
Abstract
During intraerythrocytic development, the human malaria parasite Plasmodium falciparum alters the mechanical deformability of its host cell. The underpinning biological processes involve gain in parasite mass, changes in the membrane protein compositions, reorganization of the cytoskeletons and its coupling to the plasma membrane, and formation of membrane protrusions, termed knobs. The hemoglobinopathies S and C are known to partially protect carriers from severe malaria, possibly through additional changes in the erythrocyte biomechanics, but a detailed quantification of cell mechanics is still missing. Here, we combined flicker spectroscopy and a mathematical model and demonstrated that knob formation strongly suppresses membrane fluctuations by increasing membrane-cytoskeleton coupling. We found that the confinement increased with hemoglobin S but decreases with hemoglobin C in spite of comparable knob densities and diameters. We further found that the membrane bending modulus strongly depends on the hemoglobinopathetic variant, suggesting increased amounts of irreversibly oxidized hemichromes bound to membranes.
Similar content being viewed by others


Introduction
Human red blood cells undergo repeated shape deformations during passage through the vascular system. The ability to temporarily change the cell shape in response to a mechanical stress allows erythrocytes to navigate in capillaries whose diameter is one third of their own size1. This capability relies on the fluid nature of the plasma membrane, the high extensibility and, possibly, plasticity of the underlying spectrin/actin network, a homogeneous cytoplasm consisting predominantly of hemoglobin, and a unique cell geometry as defined by a biconcave discoid shape and a high surface area-to-volume ratio2. The elasticity of the spectrin/actin network is essential to protect the membrane from rupture and to ensure mechanical stability. Mechanical stability is achieved by the spring-like spectrin tetramers that are connected in hexagonal and pentagonal arrays via actin-containing junctional complexes. The network is anchored to the membrane through the anion exchanger 1 (AE-1; formerly called band 3)-ankyrin and the protein 4.1-glycophorin C macrocomplexes3,4.
Hereditary red blood cell disorders and infection with the human malaria parasite Plasmodium falciparum interfere with this structural organization and, hence, with red blood cell function5. For instance, hemoglobin S and C, which deviate from wild-type hemoglobin A by a single amino acid substitution of valine and lysine, respectively, for glutamic acid at position 6 in the ß-globin chain, affect cytoskeletal architecture via a mechanism that involves increased oxidative stress6,7. The reactive oxygen species interfere with junctional complex formation and spectrin assembly, among other things6,7. As a result, erythrocytes containing homozygously hemoglobin S have a two- to threefold higher shear elasticity compared with wild-type erythrocytes8,9. Erythrocytes containing hemoglobin C are also thought to be stiffer, although robust quantitative data are still missing.
The protozoan parasite P. falciparum affects red blood cell deformability in various ways. Firstly, the infected erythrocyte swells during the 48 h intraerythrocytic development of the parasite due to the influx of Na+ and accompanying water via parasite-induced channels in the erythrocyte plasma membrane, thereby changing the shape of the red blood cell from biconcave discoidal to almost spherical10,11,12. Secondly, growth of the parasite creates a solid mass that hampers mechanical deformation13,14. Thirdly, the parasite remodels the membrane skeleton and, in addition, inserts proteins of its own design into the erythrocyte plasma membrane15. For instance, the parasite mines the actin from the junctional complexes in order to establish an actin network required for vesicular trafficking of adhesins and other parasite-encoded virulence factors to the erythrocyte plasma membrane16. The adhesins are presented on membrane protrusions, termed knobs, that are anchored to the membrane skeleton17 and which are thought to contribute to strain hardening18.
Although there is compelling evidence indicating a loss in deformability in P. falciparum-infected erythrocytes18,19, the detailed roles of the membrane, the cytoskeleton, and their coupling are unclear. Moreover, it is largely unknown how hemoglobinopathies influence the mechanics of parasitized erythrocytes. In fact, it is unlikely that the combination of a hemoglobinopathy and a P. falciparum infection contribute in an additive manner to membrane stiffening, since both hemoglobin S and C affect numerous physiological functions of the parasite relevant for membrane mechanics. This includes export of proteins to the erythrocyte surface, host-actin reorganization, and knob formation, with infected hemoglobinopathic erythrocytes displaying reduced anterograde protein trafficking, impaired host actin remodeling and fewer, though grossly enlarged knobs16,20,21,22,23. Given that the hemoglobinopathies S and C protect heterozygous carriers form severe malaria24, a better quantitative understanding of the membrane mechanics might provide novel insights into the underlying protective mechanism, particularly since parasitized HbAS and HbAC erythrocytes display a reduced capacity to cause pathology-mediating cytoadhesive interactions with the microvascular endothelial lining21,22.
Different biophysical techniques have been applied to quantitatively characterize the mechanical properties of wild-type erythrocytes25,26. This includes shear flow experiments27, micropipette aspiration28, and optical tweezers29 probing the mechanical properties of the composite shell of the plasma membrane and the spectrin/actin cytoskeleton. These approaches have also been applied to assess the membrane mechanics of P. falciparum-infected erythrocytes13,19,30. However, they have merely yielded the effective shear modulus of the composite system, while largely ignoring how the coupling of plasma membrane and the cytoskeleton is changed. A more informative approach to assess the cell mechanics is flicker spectroscopy, which relies on the analysis of thermally or nonthermally activated shape fluctuations31. In order to analyze erythrocyte mechanics by flicker spectroscopy32, it has been suggested that the spectrin/actin network acts as a wall that confines the fluctuations of the plasma membrane33,34. This concept allows three mechanical parameters from the fluctuation spectrum to be extracted, namely the bending modulus and the tension of the membrane as well as the membrane confinement parameter characterizing the interaction between the plasma membrane and the spectrin-actin network. By extending this approach into the dynamical domain and measuring the relaxation rates, one can, in addition, extract the cytosolic viscosity35. This procedure has been used to show that, in wild-type erythrocytes, the bending modulus and the tension depend strongly on cell shape, but less on oxidation level36. Flicker spectroscopy has recently also been applied to erythrocytes decorated with a P. falciparum-encoded protein, termed erythrocyte binding antigen 175 that is required for parasite entry37. However, it has not yet been applied to P. falciparum-infected erythrocytes per se.
Here we have used flicker spectroscopy to quantify changes in red blood cell mechanics during parasite development in wild-type erythrocytes containing hemoglobin A and in erythrocytes containing hemoglobin S or C. We show that the development of the knobs plays an essential role as they increasingly confine the fluctuations of the plasma membrane in the vicinity of the spectrin/actin network. Further, we have combined our experimental data with a mathematical model to better understand the underlying molecular changes. In addition, we show that the membrane bending modulus increased in the hemoglobinopathic erythrocytes, possibly due to the increased level of intrinsic oxidative stress.
