
Abstract
Gout is a common arthritis caused by elevated serum uric acid (SUA) levels. Here we investigated loci influencing SUA in a genome-wide meta-analysis with 121,745 Japanese subjects. We identified 8948 variants at 36 genomic loci (P<5 × 10–8) including eight novel loci. Of these, missense variants of SESN2 and PNPLA3 were predicted to be damaging to the function of these proteins; another five loci—TMEM18, TM4SF4, MXD3-LMAN2, PSORS1C1-PSORS1C2, and HNF4A—are related to cell metabolism, proliferation, or oxidative stress; and the remaining locus, LINC01578, is unknown. We also identified 132 correlated genes whose expression levels are associated with SUA-increasing alleles. These genes are enriched for the UniProt transport term, suggesting the importance of transport-related genes in SUA regulation. Furthermore, trans-ethnic meta-analysis across our own meta-analysis and the Global Urate Genetics Consortium has revealed 15 more novel loci associated with SUA. Our findings provide insight into the pathogenesis, treatment, and prevention of hyperuricemia/gout.
Similar content being viewed by others

Introduction
Serum uric acid (SUA) is reported to have an antioxidative effect1,2, whereas elevated SUA, or hyperuricemia, results in crystal deposition and causes gout3. Gout is a common disease characterized by noninfectious acute arthritis. Both gout and hyperuricemia can result from an unhealthful lifestyle4,5,6, but recent genetic studies, including genome-wide association studies (GWASs), have also revealed a genetic contribution to the development of these conditions, with this contribution being larger than that for other common diseases7,8,9,10,11,12. Moreover, epidemiologic studies have revealed their relationship among other diseases such as cardiovascular diseases13,14, indicating the importance of elucidation of the pathophysiology of these conditions. To date, several GWASs of SUA have been performed with Caucasian populations15,16,17,18,19,20,21,22,23,24 as well as Asian populations including Japanese subjects25,26. Although there are genetic differences between Caucasian and Asian populations, they have many shared associated genes3,27 that exert major effects, such as ABCG2, SLC2A9, and SLC22A12, all of which are well-known representative urate transporters in humans and which are important as therapeutic target molecules for gout and hyperuricemia. Therefore, identifying new loci may not only help elucidate the pathophysiology of these diseases, but may also reveal their target molecules, taking into account the fact that these diseases have a broader genetic basis than other common diseases as described above. Furthermore, the gene expression patterns to which the identified loci contribute should enable us to estimate effective pathways for drug delivery. In the present study, we have investigated the genetic loci that influence SUA with more than 120,000 Japanese individuals in a genome-wide meta-analysis and have compared our findings with those of previous GWASs24,28. We identified 36 loci for SUA, including eight previously unreported loci, that suggest key cellular processes which contribute to elevated serum uric acid levels, followed by the identification of 15 more loci by trans-ethnic meta-analysis.
Results
Genome-wide meta-analysis
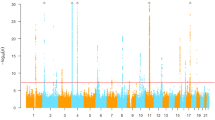
We performed a genome-wide meta-analysis based on three Japanese cohorts including those of the Japan Multi-institutional Collaborative Cohort (J-MICC) Study29,30, the Kita-Nagoya Genomic Epidemiology (KING) Study31,32, and the BioBank Japan (BBJ) Project33,34. Detailed information regarding the baseline characteristics of the study subjects, genotyping arrays, and imputation is summarized in Supplementary Tables 1 and 2. We performed a genome-wide meta-analysis for SUA with data sets encompassing 121,745 Japanese subjects. Intercepts of linkage disequilibrium (LD) score regression and the genomic control lambda for each study are shown in Supplementary Table 2. The intercepts of LD score regression and the genomic control lambda for our meta-analysis were 1.043 and 1.165, respectively. Genomic control adjustment was not applied for genomic control at the level of each study because intercepts of LD score regression did not show inflation of test statistics. The quantile–quantile (Q–Q) plot for P values is shown in Supplementary Fig. 1. The results of the meta-analysis identified 8948 variants at 36 genetic loci with a P value of <5 × 10–8 for SUA (Fig. 1). Among these 36 genetic loci, 8 were not previously reported, 10 were recently identified in a GWAS for SUA in Japanese performed by BBJ28, and 18 were previously identified by other GWASs for SUA15,16,17,19,20,24,25,26. The eight novel loci were the following: rs74896528 of SESN2, rs10188118 of LOC105373352 - TMEM18, rs6774054 of TM4SF4, rs11952102 of MXD3-LMAN2, rs16898823 of PSORS1C1-PSORS1C2, rs8024067 of LINC01578, rs6031598 of HNF4A, and rs2281293 of PNPLA3.

