
Abstract
The hydrogenation of nitriles to amines represents an important and frequently used industrial process due to the broad applicability of the resulting products in chemistry and life sciences. Despite the existing portfolio of catalysts reported for the hydrogenation of nitriles, the development of iron-based heterogeneous catalysts for this process is still a challenge. Here, we show that the impregnation and pyrolysis of iron(II) acetate on commercial silica produces a reusable Fe/Fe–O@SiO2 catalyst with a well-defined structure comprising the fayalite phase at the Si–Fe interface and α-Fe nanoparticles, covered by an ultrathin amorphous iron(III) oxide layer, growing from the silica matrix. These Fe/Fe–O core–shell nanoparticles, in the presence of catalytic amounts of aluminium additives, promote the hydrogenation of all kinds of nitriles, including structurally challenging and functionally diverse aromatic, heterocyclic, aliphatic and fatty nitriles, to produce primary amines under scalable and industrially viable conditions.

Similar content being viewed by others


Main
Catalysis plays a decisive role in many basic and applied chemical processes and is involved in the industrial production of more than 90% of fine and bulk chemicals as well as polymeric materials and many other everyday products1,2,3,4,5. In addition, the synthesis of bioactive compounds for life sciences and the success of sustainable energy technologies, including green fuels, depend on efficient catalytic processes1,2,3,4,5. Indeed, the success of organic synthesis in the last 100 years relied to a large extent on the discovery and application of suitable catalysts1,2,3,4,5. In this respect, specifically, the development of practical and cost-efficient hydrogenation strategies constitutes a key achievement of the last century6. Nowadays, a plethora of molecularly defined metal complexes as well as heterogeneous materials are frequently applied for the selective hydrogenation of alkynes7,8, olefins7,9,10, carbonyl compounds11,12, nitroarenes13,14 as well as (hetero)arenes7,15,16,17. Among these hydrogenation reactions, the transformation of nitriles to primary amines is particularly valuable because the latter products are privileged compounds in chemistry, medicine and biology18,19,20,21,22. In general, primary amines constitute important precursors and central intermediates in the chemical, pharmaceutical, agrochemical and materials industries18,19,20,21,22. After the original discovery of benzonitrile hydrogenation in the presence of nickel in 1905 (ref. 23), many catalysts were developed for this and related reactions (Fig. 1)24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42. These achievements were mainly possible due to the design of precious metal systems, which allow reactions to be performed at low temperature and pressure24,30,31,33,34,35,36,37,38. However, despite their tremendous success, their limited availability and higher price constitute major drawbacks. Thus, state-of-the-art catalysts for nitrile hydrogenation in industry continue to be Raney nickel26,27,31,33 and copper chromite29, which demand harsh conditions and suffer from toxicity issues. To solve these problems, alternative nickel- and cobalt-based heterogeneous catalysts have been reported in recent years35,39,40,41.

The hydrogenation of nitriles using heterogeneous catalysts has a long history that covers more than a century.
From a sustainability point of view, iron would be an ideal catalyst system for nitrile hydrogenation because of its abundance (at 4.7% it is the second-most abundant metal in the earth crust), low price and low toxicity14,17,42,43,44. Indeed, in the last decade, using sophisticated/synthetically demanding PNP pincer ligands, active iron complexes have been developed to hydrogenate nitriles44. In general, however, these metal complexes are less stable and difficult to use. In contrast, heterogeneous catalysts show improved stability and can be easily reused14,17,22,45,46,47. Unfortunately, to the best of our knowledge, analogous supported active iron centres for the hydrogenation of nitriles have not been created yet.
Here, we report a stable and convenient iron-based nanocatalyst obtained by the pyrolysis of iron acetate on commercial silica that shows excellent activity and selectivity for the hydrogenation of a large number of nitriles to produce primary amines in the presence of catalytic amounts of aluminium additives.
Results
Synthesis and characterization of Fe-based nanocatalysts
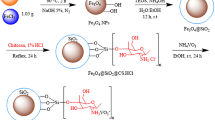
In the last decade, the excellent catalytic performance of carbon-encapsulated core–shell 3d metal nanoparticles has been demonstrated in a variety of hydrogenation reactions14,16,17,22,39,40,46. These catalysts were prepared by the immobilization and pyrolysis of metal complexes or metal organic frameworks on inorganic supports (for example, carbon, SiO2 and Al2O3)14,16,17,22,39,40,46. In this context, we developed activated Fe2O3 nanoparticles, surrounded by nitrogen-doped graphene, as selective catalysts for the hydrogenation of nitroarenes to anilines14. In addition, supported Fe-based nanoparticles have also been found active for the hydrogenation of quinolines17. However, these and related Fe materials showed no activity for more challenging substrates, including nitriles (Supplementary Table 1, entries 1 and 2). To identify potential iron-based heterogeneous catalysts for nitrile hydrogenation, we prepared a series of iron nanoparticles supported on various supports. Specifically, commercially available neutral, acidic and basic inorganic supports, for example, Vulcan XC72R carbon powder, Aerosil silica (SiO2), γ-Al2O3 and MgO, were impregnated with iron(II) acetate. Subsequently, these materials were pyrolysed at 800 °C under reductive (H2) conditions. A schematic illustration of the synthetic procedure with the SiO2 support is presented in Fig. 2. Hereafter, these materials are denoted as Fe(OAc)2-support-x, where x denotes the pyrolysis temperature.