Results
Dramatic changes in erythrocyte membrane mechanics during P. falciparum development
We first applied flicker spectroscopy to investigate uninfected wild-type erythrocytes (HbAA). The cell contours were extracted from phase contrast videos of flickering erythrocytes. In Fourier space, one expects the mean squared displacement (MSD) of a two-dimensional membrane <h(qx, qy)2 > = kBT/(γ + σq2 + κq4), where q = (qx2 + qy2)1/2 is the two-dimensional wavevector, γ the membrane confinement parameter, σ the surface tension, and κ the bending modulus. One sees from this equation that fluctuations of small wavelength (large q) are suppressed by bending and that fluctuations of large wavelength (small q) are suppressed by confinement and tension. Since we recorded the positions of membrane contours at the plane of equator, the equation for the MSD has to be transformed back to real space in regard to qy, (Equation 2 in the “Methods” section)36. For small and large qx, the MSD should scale with qx−1 and qx−3, respectively. For very small qx, it should level off due to confinement. Figure 1a depicts the measured and fitted fluctuation spectrum for uninfected wild-type erythrocytes (U). The experimental data tend to obey the expected scaling laws, although a clear plateau due to confinement is not apparent. Fitting Equation 2 to the data points yielded values for the membrane bending modulus of κ = (2.7 ± 0.6) × 10−19 N m, the surface tension of σ = (0.7 ± 0.2) × 10−6 N m−1, and the membrane confinement of γ = (0.5 ± 0.2) × 106 N m−3 (n = 42). These values are in good agreement with previous determinations36,38. In particular, the bending modulus of κ ~ 70 kBT is larger than one would expect for bare lipid bilayers (10–20 kBT)39, indicating a high protein content. The measured surface tension σ is well below the lysis tension of a lipid bilayer ~7 × 10−3 N m−140. The membrane confinement γ corresponds to a shear modulus µ = γ r2 = 8 × 10−6 N m−1, if one takes r = 4 µm as the linear dimension of the erythrocyte projected on a two-dimensional plane. Again this value agrees well with the values reported previously41.

Membrane fluctuation spectra obtained from uninfected and P. falciparum-infected erythrocytes at the ring and trophozoite stage. a Mean squared displacement (MSD) as a function of the wavenumber qx. b The corresponding power spectrum density (PSD) as a function of frequency f. U (red); uninfected, R (orange); ring phase, and T (yellow); trophozoite. Representative spectra from single cell measurements are shown. The two different power law exponents predicted by the theoretical model for the low and high values of the wavenumber qx and the frequency f respectively, are presented to guide the eye
In Fig. 1b we extended the contour analysis to the temporal domain and plotted the power spectrum density (PSD) against the frequency f. By fitting Equation 4 to the data points and taking the formerly determined values for κ, σ, and γ, this analysis allowed us to obtain the apparent red blood cell viscosity of ηRBC = (1.2 ± 0.7) × 10−2 N m−2 s. This value is consistent with previous determinations by flicker spectroscopy36, although it exceeds the viscosity of a hemoglobin solution (0.32 mg/mL) by a factor of two42.
We next investigated wild-type erythrocytes infected with the P. falciparum line FCR3 at the ring (observed time window: 10–18 h post invasion) and trophozoite (observed time window: 24–32 h post invasion) stages. These two stages represent different morphological and pathophysiological states of the parasite. Ring-stage-infected erythrocytes circulate through the vascular system, whereas trophozoites are adhesive and sequester in the microvasculature due to the presence of knob-anchored, surface-presented adhesins43. As seen in Fig. 1, the intracellular development of P. falciparum is associated with a progressive dampening of the shape fluctuation amplitude and the PSD (R and T denote ring and trophozoite stages, respectively). The dampening effect coincided with drastic changes in surface morphology, as indicated by the Gaussian surface roughness (RMS) that changed from 90 ± 34 nm in uninfected erythrocytes to 137 ± 44 nm in rings and to 169 ± 47 nm in trophozoites (Supplementary Table 1), as determined from phase contrast images. Although the absolute values for uninfected erythrocyte and trophozoites seem larger than what we previously reported using coherent diffraction X-ray imaging44 due to the difference in instrumental resolution, the increase in the surface roughness is qualitatively in good agreement.
All four mechanical parameters significantly increased as the parasite developed (p < 0.05 according to Welch t-test) (Fig. 2; Table 1). For instance, the surface tension σ increased fourfold to (3.3 ± 1.6) × 10−6 N m−1 in trophozoites, as compared with uninfected erythrocytes (Fig. 2b). Even more pronounced was the increase in the membrane confinement γ that rose tenfold to (5.3 ± 3.9) × 106 N m−3 (Fig. 2c). In comparison, the change in the bending modulus κ was subtler to (3.4 ± 1.4) × 10−19 N m (Fig. 2a). We further noted a significant, sevenfold rise to (8.8 ± 5.2) × 10−2 N m−2 s in the apparent red blood cell viscosity ηRBC between uninfected erythrocytes and trophozoites (p < 0.05) (Fig. 2d). In summary, the flicker spectroscopy confirmed the expected stiffening during parasite development. However, this effect arose less from a change in bending modulus, but rather from changes in confinement and tension, as evidenced by the dampening of the fluctuations in the low qx regime.

