysis and additionally adjusted for sex in the overall analysis, as previously46. Power calculations were conducted overall and by sex. MR studies require larger sample sizes than conventional observational studies, because the sample size needed for MR is the sample size for the conventional observational study divided by the variance in the exposure explained by the genetic predictors31. Specifically, for cardiovascular events which are the binary outcomes, the sample size was calculated based on the effect size (log OR here), the ratio of cases to non-cases in the study, and the variance explained by the genetic predictors57.
In sensitivity analysis, we conducted sex-specific analysis for all outcomes except LDL cholesterol and ApoB for which sex-specific information is not available. To examine whether effect sizes were larger in men than women, we assessed differences by sex using a z-test for the difference in sex-specific estimates (log OR or beta-coefficients) using a one-tailed p value58.
Given potential bias from invalid instruments when using multiple genetic variants, we also conducted a sensitivity analysis using different statistical methods with different assumptions, i.e., a weighted median59, a mode-based estimate60 and Mendelian Randomization Pleiotropy Residual Sum and Outlier (MR-PRESSO) with 100,000 simulations59. Specifically, a weighted median is robust to invalid instruments and able to provide consistent estimation even when up to 50% of the weight is from invalid SNPs59. The mode-based estimate is based on the assumption that a plurality of genetic variants are valid instruments; i.e., there is no larger subset of invalid instruments estimating the same causal parameter than the subset of valid instruments60. MR-PRESSO can detect and as necessary correct for potentially pleiotropic outliers61. Given the limited number of SNPs, we did not use MR Egger because it is based on the Instrument Strength Independent of Direct Effect (InSIDE) assumption and thereby is more sensitive to outliers and less efficient than other methods such as the weighted median, the mode-based estimate and MR-PRESSO used here62.
All statistical analyses were conducted using R version 3.4.4 (R Foundation for Statistical Computing, Vienna, Austria) and the R package “MendelianRandomization”63.
Ethical approval
The UK Biobank has already received ethical approval from the Research Ethics Committee and participants provided written informed consent. The analysis of other publicly available data or summary statistics does not require additional ethical approval.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The main outcomes are from the UK Biobank under application (#42468). The data is available from the UK Biobank upon request. Data on coronary artery disease/myocardial infarction have also been contributed by CARDIoGRAMplusC4D investigators and have been downloaded from www.CARDIOGRAMPLUSC4D.ORG. Genetic associations with lipids were obtained from the Global Lipids Genetics Consortium Results, downloaded from http://csg.sph.umich.edu//abecasis/public/lipids2013/. Genetic associations with apolipoprotein B were obtained from the GWAS of Kettunen et al.56, downloaded from http://www.computationalmedicine.fi/data#NMR_GWAS. Genetic associations with blood pressure and reticulocyte count were from the UK biobank GWAS results, downloaded from http://www.nealelab.is/uk-biobank/, the results of the GWAS and heritability analyses conducted by the Neale Lab. All the data sources were shown in Fig. 2. The summary data are publicly available.
Code availability
“ukbmd5”, “ukbunpack”, “ukbconv” and “ukbgene” were used for data validation, unpacking, format conversion and obtaining genetic data. They are available for the approved UK Biobank application (#42468), in the online system (https://bbams.ndph.ox.ac.uk/ams/). Software code in R for implementing the Mendelian Randomization analysis, is publicly available (https://cran.r-project.org/web/packages/MendelianRandomization/MendelianRandomization.pdf) without restriction63.
References
Murray, C. J. et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2197–2223 (2012).
Roth, G. A. et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J. Am. Coll. Cardiol. 70, 1–25 (2017).
Ezzati, M. et al. Contributions of risk factors and medical care to cardiovascular mortality trends. Nat. Rev. Cardiol. 12, 508–530 (2015).
Lemaitre, J. F. et al. Early-late life trade-offs and the evolution of ageing in the wild. Proc. Biol. Sci. 282, 20150209 (2015).
Schooling, C. M. Could androgens be relevant to partly explain why men have lower life expectancy than women? J. Epidemiol. Community Health 70, 324–328 (2016).
Byars, S. G. et al. Genetic loci associated with coronary artery disease harbor evidence of selection and antagonistic pleiotropy. PLoS Genet. 13, e1006328 (2017).
Schooling, C. M. & Ng, J. Reproduction and longevity A Mendelian randomization study of gonadotropin-releasing hormone and ischemic heart disease. SSM Popul. Health 8, 100411 (2019).
Schooling, C. M. Practical applications of evolutionary biology in public health. Lancet 390, 2246 (2017).