Synthesis of Fe/Fe–O core–shell nanoparticles by the impregnation and pyrolysis of iron(II) acetate on SiO2. DMF, N,N-dimethylformamide.
As a benchmark reaction, the hydrogenation of 4-chlorobenzonitrile (1) to 4-chlorobenzylamine (2) was chosen (Fig. 3), not only to identify an active catalyst system, but also a selective one. Notably, 1 easily undergoes reductive dehalogenation in the presence of many known hydrogenation catalysts. To our surprise, during initial control experiments, we observed some activity (26% yield of 2) and high selectivity (>90%) for the primary amine in the presence of Fe(OAc)2-SiO2-800 (Fig. 3). To improve the conversion and yield, we varied the reaction conditions (temperature, solvent, catalyst loading) and investigated the influence of additives (Supplementary Tables 1–3). Applying higher catalyst loadings (up to 12.8 mol%), the product yield increased up to 50%, and the selectivity for the primary amine remained very good. Gratifyingly, in the presence of aluminium triisopropoxide, the yield of 4-chlorobenzylamine (2) dramatically increased to 96%. Following this excellent result, several other metal alkoxides, aluminium compounds as well as Lewis acids and bases were tested as additives (Supplementary Table 2). Surprisingly, only a few additives, for example, p-toluenesulfonic acid, showed a positive effect on the reaction, while most, for example, bases, had a negative impact. Optimal results, with an almost quantitative yield of 2, were achieved in the presence of inexpensive aluminium foil (Fig. 3 and Supplementary Fig. 1). In this case, the aluminium foil completely dissolved in the solvent (isopropanol), which explains the similar positive effect of aluminium foil and aluminium triisopropoxide. Control experiments proved that this dissolution only takes place in the presence of ammonia (Supplementary Table 4). To elucidate the crucial role of aluminium additives, we performed kinetic investigations of the model reaction in the absence and presence of aluminium foil and aluminium triisopropoxide (Supplementary Figs. 2 and 3). Surprisingly, all the reactions needed a preactivation time (3–9 h) to start. Only in the presence of aluminium triisopropoxide was this preactivation drastically reduced. Based on these results and the testing of the different additives, we assume that both aluminium triisopropoxide and aluminium foil are converted under the reaction conditions to an active Lewis acid co-catalyst that activates the nitrile group. These Lewis acidic centres can probably also be generated on the silica support close to the nanoparticles by reaction with Si–OH sites on the surface. Notably, catalytic (substoichiometric) amounts (20 mol%) of the aluminium additives were sufficient to achieve improved yields.

Hydrogenation of 4-chlorobenzonitrile. Reaction conditions: 0.5 mmol 4-chlorobenzonitrile, 40 mg catalyst (8.5 mol% Fe), 50 bar H2, 5–7 bar NH3, 3 ml i-PrOH, 120 °C, 24 h. Gas chromatography (GC) yields are given using n-hexadecane as standard. aWith 60 mg catalyst. bWith 20 mol% Al(i-OPr)3. cWith 20 mol% (3 mg) Al foil.