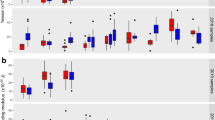
Changes in biomechanical parameters in uninfected and P. falciparum-infected HbAA erythrocytes. a Bending modulus κ, b surface tension σ, c membrane confinement γ, d and apparent red blood cell viscosity ηRBC. U (red); uninfected, R (orange); ring phase, and T (yellow); trophozoite. Individual data points represent a single determination and n the total number of data points obtained using blood of N different donors. Box plots (gray) were laid over the data points. The 25–75 percentile ranges are highlighted in red and replotted in the following figures. The gradient images of representative cells at the corresponding infection stages are presented as insets of panel a. The cytoplasmic area occupied by the parasite is highlighted by the dotted line. *p < 0.05 according to Welch t-test. Scale bar, 5 µm
Hemoglobin S and C affect membrane mechanics of P. falciparum-infected erythrocytes mainly via changes in bending
We next performed flicker spectroscopy for erythrocytes containing the hemoglobinopathies S and C in their heterologous form with wild-type hemoglobin. The values for κ, σ, γ, and ηRBC obtained for uninfected and infected erythrocytes were compared with those of wild type (Table 1). Uninfected HbAS and HbAC erythrocytes displayed surface tensions σ and membrane confinements γ that were significantly higher than those of wild-type erythrocytes (p < 0.05) (Table 1). On the other hand, the bending moduli and the apparent red blood cell viscosities ηRBC of all erythrocyte variants were comparable (Table 1). The comparable level of viscosity (Individual data points presented in Supplementary Fig. 1) seems consistent with recent reports on the mean corpuscular hemoglobin concentrations between uninfected HbAS, HbAC, and HbAA erythrocytes12. Infection with P. falciparum further altered the membrane properties, leading to gradually increasing mechanical parameters in HbAS and HbAC erythrocytes as the parasite matured, in a manner similar to parasitized wild-type HbAA erythrocytes (Table 1; Fig. 3). Importantly, however, there were clear differences between infected wild-type and hemoglobinopathic erythrocytes as well as between infected HbAS and HbAC erythrocytes. At the trophozoite stage, the bending modulus κ significantly increased by a factor of 1.7 and 1.6 in parasitized HbAS and HbAC erythrocytes, respectively, compared with infected wild-type erythrocytes (Table 1; Fig. 3a) (p < 0.05). Importantly, the surface tension σ and membrane confinement γ showed remarkable differences between HbAS and HbAC erythrocytes at the trophozoite stage (p < 0.05). Both σ and γ of parasitized HbAC erythrocytes were much larger compared with those of parasitized HbAS erythrocytes (Table 1; Fig. 3c). These results suggest that the two different hemoglobinopathies mainly affect membrane bending through a local mechanism at large qx (small wavelength), while the difference at small qx (large wavelength) was more prominent.

Changes in biomechanical parameters in uninfected and P. falciparum-infected HbAS and HbAC erythrocytes. a Bending modulus κ of U (dark blue); uninfected, R (blue); ring phase, and T (light blue); trophozoite HbAS erythrocytes. b Bending modulus κ of U (dark gray); uninfected, R (gray); ring phase, and T (white); trophozoite HbAC erythrocytes. Corresponding data for the c and d surface tension σ and e and f membrane confinement γ. Individual data points represent a single determination and n the total number of data points obtained using blood of N different donors. Box plots (gray) were laid over the data points. The 25–75 percentile ranges of the corresponding HbAA results are highlighted in red for visual comparison. *p < 0.05 according to Welch t-test
Knobs mainly affect large wavelength fluctuations
Previous studies have shown that parasitized erythrocytes containing hemoglobin S or C harbor fewer, but bigger, knobs on their surface as compared with parasitized wild-type erythrocytes16,21,22,45. We confirmed these findings for the FCR3 strain used in this study, using scanning electron microscopy. Wild-type erythrocytes infected with the FCR3 strain displayed as many as 14 ± 4 knobs per µm2 at the trophozoite stage, whereas HbAS and HbAC erythrocytes infected with the same parasite line displayed only 3 ± 2 knobs per µm2 and those few knobs presented were enlarged (Supplementary Fig. 2). To elucidate the impact of the knobs on cell mechanics in general, we investigated a knobless parasite line derived from the FCR3 strain by a chromosomal breakage and healing event within the knob-defining kahrp gene, resulting in a truncated nonfunctional kahrp gene and the loss of ~100 kbp of DNA from the affected chromosome 246. Scanning electron microscopy confirmed the knobless phenotype (Fig. 4a). Accordingly, erythrocytes infected with the knobless parasite line displayed a reduced Gaussian roughness of 114 ± 31 nm, compared with erythrocytes infected with the knob-forming FCR3 line. Importantly, the knobless parasites revealed a significantly lower membrane confinement γ (by a factor of two), as compared with an age-matched control group of the knobby FCR3 strain (p < 0.05) (Table 1 and Fig. 4e). Similarly, the surface tension σ was distinctly lower (Table 1 and Fig. 4d). On the other hand, the bending modulus κ (Table 1 and Fig. 4c) and the apparent viscosity ηRBC values were similar (Table 1 and Fig. 4f). These findings suggest that the knobs mainly change large wavelength fluctuations due to their anchoring to the membrane skeleton, but that they have little effect on the small wavelength fluctuations governed by bending.

Biomechanical properties of HbAA erythrocytes infected with a knobless, FCR3-derived mutant at the trophozoite stage. a Representative SEM image and b representative gradient map calculated from a phase contrast image. c Bending modulus κ, d surface tension σ e membrane confinement γ, and f apparent red blood cell viscosity ηRBC. Individual data points represent a single determination and n the total number of data points obtained using blood of N different donors. Box plots (gray) were laid over the data points. The 25–75 percentile ranges of the corresponding HbAA results are highlighted in red for visual comparison. *p < 0.05 according to Welch t-test. Scale bar, 2 µm
Both density and spring constants of knobs have a strong effect on the fluctuation spectrum
As described above, our experimental data implied that in general knobs strongly affect the membrane confinement γ and that this parameter significantly differed between parasitized HbAS and HbAC erythrocytes, in spite of comparable knob densities and knob morphologies (Supplementary Fig. 2) (p < 0.05). To identify possible causes for these surprising differences, we used a mathematical model to investigate the effect of the coupling between the lipid bilayer and the underlying cytoskeleton on the fluctuation spectrum. We assumed that the membrane is free to fluctuate only in the regions between the connections to the cytoskeleton, which are progressively remodeled as the parasite develops (Fig. 5a). The model takes into account the density, ρ, of membrane–cytoskeleton connectors and their strength as defined by a spring constant k (Fig. 5b). Thus, the model did not distinguish between the molecular nature of the connectors, but focused on their effect on the membrane fluctuations. The reported spatial heterogeneity in the network was considered by employing a mixture of hexagonal and triangular lattices (Fig. 5c). The fluctuation spectrum was then calculated numerically for a membrane with a given spatial distribution of pinning sites47,48. Note that the model calculates the fluctuations of a square membrane patch of ~1 × 1 µm in size with a resolution of 10 nm, and not for an entire red blood cell.