Lin, X., Yao, Y., Wang, B., Emlen, D. J. & Lavine, L. C. Ecological trade-offs between migration and reproduction are mediated by the nutrition-sensitive insulin-signaling pathway. Int. J. Biol. Sci. 12, 607–616 (2016).
Schooling, C. M., Kowk, M. K., Zhao, J. V. & Au Yeung, S. L. Re: Sulfonylureas as second line drugs in type 2 diabetes and the risk of cardiovascular and hypoglycaemic events: population based cohort study. BMJ 362, k2693 (2018).
Mehran, A. E. et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 16, 723–737 (2012).
Shanik, M. H. et al. Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care 31(Suppl 2), S262–S268 (2008).
Gill, G. et al. Essential medicines and access to insulin. Lancet Diabetes Endocrinol. 5, 324–325 (2017).
Goldner, M. G., Knatterud, G. L. & Prout, T. E. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. 3. Clinical implications of UGDP results. JAMA 218, 1400–1410 (1971).
Livingstone, S. J. et al. Estimated life expectancy in a Scottish cohort with type 1 diabetes, 2008-2010. JAMA 313, 37–44 (2015).
Tikkanen, E. et al. Genetic support for the causal role of insulin in coronary heart disease. Diabetologia 59, 2369–2377 (2016).
Zhan, Y. et al. Exploring the causal pathway from telomere length to coronary heart disease: a Network Mendelian Randomization Study. Circ. Res. 121, 214–219 (2017).
Yaghootkar, H. et al. Genetic evidence for a normal-weight “metabolically obese” phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes 63, 4369–4377 (2014).
Douros, A. et al. Sulfonylureas as second line drugs in type 2 diabetes and the risk of cardiovascular and hypoglycaemic events: population based cohort study. BMJ 362, k2693 (2018).
Braffett, B. H. et al. Association of insulin dose, cardiometabolic risk factors, and cardiovascular disease in type 1 diabetes during 30 years of follow-up in the DCCT/EDIC Study. Diabetes Care 42, 657–664 (2019).
Lawlor, D. A., Harbord, R. M., Sterne, J. A. C., Timpson, N. & Davey-Smith, G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med 27, 1133–1163 (2008).
Ference, B. A. et al. Association of triglyceride-lowering LPL variants and LDL-C-lowering LDLR variants with risk of coronary heart disease. JAMA 321, 364–373 (2019).
Suldow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2007).
Burch, G. Erythrocytosis and ischemic myocardial disease. Am. Heart J. 62, 139–140 (1961).
Astle, W. J. et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167, 1415–1429 (2016).
Nikpay, M. et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 47, 1121–1130 (2015).
Scott, R. A. et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet. 44, 991–1005 (2012).
Nowak, C. et al. Effect of insulin resistance on monounsaturated fatty acid levels: a multi-cohort non-targeted metabolomics and Mendelian Randomization Study. PLoS Genet. 12, e1006379 (2016).
Scott, R. A. et al. Common genetic variants highlight the role of insulin resistance and body fat distribution in type 2 diabetes, independent of obesity. Diabetes 63, 4378–4387 (2014).
Burgess, S. et al. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur. J. Epidemiol. 30, 543–552 (2015).
Freeman, G., Cowling, B. J. & Schooling, C. M. Power and sample size calculations for Mendelian randomization studies using one genetic instrument. Int. J. Epidemiol. 42, 1157–1163 (2013).
Pyorala, M., Miettinen, H., Laakso, M. & Pyorala, K. Hyperinsulinemia predicts coronary heart disease risk in healthy middle-aged men: the 22-year follow-up results of the Helsinki Policemen Study. Circulation 98, 398–404 (1998).
Pyorala, K. Relationship of glucose tolerance and plasma insulin to the incidence of coronary heart disease: results from two population studies in Finland. Diabetes Care 2, 131–141 (1979).
Davies, N. M., Holmes, M. V. & Davey Smith, G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 362, k601 (2018).
Pearl, J. Causality (Cambridge University Press, 2000).
Burgess, S., Davies, N. M. & Thompson, S. G. Bias due to participant overlap in two-sample Mendelian randomization. Genet. Epidemiol. 40, 597–608 (2016).
Tchetgen Tchetgen, E. J., Walter, S. & Glymour, M. M. Commentary: building an evidence base for mendelian randomization studies: assessing the validity and strength of proposed genetic instrumental variables. Int. J. Epidemiol. 42, 328–331 (2013).
Schooling, C. M. Selection bias in population-representative studies? A commentary on Deaton and Cartwright. Soc. Sci. Med. 210, 70 (2018).