Under the optimized conditions, other supported catalysts, such as Fe(OAc)2-C-800, Fe(OAc)2-Al2O3-800 and Fe(OAc)2-MgO-800 (Supplementary Figs. 4–6), did not show any activity (Fig. 3). In these samples we did not observe needle-like well-developed α-Fe nanoparticles growing from the matrix, as we did in the case of the optimal catalyst, Fe(OAc)2-SiO2-800. In contrast, the iron nanoparticles were highly aggregated and/or encapsulated within the matrix (Supplementary Figs. 4–6). Similarly, Fe(OAc)2 on SiO2 pyrolysed at 400 °C (Fe(OAc)2-SiO2-400) was completely inactive (Fig. 3). This is explained by a not fully developed active Fe nanostructure at low pyrolysis temperature, which is evident from the powder X-ray diffraction (PXRD) pattern (Supplementary Fig. 7) and transmission electron microscopy (TEM) image (Supplementary Fig. 8) of the Fe(OAc)2-SiO2-400 sample. By contrast, Fe(OAc)2-SiO2-600 and Fe(OAc)2-SiO2-1,000 exhibited comparable activities to that of Fe(OAc)2-SiO2-800, providing 93 and 94% yields of the desired product, respectively. This correlates well with the similar size and well-developed core–shell structure of the Fe(OAc)2-SiO2-600 and Fe(OAc)2-SiO2-1,000 samples (see the TEM images in Supplementary Figs. 9 and 10) compared with Fe(OAc)2-SiO2-800 (Fig. 4c). As expected, iron(II) acetate, unpyrolysed Fe(OAc)2-SiO2 and Al additives alone were completely inactive in the reaction (Fig. 3). Additionally, we prepared control samples, including pure amorphous Fe2O3 nanoparticles (NPs), fayalite (Fe2SiO4) NPs and matrix-free Fe–Fe2O3 core–shell NPs with a very thin oxidic shell (Supplementary Figs. 11–13), and investigated their performance in the model reaction. Notably, the fayalite and Fe2O3 NPs were completely inactive, whereas the Fe–Fe2O3 core–shell NPs gave 30% yield (Supplementary Table 5, entries 1–3). This confirmed the crucial role of the Fe–Fe2O3 core–shell superstructure in triggering the catalytic process. We believe that the active material involves Fe centres and/or the Fe–O atomic interface48. The high activity of the catalyst incorporating the SiO2 matrix (Fe(OAc)2-SiO2-800) strongly indicates that the matrix regulates the size of the iron oxide crystallites49,50. Indeed, it has already been reported that the Cu–O–SiOx interface in a silica-supported copper (Cu@SiO2) catalyst plays a key role in H2 dissociation to form Cu–Hδ− and SiO–Hδ+ species51. Thus, we believe that the silica in Fe(OAc)2-SiO2-800 would contribute to the catalytic activity by forming such an active metal–support (Fe–O–SiOx) interface.

a–i, TEM (a–c), HRTEM (d) and HAADF-STEM (e) images and elemental mapping of iron (f), iron and oxygen (g), iron and silicon (h), and iron, silicon and oxygen (i) for the Fe(OAc)2-SiO2-800 catalyst.
Next, we conducted a detailed characterization of the most active catalyst Fe(OAc)2-SiO2-800. TEM analysis revealed the formation of core–shell structures with globular and rod-shape morphologies, with the needle diameters ranging from 10 to 30 nm and lengths up to 100 nm (Fig. 4a–c). Energy-dispersive X-ray spectroscopy (EDS) of this material showed the presence of Si, O and Fe elements (Supplementary Fig. 14). The high-resolution TEM image (HRTEM; Fig. 4d) confirms that the metallic part of the catalyst is composed of an α-Fe core growing from the SiO2 matrix. Indeed, the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and elemental mapping images clearly verify that the Fe core nanoparticles are growing from the SiO2 matrix and are covered by a layer of ultrathin iron oxide with a thickness of a few nanometres (Fig. 4f–i). Based on this assignment, the most active Fe(OAc)2-SiO2-800 catalyst is abbreviated to Fe/Fe–O@SiO2 in the following text. A representative HAADF-STEM image of a globular particle and typical depth profile plot showing the intensity distribution of the Si, O and Fe elements at various distances from the surface are shown in Fig. 5a,b, respectively. The depth profiles confirm that the thickness of the oxidic Fe-O shell is less than 5 nm. Clearly, the catalyst surface is composed of iron nanoparticles, which grow from the SiO2 matrix, stabilized by an extremely thin iron oxide shell.

a–d, HAADF-STEM image (a), depth profiles showing the intensity distribution of Fe, Si and O (b), Mössbauer spectrum recorded at 300 K (c) and EPR spectrum (X-band 9.090 GHz) recorded at 77 K (d).
Furthermore, we performed very detailed chemical mapping with a focus on the iron-containing surface components that are responsible for the catalytic activity. All the identified Fe-bearing surface-active phase was composed of Fe nanoparticles covered with a very thin shell of iron oxide, irrespective of the size and morphology (globular, needle-like) of the Fe NPs (Supplementary Fig. 15).