Graphic representation of key parameters of the numerical model. a Simplified model of the membrane skeleton, including the junctional complexes and AE-1-ankyrin junctions (left, uninfected) and additional anchoring structures via the knob components Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) and knob-associated histidine rich protein (KAHRP) (right, trophozoite). b The structure of a singular anchoring point is depicted, with spectrin filaments indicated in yellow and connections to the lipid bilayer indicated in orange. The whole complex can be modeled by a vertical spring with a spring constant k. c The model treats the membrane–spectrin connections as springs that are distributed in a hexagonal array. Inhomogeneities are introduced by removing some midpoints of hexagons as indicated in the left panel. The other three arrays show examples of spring distributions for densities of 1096, 257, and 44 springs per µm2 (from left to right)
We initially varied the density ρ of the connectors from 44 to 1096 springs µm−2, while fixing the spring constant k at 13.2 nN m−1. This range of the connector density was chosen for the following reasons. Previous studies have estimated the number of junctional complexes per red blood cell to be around 35,0003. Given a surface area of 136 µm2 for a red blood cell49, this converts to 257 junctional complexes per µm2. Given that junctional complexes connect between five to eight spectrin filaments, as opposed to the AE-1-ankyrin complexes that bind a single spectrin filament, it is plausible to consider only the connections via the junctional complexes while neglecting the AE-1-ankyrin bridges. Infections with the malaria parasite result in a disassembling of junctional complexes and concomitantly in the formation of knobs, which we took into account by varying the junctional complex connector density of uninfected wild-type erythrocytes by a factor of five in both directions. A spring constant k of 13.2 nN m−1 was chosen given the spring constant of an individual spectrin filament of ksp = 2 µN m−1 and taking into account that spectrin filaments are arrayed in hexagonal lattices (for further details see methods)50. The corresponding fluctuation spectrum calculated from the model revealed that the MSD decreased with increasing connector density in the small wavenumber q regime, indicating a correlation between the connector density ρ and the membrane confinement (Fig. 6a). In the higher q regime, the spectra collapse, consistent with the connectors not affecting the small wavelength fluctuations at equal bending rigidity (Fig. 6a). The global shapes of the calculated spectra seem consistent with that of the experimental ones.

Simulated effect of the spring density and the spring constant on the membrane confinement. Simulated mean square displacement as a function of the wavenumber q calculated a for different connector densities ρ (see Fig. 5c) and b for different spring constants k. The discrete data points represent the results of numerical calculations and the continuous lines are the corresponding fit of the continuum theory. Membrane confinement γ as a function of c the density ρ of connectors d the spring constant k. The arrows indicate values calculated for uninfected red blood cells
We next varied the spring constant k from 1.32 × 10−10 N m−1 to 6.0 × 10−5 N m−1, while keeping the connector density constant at 257 springs per µm2. An increase in the spring constant k strongly affected the MSD, resulting in a major dampening in the low q regime and no effects in the high q regime (Fig. 6b). In order to describe the global effects of changes on connector density ρ and spring constant k, we also calculated the effective membrane confinement γ corresponding to the microscopic models. The correlations of the membrane confinement γ with the connector density ρ and the spring constant k were almost linear (for detailed explanation see supplementary information) over the range of values examined (Fig. 6c, d, respectively).
Finally, we simulated the effect of the knob density and the knob size on the membrane confinement. Knobs were modeled as clusters of springs, with the number of springs per cluster ranging from 13 to 85, corresponding to a projected knob surface area of 3200–39,200 nm2. This range covers experimentally determined knob surface areas obtained from scanning electron microscopy images of HbAA, HbAS, and HbAC erythrocytes (Supplementary Fig. 2). The knob-defining springs with an assumed spring constant k of 26.4 nN m−1, were laid over a thinned out hexagonal lattice in order to capture the situation in an infected erythrocyte. The model assumes a direct correlation between the knob surface area and the number of membrane–cytoskeleton connectors. We found that the membrane confinement directly correlated with the overall number of knob-defining springs and not with their distribution, suggesting that a few but large knobs can compensate for many small knobs (Fig. 7). In summary, these simulations demonstrate that the changes in connector density and spring constants that occur during the progressive establishment of the knob structure recapitulate damping effects on the fluctuation spectrum as observed experimentally.

Influence of knobs size and density on the membrane confinement. Spring positions of two realizations for a 1 µm × 1 µm membrane patch. Knobs are represented by a clusters of 13 springs and b 49 springs. The background represents a membrane with a thinned out anchor density of 102 µm−2. c The relative membrane confinement γ/γ0 as normalized to an array without knobs is shown as a function of the cumulative number of knob-specific springs N. Each data point represents the mean value obtained from ten individual simulations. Three different knob sizes as defined by spring number were considered. The spring numbers are indicated
Discussion
During intraerythrocytic development, P. falciparum modifies the proteinaceous composition of the red blood cell membrane13. In addition, the parasite reorganizes the membrane skeleton and it establishes new junctions between the host cell membrane and the remodeled actin/spectrin network5,16,18. All of these changes alter the mechanical properties of the host cell membrane, which, in turn, affects the cytoadhesion and clearance mechanisms of red blood cells in the spleen. For example, previous studies demonstrated that the adhesion of HbAS and HbAC trophozoites is weaker compared with that of infected HbAA trophozoites21,22. Moreover, using a model spleen system, it has been shown that the rate of splenic clearance is >90% for both HbAA and HbAS trophozoites51. This finding suggests that the change in mechanical properties caused by the malaria infection is dominant, irrespective of the hemoglobin types. Using flicker spectroscopy and a mathematical model, our study, quantified these biomechanical changes as a function of parasite development and hemoglobin variants. In particular, it measured defined mechanical parameters, which allowed us to associate them with causative biological processes.
It is well established that the parasite inserts proteins of its own design, such as solute channels and adhesion molecules, into the plasma membrane of the host erythrocyte52,53. As a result, the plasma membrane could lose flexibility. Indeed, we have measured a clear increase in the bending modulus κ (Table 1 and Fig. 2). Interestingly, the increase was more pronounced in infected HbAS and HbAC erythrocytes, as compared with parasitized wild-type red blood cells (Table 1, Fig. 3). We explain this finding by enhanced amounts of membrane cross-linked, irreversibly oxidized haemichromes that are characteristic of hemoglobinopathic erythrocytes due to an increased reactivity of hemoglobin S and C with oxygen6,7,20. The metabolic activity of the parasite contributes to oxidative stress and was shown to promote the formation of oxidized haemichromes in HbAS and HbAC erythrocytes20. Though there has been no study on the splenic clearance of infected HbAC erythrocytes, the change in the mechanical properties of HbAC trophozoites suggests that the clearance rate should be comparable to those of HbAA and HbAS trophozoites.
Another parameter of global character is the apparent viscosity of the cytosol. Note, that the values we calculated from fluctuation spectra were twofold higher than those of hemoglobin solutions measured by a viscometer42, although they were consistent with previous determinations using flicker spectroscopy36. It is widely accepted that extracting this value from flicker spectroscopy is challenging, often leading to a strong overestimation of the actual values for the cytosolic viscosity35,54. Hence, we only regarded the qualitative changes to the apparent red blood cell viscosity during the infection with P. falciparum.