Manhattan plot for the meta-analysis of SUA. The horizontal line represents the genome-wide significance level (α = 5 × 10−8). Eighteen loci shown in orange were also recently identified by BBJ as being associated with SUA, 10 loci in blue were also identified by other studies and those in red indicate eight novel loci identified in the present study. Blue triangles represent loci containing SNPs with P values of <1 × 10−20. SUA serum uric acid, BBJ BioBank Japan
Sentinel single-nucleotide polymorphisms (SNPs) with the lowest P values for SUA at each of the 36 loci are shown in Table 1. Association results of each study are shown in Supplementary Data 1. We determined the effect allele frequencies (EAFs) of these sentinel SNPs for each population in 1000 Genomes phase 3 (Supplementary Data 2). The EAFs indicated that rs74896528 of SESN2 at chromosome 1p35.3 is an East Asian–specific SNP. Regional association plots for the eight loci newly identified in the present study are shown in Fig. 2. The BBJ data recently revealed that SNPs located at 27 loci showed genome-wide significant associations with SUA including 10 novel loci (Table 1)28. About the 27 reported SNPs, we compared the results in our meta-analysis with the recent results by BBJ28 (Supplementary Data 3), with regional association plots for the 10 loci also identified in the present study being shown in Supplementary Figure 2. The results for these 27 SNPs, identified in our meta-analysis, revealed a higher level of significance for the association with SUA in our meta-analysis than in the BBJ study. A European GWAS for SUA was previously performed by the Global Urate Genetics Consortium (GUGC)24. We examined the publicly available data provided by the GUGC-based study for the sentinel SNPs or SNPs showing high LD (r2 of ≥0.8 in JPT of 1000 Genomes phase 3) with the sentinel SNPs at the eight novel loci identified in the present study. Three of these loci, including 5q35.3, 20q13.12, and 22q13.31, were significantly associated with SUA in the GUGC-based GWAS, with the same direction of effect size as in our study (Supplementary Table 3). The 2p25.3 locus was nominally significantly associated with SUA. Although the 3q25.1 locus was not significantly associated with SUA in the GUGC-based study, it was nominally significantly associated with gout in the same study.

Regional association plots for the eight novel loci identified in the meta-analysis of SUA. The vertical axis represents –log10(P value) for assessment of the association of each SNP with SUA. Panels a–h present plots for chromosome (chr) 1p35.3, 2p25.3, 3q25.1, 5q35.3, 6p21.33, 15q26.1, 20q13.12, or 22q13.31, respectively. Colors indicate LD (r2) between each sentinel SNP and neighboring SNPs based on JPT of 1000 Genomes phase 3. SUA serum uric acid
Functional annotations for novel loci
We searched for SNPs at the newly identified loci associated with SUA that were associated with gene expression level or amino acid substitution of protein and that were in high LD (r2 of ≥0.8 in JPT of 1000 Genomes phase 3) with sentinel SNPs and had a P value of <1 × 10–6 for SUA in our meta-analysis. We identified two nonsynonymous SNPs of SESN at the 1p35.3 locus and PNPLA3 at the 22q13.31 locus (Supplementary Table 4), and we found that six of the eight novel loci harbor variants with expression quantitative trait loci (eQTLs) for at least one tissue in the Genotype-Tissue Expression (GTEx) database35 (Supplementary Data 4). The two nonsynonymous SNPs, rs738409 (I148M) of PNPLA3, and rs74896528 (P87S) of SESN2, were predicted by SIFT, PolyPhen2 HVAR, and PolyPhen2 HDIV to be damaging or probably damaging.
Gene set enrichment analysis of SUA-associated loci
We searched for genes whose expression level was associated with SUA-associated SNPs in at least one tissue in the GTEx database. We found that 24 of the 36 loci identified in the present study harbor variants with eQTLs in at least one tissue in the GTEx database. We also identified 71 positively correlated genes whose expression level is increased by SUA-increasing alleles and 76 negatively correlated genes whose expression level is decreased by SUA-increasing alleles (Supplementary Data 5). Functional analysis of the sets of positively correlated genes and negatively correlated genes were performed with the Database for Annotation, Visualization, and Integrated Discovery (DAVID)36. For the positively correlated genes, the terms “Williams-Beuren syndrome”, “sodium”, “transport”, “sodium transport”, and “alternative splicing” were enriched (Supplementary Table 5). For the negatively correlated genes, the term “Williams–Beuren syndrome” was enriched.
Comparison between Japanese and European GWASs for SUA
SNPs located at 28 loci were recently found to show genome-wide significant associations with SUA based on data from individuals of European ancestry in the GUGC24. We examined the results obtained for these SNPs in our meta-analysis (Supplementary Data 6). Twenty-one of these 25 SNPs showed nominal or genome-wide significant associations with SUA in our meta-analysis, with the same direction of effect size in both studies.
We compared the SNP-based heritability (h2) of SUA in our Japanese meta-analysis and the GUGC-based study24. The heritability estimates were calculated from summary statistics of 1,447,573 SNPs, which were assessed in both studies and have MAF ≥1% in both studies. The h2 (standard error (SE)) estimates were 14.0 % (4.3%) for our Japanese study and 14.4% (3.9%) for the European study. Furthermore, we calculated the genetic correlation between Japanese and European studies employing the same data sets. The genetic correlation ρge (SE) was analyzed (0.591 (0.294), P value = 0.164), and was not significantly less than 1.
Trans-ethnic meta-analysis with the use of GUGC-based study
We performed the trans-ethnic meta-analysis across our meta-analysis and the GUGC-based study to carry out fine-mapping analysis and identify further novel loci associated with SUA. We observed genome-wide significant (log10 (Bayes’ factor) of >6) association signals at 59 loci (Fig. 3), of which 15 were novel. Shown in Supplementary Data 7 are sentinel SNPs with the highest log10 (Bayes’ factor) for SUA at each of these 15 novel loci (rs302684 of TRABD2B-SKINT1L, rs2765545 of CCDC18, rs715 of CPS1, rs9942075 of TFDP2, rs10471103 of INPP4B-LOC105377623, rs461660 of RAI14, rs2760181 of KIAA0319, rs6928482 of HLA-DQB1, rs10971419 of B4GALT1, rs2195525 of USP2, rs626277 of DACH1, rs2957742 of MYO9A, rs12451900 of ZBTB4, rs164009 of QRICH2, and rs1035941 of INSR).