To identify the chemical and structural character of the catalyst, we analysed the Fe/Fe–O@SiO2 sample by PXRD, Mössbauer spectroscopy, X-ray photoelectron spectroscopy (XPS) and electron paramagnetic resonance (EPR) spectroscopy. The PXRD pattern of Fe/Fe–O@SiO2 (Supplementary Fig. 16) shows strong metallic α-Fe reflections at 2θ = 52.33, 77.16 and 99.60°, corresponding to crystalline facets of the Fe (110), (200) and (211) planes, respectively (Joint Committee on Powder Diffraction Standards (JCPDS) card number 04-012-6482). Thus, α-Fe is the dominant crystalline phase involved in the catalyst superstructure. The low-crystalline SiO2 matrix is represented by a broad peak at 2θ ≈ 26°, indicating the presence of poorly crystalline cristobalite (JCPDS card number 04-008-7643). The ultrathin iron oxide layer is, in accord with expectation, not identifiable in the PXRD pattern due to its mostly amorphous nature. However, detailed PXRD analysis clearly showed additional low-intensity diffraction peaks corresponding to fayalite (Fe2SiO4, JCPDS card number 04-002-3681) and crystalline silicon (Si(0), JCPDS card number 04-014-8844). In summary, PXRD provided a complex picture of the high-temperature chemistry of the Fe–Si–O system.
These observations are in line with the XPS analysis, which confirmed the presence of just Fe, Si and O elements in the survey spectrum (Supplementary Fig. 17a). The high-resolution O1s spectrum of Fe/Fe–O@SiO2 (Supplementary Fig. 17b) identifies peaks at 530.72 and 533.14 eV, corresponding to Fe–O and Si–O bonds, respectively. Most importantly, the high-resolution Fe2p spectrum (Supplementary Fig. 17d) reveals Fe 2p3/2 peaks at 710.27 and 712.73 eV, and Fe2p1/2 peaks at 723.37 and 725.83 eV, which can be ascribed to Fe3+ species52, along with two satellite peaks at 717.41 and 730.51 eV. This is strong proof that the amorphous iron oxide phase covering the Fe(0) core corresponds to amorphous Fe2O3. The formation of iron(III) oxide was confirmed by the hyperfine parameters derived from room-temperature Mössbauer spectroscopy (Fig. 5c). The 57Fe Mössbauer spectrum of the sample shows a dominant sextet (68% of the relative spectrum area) and two doublet components. According to the Mössbauer hyperfine parameters, the sextet with zero isomer shift and a hyperfine field of 32.9 T can be unambiguously assigned to ferromagnetic α-Fe. The doublet with high isomer shift (1.18 mm s–1) and quadrupole splitting (2.64 mm s–1) clearly belongs to Fe(II) ions in the fayalite structure formed at the Si–Fe interface53, in perfect agreement with the results of the PXRD measurements. Finally, the doublet with an isomer shift of 0.35 mm s–1 is typical of high-spin Fe(III) in amorphous iron(III) oxide with disordered symmetry of the iron environment, as proved by the relatively high quadrupole splitting (1.31 mm s–1)54. It is worth mentioning that possible traces of Fe(III) ions usually involved in the fayalite structure would overlap the doublet of the iron(III) oxide phase. Finally, the EPR spectrum of Fe/Fe–O@SiO2 shows broad anisotropic signals with g factor values of gx = 2.72, gy = 2.04 and gz = 1.8 (gave = 2.19) at 77 K (Fig. 5d), which indicates the presence of ferromagnetic particles corresponding to Fe(0) with distinct size and morphology. In summary, HRTEM, HAADF-STEM, XPS, PXRD, EPR and Mössbauer spectroscopy allowed us to explore the chemical and structural character of the Fe/Fe–O@SiO2 catalyst, being composed of a SiO2 matrix, a fayalite interface (Fe2SiO4) and α-Fe–amorphous Fe2O3 core–shell nanoparticles growing from the silica matrix and representing the surface-active phase participating in the catalytic process. The EPR and Mössbauer data confirm the ferromagnetic character of the sample, predetermining the catalyst for simple magnetic separation.
Hydrogenation of benzonitriles and heterocyclic nitriles
With an active Fe-based catalyst (Fe/Fe–O@SiO2) in hand, we demonstrated its general applicability for the selective hydrogenation of all kinds of nitriles. Although in most of the reactions aluminium foil was used as an inexpensive additive, experiments performed for comparison in the presence of aluminium triisopropoxide gave similar product yields. First, we carried out the hydrogenation of a series of aromatic nitriles (Fig. 6). Simple benzonitriles as well as substituted ones bearing aromatic or alkyl groups gave the corresponding primary amines in yields of up to 96% (Fig. 6, products 3–7). For the general applicability of any new catalyst, its chemoselectivity is an important aspect. Thus, from a synthetic point of view, it is important to note that this iron-based catalyst system is highly selective for the hydrogenation of the nitrile group in functionalized and multisubstituted substrates. As an example, amino-substituted and halogenated benzylic amines were prepared, which are versatile intermediates in organic synthesis as well as for pharmaceuticals and agrochemicals. Such products, including the more sensitive 4-iodobenzylamine, were easily produced from the corresponding benzonitriles in yields of up to 97% (Fig. 6, products 8–16). In addition, trifluoromethyl-substituted benzylamines were obtained in yields of 94 and 95% (Fig. 6, products 17 and 18, respectively). We were also pleased to find that the nitrile group was also selectively hydrogenated in the presence of the more challenging C≡C, ester, boronic ester, amide, ether, trifluoromethoxy and thioether groups (Fig. 6, products 19–30). Furthermore, when multisubstituted nitriles were subjected to hydrogenation, reduction of the CN group again took place highly selectively towards the corresponding benzylic amines in yields of up to 95% (Fig. 6, products 31–46).