Flicker spectroscopy yielded values, particularly for infected erythrocytes at the trophozoite stage, that were unreasonably high despite the fact that the parasite consumes large amounts of hemoglobin during intraerythrocytic development11. It has been argued before that the spectrin/actin network rearranges on the timescale of microseconds, while the relaxation time of membranes is on the timescale of milliseconds. Thus, the cytoskeleton should act like a rigid wall in regard to fluid flow between the membrane and the spectrin/actin network34. Here, we speculate that this assumption becomes progressively less justified as the parasite matures, and that in infected erythrocytes, the erythrocyte cytoplasm can no longer be treated as a uniform viscous fluid and that the flicker-determined values of the apparent viscosity are strongly influenced by the rigid intracellular parasite compartment and, possibly, membrane profiles, such as Maurer’s clefts and vesicles, that the parasite establishes into the host cell cytoplasm14,55.
Our experimental data and the mathematical model revealed an essential influence of the knobs on erythrocyte mechanics, which expresses itself through the membrane confinement revealing dramatic changes during parasite development (Table 1 and Fig. 7). This finding is very plausible. Knobs are large supramolecular structures of ~70 nm in diameter and they are abundant, ~14 ± 4 knobs per µm2 in wild-type erythrocytes infected with the P. falciparum FCR3 strain (Supplementary Fig. 2). Knobs are anchored to various components of the cytoskeletal complexes, including spectrin, actin and ankyrin, and they harbor surface exposed adhesion molecules5,17. Thus, knobs firmly connect the lipid bilayer with the underlying membrane skeleton and strongly modulate their fluctuations.
We further noted a knob-independent effect on the membrane confinement as revealed by erythrocytes infected with a knobless parasite. The four- to fivefold increase in the membrane confinement in relation to the uninfected control can be explained by the reorganization of actin/spectrin network by the parasite. The parasite mines the actin from the junctional complexes to generate long cortical actin filaments that serve as cables for vesicular transport of parasite-encoded proteins to the erythrocyte surface16,17. Consequently, the connections holding spectrin filaments in hexagonal and pentagonal arrays are disassembled, resulting in the spreading of spectrin filaments56. An increased end-to-end distance of spectrin filaments leads to a spring hardening effect56 and, hence, contributes to an enhanced membrane confinement (Fig. 6). The surface tension largely followed the membrane confinement, which is expected since both parameters depend on the coupling between lipid bilayers and cytoskeletons.
Previous studies have shown that parasitized HbAS and HbAC erythrocytes have fewer but larger knobs than wild-type erythrocytes20,21,22. For instance, they possess on average only 3 ± 2 knobs per µm2 and the knobs formed have a basal diameter of ~200 nm, corresponding to an average surface area of ~31,000 nm2 (Supplementary Fig. 2). Simulating the effect of knobs on the membrane confinement revealed that both the knob size and the knob density affected this parameter. Thus, the almost comparable membrane confinements of parasitized wild-type and HbAC erythrocytes at the trophozoite stage could be explained by the compensation of the effect of larger knob size by the lower knob density.
Surprisingly, our measurements revealed a lower membrane confinement for parasitized HbAS erythrocytes than HbAC erythrocytes. This finding is rather unexpected, since both types of infected erythrocytes possess comparable knob densities and knob sizes. Although the underpinning molecular causes are still unclear, our simulations provided a possible explanation by showing that not only the number of connectors but also their strength affected the membrane confinement (Fig. 6b). Thus, the differences in membrane confinement between parasitized HbAS and HbAC erythrocytes might point towards differences in the anchoring of the knobs to the membrane skeleton. Further work is needed to verify this hypothesis and identify molecular processes that might differently affect the interactions between knob components and the membrane cytoskeleton in red blood cells containing hemoglobin S and C.
The membrane fluctuations investigated here can have both thermal and nonthermal (active) causes26. Possible nonthermal causes for membrane fluctuations include phosphorylation events in the spectrin–actin network or in lipid-related molecules like PIP2, known to lead to changes in the shear modulus; the movement of ions through membrane channels and pumps, which transfers momentum to both the membrane and the surrounding fluid; and other membrane-related processes, such as the activity of flippases, floppases and scramblases, or endo- and exocytosis. It is plausible that the parasite changes all of these processes, in particular because it strongly affects ionic currents through the membrane57. Recently a four-bead optimal tweezer setup58 showed that slow fluctuations (below 10 Hz) are mainly active in nature, while fast fluctuations (above 10 Hz) are exclusively thermal in nature. We expect that such an approach can potentially be used to overcome the time resolution of flicker spectroscopy to dissect the active process during parasite development, shedding light on the interplay between the changes in mechanics and activity.
Methods
Ethical clearance
The study was approved by the ethical review boards of Heidelberg University, Germany, Mannheim University, Germany, and the Biomolecular Research Center (CERBA/Labiogene) at the University of Ouagadougou, Burkina Faso. Written informed consent was given by all blood donors.
Blood phenotyping
All blood samples were routinely screened by HPLC in the diagnostic hematology laboratory of Heidelberg University Hospital. The phenotype was subsequently confirmed by PCR analysis.
P. falciparum culture
Uninfected HbAA, HbAS, and HbAC erythrocytes were stored in RPMI 1640 medium supplemented with 2 mM l-glutamine, 25 mM Hepes, 100 μM hypoxanthine, and 20 μg mL−1 gentamicin at 4 °C until usage. The P. falciparum strain FCR3 was kept in continuous culture59. Briefly, blood cultures were grown at a hematocrit of 5% and a parasitemia below 5% under controlled atmospheric conditions (3% CO2, 5% O2, and 92% N2, 95% humidity) at 37 °C. Parasites in HbAA, HbAS, and HbAC cultures were synchronized within a time window of 8 h, by sorbitol treatment60 at time point t = 0 h and a repetition at t ~ 10 h. Trophozoite stage parasites were enriched by magnetic column isolation61 at t ~ 22 h, starting flicker spectroscopy experiments at t ~ 24 h, with an expected parasite age of 24–32 h post invasion. Ring-stage parasites from the following cell cycle were prepared at t ~ 57 h and subjected to flicker spectroscopy at t ~ 58 h, with an expected parasite age of 10–18 h post invasion. Details on the molecular characterization of the knobless FCR3 mutant are summarized in the supplementary information (Supplementary Fig. 3).