Priscilla M. Lopez, S.V. Subramanian, C. Mary Schooling. Effect measure modification conceptualized using selection diagrams as mediation by mechanisms of varying population-level relevance. Journal of Clinical Epidemiology 113 123–128 (2019)
Vikram, N. K., Misra, A., Pandey, R. M., Luthra, K. & Bhatt, S. P. Distribution and cutoff points of fasting insulin in Asian Indian adolescents and their association with metabolic syndrome. J. Assoc. Physicians India 56, 949–954 (2008).
Czech, M. P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 23, 804–814 (2017).
Astley, C. M. et al. Genetic evidence that carbohydrate-stimulated insulin secretion leads to obesity. Clin. Chem. 64, 192–200 (2018).
Reaven, G. M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37, 1595–1607 (1988).
Adeli, K. & Theriault, A. Insulin modulation of human apolipoprotein B mRNA translation: studies in an in vitro cell-free system from HepG2 cells. Biochem. Cell Biol. 70, 1301–1312 (1992).
Wong, L. et al. Effects of sulfonylurea as initial treatment on testosterone of middle-aged men with type 2 diabetes: a 16-week, pilot study. J. Diabetes Investig. 6, 454–459 (2015).
Luo, S., Au Yeung, S. L., Zhao, J. V., Burgess, S. & Schooling, C. M. The association of genetically-predicted testosterone with thromboembolism, heart failure and myocardial infarction: a Mendelian randomization study using UK Biobank. BMJ 364, l476 (2019).
Boden, G., Vaidyula, V. R., Homko, C., Cheung, P. & Rao, A. K. Circulating tissue factor procoagulant activity and thrombin generation in patients with type 2 diabetes: effects of insulin and glucose. J. Clin. Endocrinol. Metab. 92, 4352–4358 (2007).
Khan, A. A., Chung, M. J., Novak, E. & Brown, D. L. Increased hazard of myocardial infarction with insulin-provision therapy in actively smoking patients with diabetes mellitus and stable ischemic heart disease: the BARI 2D (bypass angioplasty revascularization investigation 2 diabetes) trial. J. Am. Heart Assoc. 6, e005946 (2017).
Zhao, J. V. & Schooling, C. M. Coagulation factors and the risk of ischemic heart disease: a Mendelian randomization study. Circ. Genom. Precis. Med. 11, e001956 (2018).
Song, C., Burgess, S., Eicher, J. D., O’Donnell, C. J. & Johnson, A. D. Causal effect of plasminogen activator inhibitor type 1 on coronary heart disease. J. Am. Heart Assoc. 6, e004918 (2017).
Schooling, C. M. & Leung, G. M. A socio-biological explanation for social disparities in non-communicable chronic diseases: the product of history? J. Epidemiol. Community Health 64, 941–949 (2010).
Dongerkery, S. P., Schroeder, P. R. & Shomali, M. E. Insulin and its cardiovascular effects: what is the current evidence? Curr. Diab Rep. 17, 120 (2017).
Wang, Q., Holmes, M. V., Davey Smith, G. & Ala-Korpela, M. Genetic support for a causal role of insulin resistance on circulating branched-chain amino acids and inflammation. Diabetes Care 40, 1779–1786 (2017).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Global Lipids Genetics, C. et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45, 1274–1283 (2013).
Kettunen, J. et al. Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat. Commun. 7, 11122 (2016).
Burgess, S. Sample size and power calculations in Mendelian randomization with a single instrumental variable and a binary outcome. Int. J. Epidemiol. 43, 922–929 (2014).
Paternoster, R., Brame, R., Mazerolle, P. & Piquero, A. Using the correct statistical test for the equality of regression coefficients. Criminology 36, 859–866 (1998).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Hartwig, F. P., Smith, G. D. & Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 46, 1985–1998 (2017).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Slob, E. A. W. & Burgess, S. A comparison of robust Mendelian randomization methods using summary data. https://www.biorxiv.org/content/biorxiv/early/2019/2003/2015/577940.full.pdf (2019).
Yavorska, O. O. & Burgess, S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int. J. Epidemiol. 46, 1734–1739 (2017).
Acknowledgements
The authors would like to thank the UK Biobank for approving our application. The authors would like to thank all investigators of the GWAS applied in this study, for sharing these valuable data. The authors thank Dr. Au Yeung SL for facilitating the storage of UK Biobank genetic data.
Author information
Authors and Affiliations
Contributions
C.M.S. wrote the original draft and J.V.Z. rewrote, J.V.Z. conducted the analysis with the help of C.M.S. and S.L., J.V.Z. drafted the manuscript, C.M.S. and S.L. critically revised it. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, J.V., Luo, S. & Schooling, C.M. Sex-specific Mendelian randomization study of genetically predicted insulin and cardiovascular events in the UK Biobank. Commun Biol 2, 332 (2019). https://doi.org/10.1038/s42003-019-0579-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-019-0579-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