Hydrogenation of (hetero)aromatic nitriles. Reaction conditions: 0.5 mmol nitrile, 40 mg Fe/Fe–O@SiO2 (8.5 mol% Fe), 3 mg Al foil (20 mol%), 5–7 bar NH3, 50 bar H2, 3 ml i-PrOH, 120 °C, 24 h. Isolated yields are given. aYields were determined by GC using n-hexadecane as standard. bWith 50 mg Fe/Fe–O@SiO2. cWith 50 mg Fe/Fe–O@SiO2 and 5 mg Al foil. dAt 135 °C. eWith 60 mg Fe/Fe–O@SiO2 and 5 mg Al foil. fWith 50 mg Fe/Fe–O@SiO2 at 135 °C. gMethyl 4-cyanobenzoate was used as substrate. Transesterification product with i-PrOH. Yields in parentheses refer to the reaction performed in the presence of 20 mol% Al(i-OPr)3. Products were isolated as free amines and converted to their hydrochloride salts for NMR and high-resolution mass spectrometry (HRMS) analysis.
Heterocyclic amines constitute versatile intermediates in the pharmaceutical and agrochemical industries. In this respect, the selective hydrogenation of cyano-substituted heterocycles, for example, quinolines, indoles, pyrroles, benzodioxoles, benzodioxanes, furans, morpholines and phthalanes, is of particular interest. The corresponding heterocyclic amines were obtained (except for 3-cyanofuran) in yields of 85–94% (Fig. 6).
Hydrogenation of aliphatic nitriles
Compared with aromatic nitriles, the hydrogenation of aliphatic nitriles is in general more challenging. Importantly, Fe/Fe–O@SiO2 exhibited high activity and selectivity for these substrates, including dinitriles, under identical conditions (Fig. 7). Initially, several benzylic cyanides were hydrogenated to the corresponding primary amines in excellent yields (Fig. 7, products 57–72). Interestingly, the 2-arylethylamino motif is a common scaffold in many central nervous system-active compounds. Here, a variety of substituted derivatives were smoothly hydrogenated and furnished the corresponding primary amines in yields of up to 98% (Fig. 7, products 57–68). Phenylpropylamines are another important class of pharmaceutically relevant amines. For example, the parent compound (phenylpropylamine) is used in the synthesis of carboxypeptidase B-type enzyme inhibitors, muscarinic receptor antagonists and potential anticancer agents. Here, it was prepared in 94% yield from the corresponding nitrile (Fig. 7, product 69).

Hydrogenation of aliphatic nitriles. Reaction conditions: 0.5 mmol nitrile, 40 mg Fe/Fe–O@SiO2 (8.5 mol% Fe), 3 mg Al foil (20 mol%), 5–7 bar NH3, 50 bar H2, 3 ml i-PrOH, 120 °C, 24 h. Isolated yields are given. aWith 50 mg Fe/Fe–O@SiO2. bAt 135 °C. cYields were determined by GC using n-hexadecane as standard. dWith 80 mg Fe/Fe–O@SiO2 and 6 mg Al foil at 135 °C. eWith 80 mg Fe/Fe–O@SiO2 and 6 mg Al foil at 120 °C. Yields in parentheses refer to the reaction performed in the presence of 20 mol% Al(i-OPr)3. Products were isolated as free amines and converted to their hydrochloride salts for NMR and HRMS analysis.