Flicker spectroscopy
Cells at different infectious stages (uninfected, ring, and trophozoite stage) were resuspended in RPMI 1640 medium (pH 7.4) supplemented with 1 mg mL−1 bovine serum albumin (Sigma-Aldrich) at a hematocrit of ~0.1% and incubated for 30 min at 37 °C in petri dishes with glass bottom. Note that RPMI 1640 contains 2 mg mL−1 d-glucose needed to keep a constant ATP level to preserve the activity of the cytoskeleton62. The infectious stage was confirmed for each individual cell by microscopic examination (Supplementary Fig. 4). Samples were set inside a temperature-controlled chamber (37 °C) mounted on an Axio Observer Z1 microscope (Zeiss) equipped with a ring aperture, a ×100 oil-immersion objective lens (N.A. = 1.4), and an ORCA-Flash4.0 LT camera (Hamamatsu). Five hundred phase-contrast images of erythrocytes were collected by setting the exposure time and time interval at 25 ms each. Compared with the phase-contrast images, the gradient images give a much better contrast to visualize both parasites surrounded by vacuole membranes and cell membranes (Supplementary Fig. 5).
Analysis of fluctuation spectra
The cell rim position rrim(θ, t) was obtained from gradient images transformed into a polar coordinate (0 < θ < 2π) with a step size of Δθ = 2π/256, where rrim(θ, t) = 0 corresponds to the center of mass of each cell (Supplementary Fig. 6).
The deviation of rrim(θ, t) from the averaged position was Fourier-transformed into the mean square displacement (MSD) as a function of the wavenumber q:
< >stands for the time average over all frames. The theoretically predicted mean square displacement is described as a function of qx, which is in the continuous wavenumber corresponding to the experimental q:
L is the contour length L = 2π < r > and < r > the mean radius of the cell. σ is the effective surface tension and κ the bending modulus. γ is the membrane confinement, which is proportional to the shear modulus μ of the membrane–cytoskeleton complex. As this equation is not valid for a closed membrane system at small values of qx, the analysis was performed only above qx ≥ 4, where the difference between the MSD of a planar and a spherical membrane is ≤15%36,38. Equation 2 predicts that the fluctuation spectrum should exhibit two different power law dependences: the region dominated by the tension (σ > > κqx2) following <u(qx, y = 0)2> ~ qx−1, and the region dominated by the bending modulus (σ < < κqx2) following <u(qx, y = 0)2> ~ qx−3 36. To shed light on the dynamics of the system we applied the spherical harmonics approach35,63
where Ω is the solid angle, <r> the mean radius of the cell, ulm the amplitude corresponding to the l, m mode, and Ylm the spherical harmonics. The theoretical PSD as a function of the angular frequency ω is given by:
<|ulm|2> is the mean squared fluctuation amplitude for the l,m mode following:
and ωl is the associated characteristic decay frequency:
Z(l) is defined as:
Equation 4 predicts that the PSD for low frequencies is dominated by the membrane tension σ, following a power law f −1, while the PSD for high frequencies is dominated by the bending modulus κ following f −5/3 35. The apparent RBC viscosity ηRBC was obtained from equation 4 using the three principal parameters bending modulus κ, surface tension σ, and cytoskeletal membrane confinement γ obtained from equation 2, and using a buffer viscosity of 0.8 × 10−3 N m−2 s64. The data were analyzed using self-written macros in MatLab.
Numerical model
In order to better understand the experimentally observed differences in mechanical parameters during the parasite maturation, we employ numerical calculations connecting the membrane continuum theory with structural changes of the RBC spectrin network on the microscopic scale. Specifically, the confinement parameter γ can be well explained with discrete connections between the phospholipid bilayer and the spectrin network where the anchor points are modeled as springs with spring constant k in the vertical direction (see Fig. 5a). There are two major contributions that set the strength of the spring constant. Firstly, the anchoring complex itself behaves spring-like and secondly, during vertical displacements the spectrin network is stretched out of its plane which results in a force in z-direction as can be seen in Fig. 5b. Since the parasite alters the spectrin network while it grows, we consider this as the main contribution.
The red blood cell membrane is described by a Hamiltonian consisting of two distinct contributions similarly to the approaches by Lin and Brown47,48. The elastic contribution is given by the Canham–Helfrich Hamiltonian, in Monge gauge, which takes into account the bending of the bilayer κ and a surface tension σ:
where h(r) is the normal displacement (z-direction) of the membrane from its equilibrium position in the xy-plane. The expression needs to be integrated over the whole membrane patch with the dimensions Lx and Ly. The contribution from the springs consists of a sum over the discrete attachment sites (α = 1, …, N):
where kα is the spring constant at position rα. For the simplicity the spring constants kα are set to be equal to k for a given realization unless stated otherwise. After Fourier transforming the above equations, an expression for the mean squared amplitudes can be derived using the equipartition theorem. Details about the calculation can be found the work by Lin and Brown65. The numerical treatment which is necessary because of the discrete attachment sites, limits the size of the membrane patch. Here we calculate the fluctuations for square patches of 1 µm × 1 µm size with a resolution of 10 nm. By fitting the corresponding formula34 to the MSD, the values of the relevant parameters can be extracted similarly to the analysis of the experimental data.
The springs, which mainly present junctional complexes, are arranged in a hexagonal array if not otherwise stated (Fig. 5c). This mimics the hexagonal structure of the red blood cell cytoskeleton. However, since the cytoskeleton has been found to have irregularities in form of large voids66, we take away 49% of the hexagonal midpoints in each configuration (Fig. 5c).
Estimation of effective spectrin spring constant
For the numerical calculations we are interested in an effective spring constant for out of plane motion. In order to estimate a realistic value, we neglect the strain hardening property of the spectrin filaments and model it as a simple spring. An individual spectrin filament has been estimated to have a spring constant of ~ksp = 2 × 10−6 N m−1 when extended along its contour length50 and we assume that an average of six filaments are attached to a junction4. By a simple force balance equation, the following formula can be derived:
where α is the rest length of the spectrin filament and z the typical displacement orthogonal to the membrane (z/α ~ 0.0465).
Statistics and reproducibility
The presented data were obtained during at least four different experimental sessions, using blood from at least three different donors of each erythrocyte variant. Statistical significance was analyzed using the Welch t-test and the one-sided Anova test, within the MatLab software package.
Please note, that values for all four mechanical parameters obtained for erythrocytes from different donors exhibited no significant differences according to one-sided Anova test (p < 0.05) for HbAA, HbAS, and HbAC for the uninfected, ring, and trophozoite state.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that the main data supporting the findings in this study are available within this article (and the supplementary information). Data of individual determinations of the mechanical parameters presented in Figs. 2, 3, and 4 and the corresponding statistical analyses are available at https://doi.org/10.17632/bfn8p44vcr.1.
Code availability
The utilized computer code is deposited at https://doi.org/10.5281/zenodo.2657921.
References
Fung, Y., Skalak, R. Biomechanics: mechanical properties of living tissues. (Springer Science & Business Media, Berlin, Germany, 2013).