Manhattan plot for the trans-ethnic meta-analysis of SUA. The horizontal line represents the genome-wide significance level (log10 (Bayes’ factor) = 6). Loci shown in blue were identified by our Japanese meta-analysis and other studies and those in red indicate 15 novel loci identified in the trans-ethnic meta-analysis. Blue triangles represent loci containing SNPs with log10 (Bayes’ factor) of >20. SUA serum uric acid
Discussion
In a genome-wide meta-analysis performed with 121,745 Japanese subjects, we have here identified eight novel loci significantly associated with SUA. Moreover, five of these loci were replicated in Caucasian populations.
Gout, which develops as a consequence of hyperuricemia, is a form of arthritis known from the time of ancient Egypt37, and modern Japanese are genetically known to be more susceptible to hyperuricemia and gout,10,38. To our knowledge, the present study is the largest genome-wide meta-analysis performed for SUA to date, and it thus provides important insight into the genetic background of hyperuricemia and gout.
Uric acid or urate is an end metabolite of purines such as adenosine derived from ATP and guanine derived from DNA. Urate is produced predominantly in the liver and is excreted by the kidneys and the intestine9,39,40. Genes for urate transporters and proteins associated with cell metabolism might therefore be expected to be associated with SUA. Indeed, urate transporter genes such as SLC22A12 (also known as URAT1), SLC2A9 (GLUT9), and ABCG2 (BCRP) have been markedly associated with SUA, hyperuricemia, and gout7,8,9,10,11,12.
Among the eight novel loci identified in our study, TMEM18, TM4SF4, MXD3, and HNF4A are related to cell metabolism or proliferation. TMEM18 is a highly conserved gene related to obesity and plays a role in the central control of appetite and body weight regulation41,42,43. TM4SF4 is associated with gallstone disease and has been implicated in both liver regeneration and pancreas development44,45. Both MXD3 and HNF4A encode transcription factors. MXD3 forms a heterodimer with the cofactor MAX and is thought to promote uncontrolled cell proliferation and tumorigenesis46,47. HNF4A is associated with nonalcoholic steatohepatitis48 and plays a role in hepatic gluconeogenesis and lipid metabolism49. In addition, HNF4A controls gene expression in pancreatic islets, with HNF4A mutations having been associated with maturity-onset diabetes of the young type 1 and hyperinsulinemic hypoglycemia50. Furthermore, three of the eight novel loci identified in the present study harbor genes related to oxidative stress and inflammation: SESN2, PSORS1C1, and PNPLA3. SESN2 encodes a highly conserved stress-inducible metabolic protein that protects cells from stressors such as hypoxia, starvation, DNA damage, and oxidative stress51,52. PSORS1C1 and PSORS1C2 encode psoriasis susceptibility 1 candidates 1 and 2, respectively. PSORS1C1 is implicated in synovial inflammation and bone destruction in rheumatoid arthritis53, which, like gout, is a common type of arthritis. Its expression is inhibited in synovial fibroblasts affected by rheumatoid arthritis, which results in a reduction in interleukin-17, osteoclastogenic factor, and interleukin-1 levels as well as attenuation of cell proliferation54. PNPLA3 encodes a membrane protein located at the surface of hepatocyte lipid droplets55. A GWAS of nonalcoholic fatty liver disease identified PNPLA3 as a major genetic determinant of fatty liver and hepatic fat content56. PNPLA3 is also associated with inflammation, fibrosis, and the development of hepatocellular carcinoma55,57. Thus, novel loci associated with SUA were also related to oxidative stress and inflammation. Given that uric acid has an antioxidative effect1,2, loci related to oxidative stress or inflammation might also be expected to be associated with SUA. However, further molecular functional analyses are required to confirm these associations. The functional relation of the last of the eight novel loci identified in the present study, LINC01578, to SUA is unknown. Indeed, LINC01578 encodes a long intergenic non-protein-coding RNA of unknown function. It is also possible that a gene located near LINC01578 is actually responsible for the observed association with SUA.
Previous candidate analyses7,8 and GWASs11,12,58 of clinically defined gout identified nonsynonymous variants of gout susceptibility genes such as ABCG2 (rs72552713, Q126X; rs2231142, Q141K) and GCKR (rs1260326, L446P). SLC22A12 (URAT1) and SLC2A9 (GLUT9) are also genetic loci that influence SUA and encode urate transporters that mediate physiological urate reabsorption in the kidney59,60. We previously showed that dysfunctional nonsynonymous variants of SLC22A12 and SLC2A9 are responsible for renal hypouricemia type 159,60 and type 243, respectively. The present study also identified missense SNPs at two loci, rs738409 (I148M) of PNPLA3 and rs74896528 (P87S) of SESN2, that are predicted to impair the function of the encoded proteins (Supplementary Table 4). The rs738409 (I148M) polymorphism of PNPLA3 is in LD with rs2281293, which showed the most significant association with SUA at this locus in our genome-wide meta-analysis. The rs2281293 SNP of PNPLA3 is also an eQTL for this gene (Supplementary Data 4). On the other hand, rs74896528 of SESN2 has not been identified as an eQTL (Supplementary Data 4), and its SNP was not reported in the previous study based on GUGC data24 because of its low frequency in Caucasian populations (Supplementary Data 2). These results suggest that this missense (P87S) variant of SESN2 (rs74896528) is a novel locus that is associated with SUA specifically in Japanese or Asian populations.