Although 3-(arylamino)propanenitriles are prepared in a straightforward manner from anilines and acrylonitrile, the hydrogenation of such substrates is difficult because retro-Michael additions can occur. However, this class of compounds was smoothly hydrogenated under our conditions to give the respective primary amines in good yields of up to 85% (Fig. 7, products 70–72). Finally, a selection of aliphatic nitriles was tested. Gratifyingly, Fe/Fe–O@SiO2 also showed good-to-excellent activity and selectivity for these demanding substrates (Fig. 7, products 73–78). Particularly interesting is the selective reduction of 5-hexenenitrile (Fig. 7, product 78). Notably, hexamethylenediamine (79), the key feedstock for the production of nylon 66, was prepared in 85% yield by direct hydrogenation of adiponitrile. Similarly, other diamines were obtained in 90–95% yield (Fig. 7, products 80 and 81).
Synthesis of fatty nitriles
With a worldwide production of fatty amines of >800,000 tons yr–1, the hydrogenation of fatty nitriles constitutes an important industrial application55. Fatty amines are valuable oleochemicals mainly used to produce fabric softeners, flotation agents, emulsifiers, corrosion inhibitors and lubricating additives55.
Until today, the industrial hydrogenation of fatty nitriles to amines has relied on well-established Raney Ni or Co catalysts as well as copper chromite55. These materials have considerable toxicity issues for biological systems. Hence, alternative Ru-, Pd- and Pt-based catalysts were developed55. Unfortunately, none of these systems is commercially viable due to the high price of the precious metals. Gratifyingly, our catalyst is capable of hydrogenating fatty nitriles in a highly selective manner. As a result, seven different fatty amines were prepared in excellent yields of 95–97% (Fig. 8, products 82–88).

Synthesis of fatty amines. Reaction conditions: 0.5 mmol nitrile, 40 mg Fe/Fe–O@SiO2 (8.5 mol% Fe), 3 mg Al foil (20 mol%), 5–7 bar NH3, 50 bar H2, 3 ml i-PrOH, 120 °C, 24 h. Isolated yields are given. aAt 135 °C. bWith 50 mg Fe/Fe–O@SiO2. Yields in parentheses refer to the reaction performed in the presence of 20 mol% Al(i-OPr)3. Products were isolated as free amines and converted to their hydrochloride salts for NMR and HRMS analysis.
To further prove the synthetic utility and practicability of our Fe catalyst, we scaled up both the catalyst preparation (up to 12 g; Supplementary Table 6) and the nitrile hydrogenation protocol (up to 20 g). Regardless of the scale of preparation (1–12 g), all the Fe materials exhibited similar activity and selectivity (Supplementary Table 7). Next, the catalytic hydrogenation reactions of four selected aromatic and aliphatic nitriles were performed using quantities of up to 20 g nitrile. Again, similar conversions and yields were obtained to those achieved with small-scale reactions using up to 100 mg nitrile (Supplementary Fig. 18).
Finally, catalyst recycling was investigated at full and half conversions, which is an important aspect for any heterogeneous catalyst. Indeed, the Fe/Fe–O@SiO2 catalyst could be reused up to the fourth run. After that, a decrease in the product yield was observed. Recycling tests performed at half conversion for 14 h showed a drop in activity from the third run onward (Supplementary Fig. 19).
Conclusions
We have presented here the development of a heterogeneous iron-based catalyst for the hydrogenation of nitriles. Key to success was the use of silica-supported Fe nanoparticles covered with an ultrathin shell of amorphous iron(III) oxide (Fe/Fe–O@SiO2). These core–shell nanoparticles were prepared by simple impregnation of iron(II) acetate on commercial silica and subsequent pyrolysis under reductive conditions. The low cost and environmentally friendly character of the catalyst, easy recycling as well as upscaling of the synthetic process represent key advantages and make the material attractive for many applications. Importantly, the developed silica-supported Fe/Fe–O core–shell material exhibited high chemoselectivity for the reduction of functionalized and structurally diverse aromatic, heterocyclic and aliphatic nitriles, including industrially relevant fatty nitriles, to produce the corresponding primary amines in good-to-excellent yields. Aluminium alkoxide species generated in situ from aluminium foil or aluminium triisopropoxide proved to be important for the co-catalytic activation of the nitrile substrate.
Methods
General considerations
All nitriles were obtained commercially from various chemical companies. Before using, the purity of all the nitriles was checked. Iron(II) acetate (99.99%, cat no. 517933-25G) was obtained from Sigma Aldrich. Silica (Aerosil OX-50) was obtained from Evonik. Carbon powder (VULCAN XC72R, with code XVC72R) was obtained from Cabot Corporation. γ-Al2O3 and MgO were obtained from Sigma-Aldrich. Al foil was obtained from Sigma-Aldrich (Mini Bin, HS23534A). For comparison purposes, Al foil used for food covering was also purchased from a local store (Kaufland, ALUFOLIE; ICP). The percentage of aluminium in Al-foil was determined by inductively coupled plasma (ICP) and it was found to be 99.97%. DMF was obtained from Acros Chemicals. Pyrolysis experiments were carried out in a Lenton tube furnace.