Tomaiuolo, G. Biomechanical properties of red blood cells in health and disease towards microfluidics. Biomicrofluidics 8, 051501 (2014).
Lux, S. E. Anatomy of the red cell membrane skeleton: unanswered questions. Blood, blood-2014-2012-512772 (2015).
Machnicka, B. et al. Spectrins: a structural platform for stabilization and activation of membrane channels, receptors and transporters. Biochim. Biophys. Acta (BBA)-Biomembr. 1838, 620–634 (2014).
Lavazec, C. Molecular mechanisms of deformability of Plasmodium-infected erythrocytes. Curr. Opin. Microbiol. 40, 138–144 (2017).
Hebbel, R. P. Beyond hemoglobin polymerization: the red blood cell membrane and sickle disease pathophysiology. Blood 77, 214–237 (1991).
Silva, D. G., Belini Junior, E., de Almeida, E. A. & Bonini-Domingos, C. R. Oxidative stress in sickle cell disease: an overview of erythrocyte redox metabolism and current antioxidant therapeutic strategies. Free Radic. Biol. Med. 65, 1101–1109 (2013).
Nash, G. B., Johnson, C. S. & Meiselman, H. Mechanical properties of oxygenated red blood cells in sickle cell (HbSS) disease. Blood 63, 73–82 (1984).
Byun, H. et al. Optical measurement of biomechanical properties of individual erythrocytes from a sickle cell patient. Acta Biomater. 8, 4130–4138 (2012).
Staines, H. M., Ellory, J. C. & Kirk, K. Perturbation of the pump-leak balance for Na+ and K+ in malaria-infected erythrocytes. Am. J. Physiol.Cell Physiol. 280, C1576–C1587 (2001).
Lew, V. L., Tiffert, T. & Ginsburg, H. Excess hemoglobin digestion and the osmotic stability of Plasmodium falciparum-infected red blood cells. Blood 101, 4189–4194 (2003).
Waldecker, M. et al. Differential time-dependent volumetric and surface area changes and delayed induction of new permeation pathways in P. falciparum-infected hemoglobinopathic erythrocytes. Cell. Microbiol. 19, 1–13 (2017).
Maier, A. G. et al. Exported proteins required for virulence and rigidity of Plasmodium falciparum-infected human erythrocytes. Cell 134, 48–61 (2008).
Hosseini, S. M. & Feng, J. J. How malaria parasites reduce the deformability of infected red blood cells. Biophys. J. 103, 1–10 (2012).
Maier, A. G., Cooke, B. M., Cowman, A. F. & Tilley, L. Malaria parasite proteins that remodel the host erythrocyte. Nat. Rev. Microbiol 7, 341–354 (2009).
Cyrklaff, M. et al. Hemoglobins S and C interfere with actin remodeling in Plasmodium falciparum-infected erythrocytes. Science 334, 1283–1286 (2011).
Cyrklaff, M., Sanchez, C. P., Frischknecht, F. & Lanzer, M. Host actin remodeling and protection from malaria by hemoglobinopathies. Trends Parasitol. 28, 479–485 (2012).
Zhang, Y. et al. Multiple stiffening effects of nanoscale knobs on human red blood cells infected with Plasmodium falciparum malaria parasite. Proc. Natl Acad. Sci. USA 112, 6068–6073 (2015).
Suresh, S. et al. Connections between single-cell biomechanics and human disease states: gastrointestinal cancer and malaria. Acta Biomater. 1, 15–30 (2005).
Cyrklaff, M. et al. Oxidative insult can induce malaria-protective trait of sickle and fetal erythrocytes. Nat. Commun. 7, 13401 (2016).
Fairhurst, R. M. et al. Abnormal display of PfEMP-1 on erythrocytes carrying haemoglobin C may protect against malaria. Nature 435, 1117–1121 (2005).
Cholera, R. et al. Impaired cytoadherence of Plasmodium falciparum-infected erythrocytes containing sickle hemoglobin. Proc. Natl Acad. Sci. USA 105, 991–996 (2008).
Kilian, N. et al. Hemoglobin S and C affect protein export in Plasmodium falciparum-infected erythrocytes. Biol. Open 4, 400–410 (2015).
Taylor, S. M., Parobek, C. M. & Fairhurst, R. M. Haemoglobinopathies and the clinical epidemiology of malaria: a systematic review and meta-analysis. Lancet Infect. Dis. 12, 457–468 (2012).
Peng, Z. et al. Lipid bilayer and cytoskeletal interactions in a red blood cell. Proc. Natl Acad. Sci. USA 110, 13356–13361 (2013).
Turlier, H., Betz, T. Fluctuations in active membranes. arXiv:180100176, (2017).
Fischer, T., Stohr-Lissen, M. & Schmid-Schonbein, H. The red cell as a fluid droplet: tank tread-like motion of the human erythrocyte membrane in shear flow. Science 202, 894–896 (1978).
Discher, D., Mohandas, N. & Evans, E. Molecular maps of red cell deformation: hidden elasticity and in situ connectivity. Science 266, 1032–1035 (1994).
Dao, M., Lim, C. T. & Suresh, S. Mechanics of the human red blood cell deformed by optical tweezers. J. Mech. Phys. Solids 51, 2259–2280 (2003).
Glenister, F. K., Coppel, R. L., Cowman, A. F., Mohandas, N. & Cooke, B. M. Contribution of parasite proteins to altered mechanical properties of malaria-infected red blood cells. Blood 99, 1060–1063 (2002).
Brochard, F. & Lennon, J. Frequency spectrum of the flicker phenomenon in erythrocytes. J. de. Phys. 36, 1035–1047 (1975).
Duwe, H.-P., Engelhardt, H., Zilker, A. & Sackmann, E. Curvature elasticity of smectic A lipid bilayers and cell plasma membranes. Mol. Cryst. Liq. Cryst. 152, 1–7 (1987).
Gov, N. & Safran, S. Red blood cell shape and fluctuations: cytoskeleton confinement and ATP activity. J. Biol. Phys. 31, 453–464 (2005).
Gov, N., Zilman, A. G. & Safran, S. Cytoskeleton confinement and tension of red blood cell membranes. Phys. Rev. Lett. 90, 228101 (2003).
Betz, T. & Sykes, C. Time resolved membrane fluctuation spectroscopy. Soft Matter 8, 5317–5326 (2012).
Yoon, Y.-Z. et al. Flickering analysis of erythrocyte mechanical properties: dependence on oxygenation level, cell shape, and hydration level. Biophys. J. 97, 1606–1615 (2009).
Koch, M. et al. Plasmodium falciparum erythrocyte-binding antigen 175 triggers a biophysical change in the red blood cell that facilitates invasion. Proc. Natl Acad. Sci. USA 114, 4225–4230 (2017).