The 28 loci identified in the European population of the GUGC study, SNPs at 21 loci showed a nominal or genome-wide significant association with SUA in our meta-analysis (Supplementary Data 6), again with the same direction of effect size. The SNP-based heritability for Japanese was 14.0%, and was similar to the 14.6% seen in Europeans. The genetic correlation between Japanese and Europeans was not significantly <1. These results suggest the possibility that most genetic causal variants of SUA are shared across ancestries.
The present study also identified 132 correlated genes whose expression levels are associated with SUA-increasing alleles (Supplementary Data 5). UniProt term enrichment analysis showed that these correlated genes are enriched in genes related to “transport” (Supplementary Table 5). A novel locus, rs6031598 of HNF4A, is correlated with the expression level of HNF4A. Of note, a noncoding genetic variant, rs1967017 of PDZK1, which encodes a scaffold protein for urate transporters61,62, has been shown to be functionally linked to HNF4-dependent PDZK1 expression63.
For SNP rs9394948 of ZNF318, ABCC10 (MRP7), an ABC transporter gene, was a positively correlated gene, and SLC22A7 (OAT2), an SLC transporter gene, was a negatively correlated gene (Supplementary Data 5). SLC22A7 encodes organic anion transporter 2 (OAT2), which mediates urate transport64 and is expressed in kidney and liver. Furthermore, for SNP rs11952102 of MXD3, RAB24, and PRELID1 were positively correlated genes, and MXD3 was a negatively correlated gene. RAB24 is localized to the endoplasmic reticulum and is thought to participate in autophagosome maturation65. RAB24 may influence SUA via autophagy, because there is a report on relationship between SUA and autophagy which is promoted by NLRP3 and results in phagocytosis of urate crystals by human osteoblasts66. PRELID1 encodes PRELI, which forms a complex with TRIAP1 and mediates intramitochondrial transport of phosphatidic acid67. It is possible that PRELI may function as a urate transporter that directly affects SUA or that it indirectly influences SUA via its function as a phosphatidic acid transporter.
In trans-ethnic meta-analysis across our own meta-analysis and the GUGC study, we have here identified 15 more novel loci significantly associated with SUA. Out of these, rs2760181 of KIAA0319 at 6p22.3 showed different direction of regression coefficients between Japanese and European studies, but showed genome-wide significant association (log10 Bayes’ factor >6). Future studies will therefore be necessary to validate our findings in independent cohorts.
The present genome-wide meta-analysis of SUA in Japan identified eight novel loci. Furthermore, trans-ethnic meta-analysis of SUA in the present study revealed 15 more novel loci associated with SUA. The present study also demonstrated that SUA is regulated by multiple “transport”-related genes, that is, not only urate transporter genes but also non-transporter genes such as PDZK1 and HNF4A. Our findings thus provide important insight into SUA regulation and the pathogenesis of hyperuricemia and gout, and they provide a potential basis for the development of new treatments for these diseases.
Methods
Study subjects and genotyping
We performed a genome-wide meta-analysis based on three Japanese cohorts including those of the J-MICC Study29,30, KING Study31,32, and BBJ Project33,34. An overview of the characteristics of the study populations is provided in Supplementary Table 1. Information regarding study-specific genotyping, imputation, and analysis tools is provided in Supplementary Table 2. Data and sample collection for the cohorts participating in the present study were approved by the respective research ethics committees. All participants provided written informed consent.
Details of cohorts
The Japan Multi-institutional Collaborative Cohort (J-MICC) Study was launched in 2005. Through March 2014, 92,642 Japanese participants aged 35 to 69 years had provided blood samples and lifestyle data based on a questionnaire after having given their informed consent29,30. The present study included 14,539 J-MICC Study participants randomly selected from the 12 targeted areas (Chiba, Shizuoka-Sakuragaoka, Shizuoka, Daiko, Okazaki, Aichi, Takashima, Kyoto, Tokushima, Fukuoka, Kagoshima, and Kyushu-KOPS (Kyushu Okinawa Population Study)). After preimputation quality control, 14,091 participants remained for the imputation process (Supplementary Table 2). SUA was measured with the uricase-peroxidase method or the uricase–3,5-dimethoxy-4-fluoroanilide (F-DAOS) method in 10,794 of the 14,091 participants. Individuals receiving treatment for hyperuricemia or gout were excluded. Finally, 10,621 participants remained for the association analysis (Supplementary Table 1). This study was approved by the ethics committees of Nagoya University Graduate School of Medicine (approval no. 939-14), Aichi Cancer Center, and all other participating institutions. All research procedures were conducted according to the Ethical Guidelines for Human Genome and Genetic Sequencing Research in Japan and the Declaration of Helsinki.
The Kita-Nagoya Genomic Epidemiology (KING) Study (ClinicalTrials.gov identifier NCT00262691) is an ongoing community-based prospective observational study of the genetic basis of cardiovascular disease and its risk factors31,32. It recruited 3975 Japanese subjects aged 50–80 years who underwent community-based annual health checkups between May 2005 and December 2007. A total of 2095 of the KING Study samples was included in the present study. SUA was measured with the uricase method (Mizuho Medy, Saga, Japan). Individuals under treatment for hyperuricemia or gout were excluded. The study was performed according to the guidelines of the Declaration of Helsinki; the study protocol was approved by the ethics committees of Aichi Gakuin University, Jichi Medical University, Nagoya University, and Kyushu University; and all participants provided written informed consent.