PXRD patterns were measured at room temperature with an Aeris diffractometer (PANalytical) in Bragg–Brentano geometry equipped with an iron-filtered Co Kα radiation source (40 kV, 15 mA, λ = 0.1789 nm) and PIXcell detector. Some samples were measured employing an X’PertPRO MPD diffractometer (PANalytical) in Bragg–Brentano geometry equipped with a Co Kα radiation source (40 kV, 30 mA, λ = 0.1789 nm), programmable divergence, diffracted beam anti-scatter slits and X’Celerator detector. The angular range of measurement was 5–105° 2θ (Fe/Fe–O@SiO2 was measured in the range 10–105° 2θ) with a step size of 0.022 and 0.033° for Aeris and X’Pert PRO MPD diffractometers, respectively. The crystalline phases in the experimental PXRD patterns were identified using the X’Pert High Score Plus software56 in conjunction with the PDF-4+57 and ICSD58 databases. The commercially available silicon standard reference material SRM 640 was used to evaluate the line positions.
Low-resolution TEM imaging of the catalyst morphology was carried out with a JEOL microscope equipped with a LaB6 emission gun, operating at 160 kV. HRTEM images were obtained with a TITAN 60-300 HRTEM microscope equipped with an X-FEG-type emission gun, operating at 80 kV. This microscope was equipped with a Cs image corrector and a HAADF-STEM instrument. The point resolution was 0.06 nm in TEM mode. Elemental mapping was performed by STEM-EDS with an acquisition time of 20 min. For the HRTEM analysis, the powder samples were dispersed in ethanol and ultrasonicated for 5 min. One drop of this solution was placed on a copper grid supporting a holey carbon film.
The XPS surface investigation was performed on a PHI 5000 VersaProbe II XPS system (Physical Electronics) with a monochromatic Al Kα source (15 kV, 50 W) and photon energy of 1,486.7 eV. Dual beam charge compensation was used for all measurements. All spectra were recorded in a vacuum of 1.3 × 10−7 Pa at 21 °C. A 200-µm-diameter spot was analysed for each sample. The survey spectra were measured with a pass energy of 187.850 eV and an electronvolt step of 0.8 eV, whereas the high-resolution spectra were recorded with a pass energy of 23.500 eV and an electronvolt step of 0.2 eV. The spectra were evaluated with the MultiPak software (ULVAC-PHI). All binding energies are referenced to the C1s carbon peak at 284.80 eV.
The transmission 57Fe Mössbauer spectra were collected employing a Mössbauer MS96 spectrometer operating in constant acceleration mode and equipped with a 40 mCi 57Co(Rh) source. The Mössbauer spectra were fitted with the MossWinn software. The isomer shifts are referenced to α-Fe at room temperature. EPR spectra were recorded on a JEOL JES-X-320 spectrometer, operating at the X-band frequency (~9.14 GHz) at 77 K, and equipped with a JEOL ES 13060DVT5 variable-temperature control apparatus.
All catalytic experiments were carried out in 300 or 100 ml autoclaves (PARR Instrument). To avoid unspecific reactions, all catalytic reactions were carried out either in glass vials, which were placed inside the autoclave, or in autoclaves fitted with a glass/Teflon vessel.
GC and GC-mass spectrometry (GC–MS) were performed on an Agilent Technologies 6890N instrument. GC conversions and yields were determined by GC using flame ionization detection (FID) on an Agilent 6890N chromatograph equipped with Agilent HP-5MS 30m column (250 mm × 0.25 μm). The mass was determined by GC-MS using Agilent 6890N chromatograph equipped with Agilent HP-5MS 30m column (250 mm × 0.25 μm) and Agilent 5973N Mass Selective Detector (MSD).
1H and 13C NMR spectra were recorded on Bruker ARX 300 and ARX 400 spectrometers using [D6]DMSO and CDCl3 solvents.
Preparation of Fe/Fe–O@SiO2 on the 1.5 g scale
A magnetic stirring bar and 280.33 mg Fe(OAc)2 were transferred to a 50- ml round-bottomed flask and 30 ml DMF was added. The reaction mixture was stirred at 50 °C to dissolve the iron acetate. To this solution, 1.2 g SiO2 (Aerosil OX 50) was added, followed by 10 ml DMF. Next, a reflux condenser was fitted to the round-bottomed flask containing the reaction mixture, which was then placed in an aluminium block preheated at 150 °C and stirred for 4 h. Next, the reflux condenser was removed and the round-bottomed flask containing the reaction products was allowed to stand without stirring or closing for 20 h at 150 °C for the slow evaporation of DMF. After evaporation of the solvent and ensuring complete drying, the solid material was cooled to room temperature and ground to a fine powder. This powder was pyrolysed at a defined temperature (400, 600, 800 or 1,000 °C) for 4 h in a tubular furnace under the flow of 20% H2/N2 (ramp: 5 °C min–1, total flow: 3 l h–1) and then cooled to room temperature.