Ito, H. et al. Quantification of the influence of endotoxins on the mechanics of adult and neonatal red blood cells. J. Phys. Chem. B 119, 7837–7845 (2015).
Pecreaux, J., Dobereiner, H. G., Prost, J., Joanny, J. F. & Bassereau, P. Refined contour analysis of giant unilamellar vesicles. Eur. Phys. J. E, Soft Matter 13, 277–290 (2004).
Kim, D. H. & Frangos, J. A. Effects of amyloid β-peptides on the lysis tension of lipid bilayer vesicles containing oxysterols. Biophys. J. 95, 620–628 (2008).
Dasanna, A. K., Schwarz, U. S., Gompper, G., Fedosov, D. A. Multiscale Modeling of Malaria-Infected Red Blood Cells. In: Handbook of Materials Modeling: Applications: Current and Emerging Materials (eds Andreoni W., Yip S.). (Springer International Publishing, Basel, Switzerland, 2018).
Cokelet, G. R. & Meiselman, H. J. Rheological comparison of hemoglobin solutions and erythrocyte suspensions. Science 162, 275–277 (1968).
Miller, L. H., Baruch, D. I., Marsh, K. & Doumbo, O. K. The pathogenic basis of malaria. Nature 415, 673–679 (2002).
Frank, V. et al. Lensless tomographic imaging of near surface structures of frozen hydrated malaria-infected human erythrocytes by coherent X-Ray diffraction microscopy. Sci. Rep. 7, 14081 (2017).
Fairhurst, R. M., Bess, C. D. & Krause, M. A. Abnormal PfEMP1/knob display on Plasmodium falciparum-infected erythrocytes containing hemoglobin variants: fresh insights into malaria pathogenesis and protection. Microbes Infect. 14, 851–862 (2012).
Lanzer, M., Wertheimer, S. P., de Bruin, D. & Ravetch, J. V. Chromatin structure determines the sites of chromosome breakages in Plasmodium falciparum. Nucleic Acids Res. 22, 3099–3103 (1994).
Lin, L. C. & Brown, F. L. Dynamics of pinned membranes with application to protein diffusion on the surface of red blood cells. Biophys. J. 86, 764–780 (2004).
Merath, R. J. & Seifert, U. Nonmonotonic fluctuation spectra of membranes pinned or tethered discretely to a substrate. Phys. Rev. E, Stat., Nonlinear, Soft Matter Phys. 73, 010401 (2006).
Turgeon, M. L. Clinical Hematology: Theory and Procedures 5th edn. (Lippincott Williams & Wilkins, Philadelphia, USA, 2011).
Boal, D. Mechanics of the cell (Cambridge University Press, Cambridge, GB, 2001).
Diakité, S. A. et al. Stage-dependent fate of Plasmodium falciparum-infected red blood cells in the spleen and sickle-cell trait-related protection against malaria. Malar. J. 15, 482 (2016).
Nguitragool, W. et al. Malaria parasite clag3 genes determine channel-mediated nutrient uptake by infected red blood cells. Cell 145, 665–677 (2011).
Smith, J. D., Rowe, J. A., Higgins, M. K. & Lavstsen, T. Malaria's deadly grip: cytoadhesion of Plasmodium falciparum-infected erythrocytes. Cell. Microbiol. 15, 1976–1983 (2013).
Prado, G., Farutin, A., Misbah, C. & Bureau, L. Viscoelastic transient of confined red blood cells. Biophys. J. 108, 2126–2136 (2015).
Lanzer, M., Wickert, H., Krohne, G., Vincensini, L. & Braun Breton, C. Maurer's clefts: a novel multi-functional organelle in the cytoplasm of Plasmodium falciparum-infected erythrocytes. Int J. Parasitol. 36, 23–36 (2006).
Lai, L., Xu, X., Lim, C. T. & Cao, J. Stiffening of red blood cells induced by cytoskeleton disorders: a joint theory-experiment study. Biophys. J. 109, 2287–2294 (2015).
Mauritz, J. M. et al. The homeostasis of Plasmodium falciparum-infected red blood cells. PLoS Comput. Biol. 5, e1000339 (2009).
Turlier, H. et al. Equilibrium physics breakdown reveals the active nature of red blood cell flickering. Nat. Phys. 12, 513 (2016).
Trager, W. & Jensen, J. B. Human malaria parasites in continuous culture. Science 193, 673–675 (1976).
Lambros, C. & Vanderberg, J. P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65, 418–420 (1979). (Allen Press, Lawrence, USA).
Ljungström, I., Moll, K., Perlmann, H., Scherf, A., Wahlgren, M. Methods in malaria research (Citeseer, 2008).
Betz, T., Lenz, M., Joanny, J.-F. & Sykes, C. ATP-dependent mechanics of red blood cells. Proc. Natl. Acad. Sci. USA 106, 15320–15325 (2009).
Milner, S. T. & Safran, S. Dynamical fluctuations of droplet microemulsions and vesicles. Phys. Rev. A 36, 4371 (1987).
Rinker, K. D., Prabhakar, V. & Truskey, G. A. Effect of contact time and force on monocyte adhesion to vascular endothelium. Biophys. J. 80, 1722–1732 (2001).
Lin, L. C. & Brown, F. L. Simulating membrane dynamics in nonhomogeneous hydrodynamic environments. J. Chem. Theory Comput. 2, 472–483 (2006).
Pan, L., Yan, R., Li, W. & Xu, K. Super-resolution microscopy reveals the native ultrastructure of the erythrocyte cytoskeleton. Cell Rep. 22, 1151–1158 (2018).
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft through the Collaborative Research Center SFB 1129. U.S.S., M.T., and M.L. are members of the cluster of excellence CellNetworks. C.L. and J.J. were supported by the HBIGS and HGS MathComp graduate schools, respectively, at Heidelberg University. M.T. thanks Nakatani Foundation for supports.
Author information
Authors and Affiliations
Contributions
M.T., U.S.S. and M.L. designed the study; B.F., J.J., C.L., C.P.S. and M.C. performed the experiments; B.F., J.J., C.L., H.I., U.S.S., M.L. and M.T. analyzed the data. B.B., S.T.S., J.S. characterized the blood samples and provided the samples; B.F., J.J., U.S.S., M.L. and M.T. wrote the paper. All authors participated in discussion and paper editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fröhlich, B., Jäger, J., Lansche, C. et al. Hemoglobin S and C affect biomechanical membrane properties of P. falciparum-infected erythrocytes. Commun Biol 2, 311 (2019). https://doi.org/10.1038/s42003-019-0556-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-019-0556-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