The BioBank Japan (BBJ) Project (http://biobankjp.org/english/index.html) was initiated in 2003 at the Institute of Medical Science, The University of Tokyo, and it has constructed a large-scale, multi-institutional, hospital-based biobank. The BBJ collected DNA, serum, and clinical information from ~200,000 Japanese patients with any of 47 target diseases between fiscal years 2003 and 200733,34. Patients were recruited from 66 hospitals of 12 medical institutes throughout Japan (Osaka Medical Center for Cancer and Cardiovascular Diseases, Cancer Institute Hospital of Japanese Foundation for Cancer Research, Juntendo University, Tokyo Metropolitan Geriatric Hospital, Nippon Medical School, Nihon University School of Medicine, Iwate Medical University, Tokushukai Hospitals, Shiga University of Medical Science, Fukujuji Hospital, National Hospital Organization Osaka National Hospital, and Iizuka Hospital). All patients were diagnosed with one or more of the 47 target diseases by physicians at the cooperating hospitals. Clinical information, including SUA measurements, was collected through interviews and reviews of medical records with the use of a standard questionnaire. The present study included 109,029 individuals aged between 18 and 85 years with valid SUA measurements as described elsewhere28. Subjects receiving urate-lowering therapy (allopurinol, febuxostat, probenecid, or benzbromarone) or with renal insufficiency (estimated glomerular filtration rate of <15 ml min–1 1.73 m–2) were excluded. We obtained written informed consent from all participants, and this study was approved by the ethics committees of RIKEN Center for Integrative Medical Sciences and the Institute of Medical Science, The University of Tokyo.
Association analysis for SNPs and SUA
Individuals taking urate-lowering drugs were excluded from the present study. SUA was adjusted for age, sex, the top 10 principal components, and study-specific covariates in a linear regression model. We then standardized the resulting residuals. The association of the z-score of the residuals with SNP allele dose was tested by linear regression analysis. The effect sizes and standard errors estimated in linear regression analysis were used in the subsequent meta-analysis.
Quality control after genotype imputation
After genotype imputation, quality control was applied to each study. SNPs with an imputation quality of r2 < 0.3 or a minor allele frequency of <0.005 were excluded. SNPs that passed quality control in both the J-MICC Study and BBJ cohorts were subjected to meta-analysis. To identify studies with inflated GWAS significance, which can result from population stratification, we computed the genomic control lambda68 and the intercept of LD score regression69. We calculated the genomic control lambda in R. A study showing a score of >1.1 for both measures was regarded as inflated. Inflation was not detected in any study included in the present meta-analysis, and so genomic control adjustment was not applied.
Meta-analysis
The meta-analysis was performed with a total of 121,745 Japanese subjects from the three cohorts (Supplementary Table 1). The association results for each SNP across the studies were combined with METAL software70 by the fixed-effects inverse-variance-weighted method. Heterogeneity of effect sizes was assessed with the I2 index. The meta-analysis included 5,864,938 SNPs and the results from at least both the J-MICC Study and BBJ Project. The genome-wide significance level α was set to a P value <5 × 10–8.
Replication study for novel loci with the GUGC-based study
To employ a replication study and compare our meta-analysis with publicly available results from Europeans conducted by the GUGC, we downloaded the summary statistics from their website. The EAF of the HapMap project phase 2 CEU samples for each SNP was added to the summary statistics of the GUGC because the results of the GUGC study did not include EAFs. We excluded variants with MAF < 0.01. P-values for the GUGC study were corrected for genomic control (lambda = 1.12 for SUA and 1.03 for gout)24. Genomic inflation did not occur in the GUGC study because the intercepts of the LD score regression, based on the raw P-values, were 1.01 for SUA and 1.09 for gout. We therefore calculated the raw P values from the corrected P values, and used the raw P values as a replication study for novel loci in our meta-analysis. For the replication of five novel loci, the significance level α was determined by dividing 0.05 by the number of loci for Bonferroni correction (α = 0.05/5 = 0.01).
Functional annotations
For prioritization of associated SNPs at the novel loci, we adopted a series of bioinformatics approaches to collate functional annotation. We first used ANNOVAR71 to obtain an aggregate set of functional annotations—including gene location and impact of amino acid substitution based on the prediction tools SIFT and PolyPhen-2—for the sentinel SNPs and SNPs in high-LD (r2 of ≥0.8 in JPT of 1000 Genomes phase 3) with the sentinel SNPs and with a P value of <1 × 10–6 for SUA. We also examined these sentinel and high-LD SNPs for identification of eQTLs in 14 tissues considered relevant to SUA regulation using the GTEx v7 database. The significant criteria for eQTL were based on the GTEx project:21 variants with a nominal P value below the gene-level threshold were regarded as significant. The gene level threshold was determined by the permutation test in the GTEx project21. UniProt term enrichment analysis for the sets of positively correlated genes and negatively correlated genes was performed with DAVID and with the threshold of a false discovery rate of <0.05 as calculated by the Benjamini–Hochberg adjustment method.
SNP-based heritability in Japanese and European samples
We estimated the SNP-based heritability of SUA for our Japanese meta-analysis and GUGC-based study24 with the use of LD score regression69. As explained in our replication study section, the EAF of the HapMap project phase 2 CEU samples for each SNP was added to the summary statistics of the GUGC because the results of the GUGC study did not include EAFs. The heritability estimates were calculated from the summary statistics of 1,447,573 SNPs, which were assessed in both studies and have MAF ≥ 1% in both studies and were not palindromic SNPs. The P values for the GUGC study were corrected for genomic control (lambda = 1.12)24. Genomic inflation did not occur in GUGC because the intercept of LD score regression based on the raw P values was 1.01. Thus, we used raw P values calculated from corrected P values. Furthermore, we calculated the genetic correlation between Japanese and Europeans using the same data sets. The genetic correlation was calculated with the use of Popcorn72.
Trans-ethnic meta-analysis with the use of GUGC-based study
For our trans-ethnic meta-analysis across our meta-analysis and the GUGC-based study, we used MANTRA v.1 software73, which has been developed for trans-ethnic meta-analysis allowing heterogeneity in allelic effects. The trans-ethnic meta-analysis was calculated from the summary statistics of 1,986,983 SNPs, which were assessed in both studies and have MAF ≥ 1% in both. In our meta-analysis, the effect sizes were calculated from a linear regression analysis in which the z-score of residual values of SUA values after adjustment for covariates was used as a dependent variable. In the GUGC project, the effect sizes were calculated from the linear regression analysis in which the SUA value was used as a dependent variable. The scale of effect size for these studies was therefore different. Thus, before the MANTRA analysis, the effect sizes and standard errors of the GUGC study were divided by the standard deviation of SUA in the GUGC study (=1.4 mg/dl) to approximate the scale of effect sizes. A prior model of the relatedness between the studies was estimated by employing a dmatcal script in the software using the allele frequency of the analyzed SNPs. We regarded log10 Bayes’ factor >6 as a significant threshold in line with the previous simulation study74.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The summary statistics of our genome-wide meta-analysis based on three Japanese cohorts is available at the National Bioscience Database Center (Research ID: hum0167.v1.meta.v1).
References
Ames, B. N., Cathcart, R., Schwiers, E. & Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc. Natl Acad. Sci. USA 78, 6858–6862 (1981).
Matsuo, H. et al. ABCG2 variant has opposing effects on onset ages of Parkinson’s disease and gout. Ann. Clin. Transl. Neurol. 2, 302–306 (2015).
Dalbeth, N., Merriman, T. R. & Stamp, L. K. Gout. Lancet 388, 2039–2052 (2016).
Choi, H. K., Atkinson, K., Karlson, E. W., Willett, W. & Curhan, G. Alcohol intake and risk of incident gout in men: a prospective study. Lancet 363, 1277–1281 (2004).
Choi, H. K., Atkinson, K., Karlson, E. W., Willett, W. & Curhan, G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N. Engl. J. Med. 350, 1093–1103 (2004).
Choi, H. K. & Curhan, G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ 336, 309–312 (2008).
Matsuo, H. et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: a function-based genetic analysis in a Japanese population. Sci. Transl. Med. 1, 5ra11 (2009).
Woodward, O. M. et al. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl Acad. Sci. USA 106, 10338–10342 (2009).
Ichida, K. et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 3, 764 (2012).
Nakayama, A. et al. Common dysfunctional variants of ABCG2 have stronger impact on hyperuricemia progression than typical environmental risk factors. Sci. Rep. 4, 5227 (2014).
Matsuo, H. et al. Genome-wide association study of clinically defined gout identifies multiple risk loci and its association with clinical subtypes. Ann. Rheum. Dis. 75, 652–659 (2016).
Nakayama, A. et al. GWAS of clinically defined gout and subtypes identifies multiple susceptibility loci that include urate transporter genes. Ann. Rheum. Dis. 76, 869–877 (2017).
Feig, D. I., Kang, D. H. & Johnson, R. J. Uric acid and cardiovascular risk. N. Engl. J. Med. 359, 1811–1821 (2008).
Edwards, N. L. The role of hyperuricemia in vascular disorders. Curr. Opin. Rheumatol. 21, 132–137 (2009).
Li, S. et al. The GLUT9 gene is associated with serum uric acid levels in Sardinia and Chianti cohorts. PLoS Genet. 3, e194 (2007).
Döring, A. et al. SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat. Genet. 40, 430–436 (2008).
Vitart, V. et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 40, 437–442 (2008).
McArdle, P. F. et al. Association of a common nonsynonymous variant in GLUT9 with serum uric acid levels in old order amish. Arthritis Rheum. 58, 2874–2881 (2008).
Dehghan, A. et al. Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet 372, 1953–1961 (2008).
Kolz, M. et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 5, e1000504 (2009).
Yang, Q. et al. Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ. Cardiovasc. Genet. 3, 523–530 (2010).
Tin, A. et al. Genome-wide association study for serum urate concentrations and gout among African Americans identifies genomic risk loci and a novel URAT1 loss-of-function allele. Hum. Mol. Genet. 20, 4056–4068 (2011).
Sulem, P. et al. Identification of low-frequency variants associated with gout and serum uric acid levels. Nat. Genet. 43, 1127–1130 (2011).
Köttgen, A. et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 45, 145–154 (2013).
Kamatani, Y. et al. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat. Genet. 42, 210–215 (2010).
Okada, Y. et al. Meta-analysis identifies multiple loci associated with kidney function-related traits in east Asian populations. Nat. Genet. 44, 904–909 (2012).
Reginato, A. M., Mount, D. B., Yang, I. & Choi, H. K. The genetics of hyperuricaemia and gout. Nat. Rev. Rheumatol. 8, 610–621 (2012).
Kanai, M. et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 50, 390–400 (2018).
Hamajima, N. & Group, J.-M. S. The Japan Multi-Institutional Collaborative Cohort Study (J-MICC Study) to detect gene-environment interactions for cancer. Asian Pac. J. Cancer Prev. 8, 317–323 (2007).
Wakai, K. et al. Profile of participants and genotype distributions of 108 polymorphisms in a cross-sectional study of associations of genotypes with lifestyle and clinical factors: a project in the Japan Multi-Institutional Collaborative Cohort (J-MICC) Study. J. Epidemiol. 21, 223–235 (2011).
Asano, H. et al. Plasma resistin concentration determined by common variants in the resistin gene and associated with metabolic traits in an aged Japanese population. Diabetologia 53, 234–246 (2010).
Nakatochi, M. et al. The ratio of adiponectin to homeostasis model assessment of insulin resistance is a powerful index of each component of metabolic syndrome in an aged Japanese population: results from the KING Study. Diabetes Res. Clin. Pract. 92, e61–e65 (2011).
Nagai, A. et al. Overview of the BioBank Japan Project: study design and profile. J. Epidemiol. 27, S2–S8 (2017).
Hirata, M. et al. Cross-sectional analysis of BioBank Japan clinical data: A large cohort of 200,000 patients with 47 common diseases. J. Epidemiol. 27, S9–S21 (2017).
GTEx Consortium et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Nuki, G. & Simkin, P. A. A concise history of gout and hyperuricemia and their treatment. Arthritis Res. Ther. 8(Suppl 1), S1 (2006).
Okada, Y. et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun. 9, 1631 (2018).
Matsuo, H. et al. ABCG2 dysfunction causes hyperuricemia due to both renal urate underexcretion and renal urate overload. Sci. Rep. 4, 3755 (2014).
Matsuo, H. et al. Hyperuricemia in acute gastroenteritis is caused by decreased urate excretion via ABCG2. Sci. Rep. 6, 31003 (2016).
Thorleifsson, G. et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 41, 18–24 (2009).
Almen, M. S. et al. The obesity gene, TMEM18, is of ancient origin, found in majority of neuronal cells in all major brain regions and associated with obesity in severely obese children. BMC Med. Genet. 11, 58 (2010).
Larder, R. et al. Obesity-associated gene TMEM18 has a role in the central control of appetite and body weight regulation. Proc. Natl Acad. Sci. USA 114, 9421–9426 (2017).
Anderson, K. R. et al. The L6 domain tetraspanin Tm4sf4 regulates endocrine pancreas differentiation and directed cell migration. Development 138, 3213–3224 (2011).
Joshi, A. D. et al. Four susceptibility loci for gallstone disease identified in a meta-analysis of genome-wide association studies. Gastroenterology 151, 351–363 e28 (2016).
Satake, N. et al. Targeted therapy with MXD3 siRNA, anti-CD22 antibody and nanoparticles for precursor B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 167, 487–499 (2014).
Barisone, G. A., Yun, J. S. & Diaz, E. From cerebellar proliferation to tumorigenesis: new insights into the role of Mad3. Cell Cycle 7, 423–427 (2008).
Baciu, C. et al. Systematic integrative analysis of gene expression identifies HNF4A as the central gene in pathogenesis of non-alcoholic steatohepatitis. PLoS ONE 12, e0189223 (2017).
Chandra, V. et al. Multidomain integration in the structure of the HNF-4alpha nuclear receptor complex. Nature 495, 394–398 (2013).
Yoon, J. C. et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–138 (2001).
Lee, J. H., Budanov, A. V. & Karin, M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 18, 792–801 (2013).
Pasha, M., Eid, A. H., Eid, A. A., Gorin, Y. & Munusamy, S. Sestrin2 as a novel biomarker and therapeutic target for various diseases. Oxid. Med. Cell Longev. 2017, 3296294 (2017).
Conigliaro, P. et al. Polymorphisms in STAT4, PTPN2, PSORS1C1 and TRAF3IP2 genes are associated with the response to TNF inhibitors in patients with rheumatoid arthritis. PLoS ONE 12, e0169956 (2017).
Ciccacci, C. et al. Polymorphisms in STAT-4, IL-10, PSORS1C1, PTPN2 and MIR146A genes are associated differently with prognostic factors in Italian patients affected by rheumatoid arthritis. Clin. Exp. Immunol. 186, 157–163 (2016).
Kawaguchi, T. et al. Risk estimation model for nonalcoholic fatty liver disease in the Japanese using multiple genetic markers. PLoS ONE 13, e0185490 (2018).
Romeo, S. et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 40, 1461–1465 (2008).
Valenti, L., Dongiovanni, P., Ginanni Corradini, S., Burza, M. A. & Romeo, S. PNPLA3 I148M variant and hepatocellular carcinoma: a common genetic variant for a rare disease. Dig. Liver Dis. 45, 619–624 (2013).
Li, C. et al. Genome-wide association analysis identifies three new risk loci for gout arthritis in Han Chinese. Nat. Commun. 6, 7041 (2015).
Enomoto, A. et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 417, 447–452 (2002).
Matsuo, H. et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am. J. Hum. Genet. 83, 744–751 (2008).
Higashino, T. et al. Common variant of PDZ domain containing 1 (PDZK1) gene is associated with gout susceptibility: A replication study and meta-analysis in Japanese population. Drug Metab. Pharmacokinet. 31, 464–466 (2016).
Ichida, K. What lies behind serum urate concentration? Insights from genetic and genomic studies. Genome Med. 1, 118 (2009).
Ketharnathan, S. et al. A non-coding genetic variant maximally associated with serum urate levels is functionally linked to HNF4A-dependent PDZK1 expression. Hum Mol Genet, 27, 3964–3973 (2018).
Sato, M. et al. Renal secretion of uric acid by organic anion transporter 2 (OAT2/SLC22A7) in human. Biol. Pharm. Bull. 33, 498–503 (2010).
Ao, X., Zou, L. & Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 21, 348–358 (2014).
Allaeys, I., Marceau, F. & Poubelle, P. E. NLRP3 promotes autophagy of urate crystals phagocytized by human osteoblasts. Arthritis Res. Ther. 15, R176 (2013).
Potting, C. et al. TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab. 18, 287–295 (2013).
Devlin, B. & Roeder, K. Genomic control for association studies. Biometrics 55, 997–1004 (1999).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Brown, B. C., Ye, C. J., Price, A. L. & Zaitlen, N. Transethnic genetic-correlation estimates from summary statistics. Am. J. Hum. Genet. 99, 76–88 (2016).
Morris, A. P. Transethnic meta-analysis of genomewide association studies. Genet. Epidemiol. 35, 809–822 (2011).
Wang, X. et al. Comparing methods for performing trans-ethnic meta-analysis of genome-wide association studies. Hum. Mol. Genet. 22, 2303–2311 (2013).
Acknowledgements
We thank all subjects for their involvement in the study; staff of the institutions participating in the J-MICC Study, BBJ Project, and KING Study for their assistance in collection of samples and clinical information; Y. Mitsuda and K. Shibata (Department of Preventive Medicine, Nagoya University Graduate School of Medicine) for technical assistance; K. Gotanda, M. Miyazawa, and R. Sugiyama (National Defense Medical College) for discussion; and N. Hamajima (Nagoya University Graduate School of Medicine) for sample collection. We thank A.P. Morris for providing us with the MANTRA software. The present study was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan including KAKENHI grants (nos. 25293145 and 15K15227); the Ministry of Health, Labor, and Welfare of Japan; the Ministry of Defense of Japan; the Japan Society for the Promotion of Science (JSPS); the Kawano Masanori Memorial Foundation for Promotion of Pediatrics; and the Gout Research Foundation of Japan. The KING Study was supported in part by Grants-in-Aid from MEXT (nos. 24390169, 16H05250, 25293144, 15K19242, 16H06277, and 18K06942) as well as by a grant from the Funding Program for Next-Generation World-Leading Researchers (NEXT Program, no. LS056). The J-MICC Study was supported by Grants-in-Aid for Scientific Research from MEXT, including those for Priority Areas of Cancer (no. 17015018) and Innovative Areas (no. 221S0001), as well as by a JSPS KAKENHI grant (no. 16H06277). This study was supported in part by funding from the BioBank Japan Project from the Japan Agency for Medical Research and Development, and the Ministry of Education, Culture, Sports, Science and Technology.
Author information
Authors and Affiliations
Contributions
M. Nakatochi, A.H., Y.O. and H. Matsuo conceived and designed the study. A.N., Y. Kawamura, M. Kubo, Y. Kamatani, and N.S. contributed to research design. M. Nakatochi, S.I., K.Y., N.K., T.M. and M.Y. collected and analyzed clinical data of the KING Study. M. Nakatochi, A.H., H.I., N.F., R.O., S.K., Y. Nishida, C.S., R.I., T. Takezaki, E.O., D.M., T. Nishiyama, S. Suzuki, N.T., Y. Kita, K.E., K.K., H.U., K.A., I.O., K. Matsuo, Y. Nakamura, H. Mikami, T. Tamura, M. Naito, and K.W. collected and analyzed clinical data of the J-MICC Study. M. Kanai, M.A., M.H., K. Matsuda, Y.M., M. Kubo, Y. Kamatani, and Y.O. collected and analyzed clinical data of the BBJ Project. M. Nakatochi, M. Kanai, Y. Kamatani, and Y.O. performed statistical analysis. A.N., Y. Kawamura, S. Shimizu, K.Y., M. Kawaguchi, M. Nakajima, M.T., and H. Matsuo analyzed data. H. Matsuo organized this collaborative study. H.N., T. Nakamura, N.K., K. Matsuda, Y.M., T.M., N.S., M.Y. and K.W. provided intellectual input and assisted with preparation of the manuscript. M. Nakatochi, M. Kanai, A.N., A.H., Y. Kawamura, Y.O. and H. Matsuo wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nakatochi, M., Kanai, M., Nakayama, A. et al. Genome-wide meta-analysis identifies multiple novel loci associated with serum uric acid levels in Japanese individuals. Commun Biol 2, 115 (2019). https://doi.org/10.1038/s42003-019-0339-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-019-0339-0
This article is cited by
-
The pathogenesis of gout: molecular insights from genetic, epigenomic and transcriptomic studies
Nature Reviews Rheumatology (2024)
-
Association between serum uric acid and prostate cancer risk in East Asian populations: a Mendelian randomization study
European Journal of Nutrition (2023)
-
Relationship between rs4349859 and rs116488202 polymorphisms close to MHC-I region and serum urate levels in patients with gout
Molecular Biology Reports (2023)
-
Assessing the causal relationships between gout and hypertension: a bidirectional Mendelian randomisation study with coarsened exposures
Arthritis Research & Therapy (2022)
-
The functions and roles of sestrins in regulating human diseases
Cellular & Molecular Biology Letters (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