Elemental analysis of Fe/Fe–O@SiO2 by ICP and CHN analysis revealed the following (wt%) distribution: Fe = 6.09%; Si = 37.73%; C = 0.11%; H = 0.41%. The Brunauer–Emmett–Teller surface area was measured to be 46.04 m2 g–1.
The same procedure was employed for the preparation of Fe(OAc)2-C-800, Fe(OAc)2-γ-Al2O3-800 and Fe(OAc)2-MgO-800.
Preparation of Fe/Fe–O@SiO2 on 6 and 12 g batches
The same procedure was used for the preparation of Fe/Fe–O@SiO2 (Fe(OAc)2-SiO2-800) on the 6 and 12 g scale with a slight modification of the pyrolysis procedure, as described in Supplementary Table 6.
General procedure for the hydrogenation of nitriles
A magnetic stirring bar and 0.5 mmol of the corresponding nitrile were transferred to a 7-ml glass vial and then 3 ml i-PrOH was added. Next, 40 mg Fe/Fe–O@SiO2 (8.5 mol% Fe) and 3 mg Al foil (the Al foil was cut into small pieces and used in the reactions) or 20.42 mg Al(i-OPr)3 (20 mol%) were added and the vial was fitted with a septum, cap and needle. Then, the reaction vials were placed in a 300-ml autoclave (eight vials containing different substrates at a time). The autoclave was closed, flushed twice with 20 bar hydrogen and then pressurized with 5–7 bar ammonia gas and 50 bar hydrogen. The autoclave was placed in an aluminium block preheated at 133 °C and the reactions were allowed to proceed for the required time under stirring. During the reactions, the inside temperature of the autoclave was measured to be 120 °C, and this temperature was considered to be the reaction temperature. After completion of the hydrogenation reactions, the autoclave was cooled to room temperature. The remaining ammonia and hydrogen were discharged and the vials containing the reaction products were removed from the autoclave. The solid catalyst was filtered and washed thoroughly with ethyl acetate. The reaction products were analysed by GC–MS. The corresponding primary amines were purified by column chromatography (silica, methanol–dichloromethane). The amines were converted to their respective hydrochloride salt and characterized by GC–MS and NMR analysis. To convert the amines to the hydrochloride salts, 1–2 ml methanolic HCl (0.5 M HCl in methanol) was added to the ethyl acetate solution of the respective amine and the mixture stirred at room temperature for 4–5 h. Then, the solvent was removed and the resulting hydrochloride salt was dried under high vacuum. For selected amines, the yields were determined by GC using the following procedure. After completing the reaction, n-hexadecane (100 µl) was added as standard to the reaction vials and the reaction products were diluted with ethyl acetate followed by filtration using a plug of silica and then analysed by GC.
Gram-scale reactions
A magnetic stirring bar and the corresponding nitrile were transferred to a glass-fitted 300-ml Parr autoclave and 15–50 ml i-PrOH was added. Next, the required amount of catalyst (Fe/Fe–O@SiO2, 8.5–10 mol%) and Al foil (20 mol%; the Al foil was cut into small pieces and used in the reactions) were added. Then, the autoclave was closed, flushed twice with 20 bar hydrogen and then pressurized with 5–7 bar NH3 followed by 50 bar hydrogen. The autoclave was placed in an aluminium block preheated at 133–147 °C (placed 30 min before counting the reaction time to achieve the reaction temperature) and the reactions were stirred for 24 h. During the reactions, the inside temperature of the autoclave was measured to be 120–135 °C. After completion of the reactions, the autoclave was cooled to room temperature. The remaining ammonia and hydrogen were discharged, and the reaction products were removed from the autoclave. The solid catalyst was filtered and washed thoroughly with methanol and ethyl acetate. The reaction products were analysed by GC–MS and the corresponding products were purified by column chromatography (silica, dichloromethane–methanol) and characterized by NMR and GC–MS analysis.
Catalyst recycling
A magnetic stirring bar and 10 mmol benzonitrile were transferred to a 100-ml autoclave and then 20 ml i-PrOH was added. Next, 900 mg catalyst (Fe/Fe–O@SiO2) and 408.50 mg Al(i-OPr)3 were added. The autoclave was closed, flushed with 20 bar hydrogen and then pressurized with 5–7 bar NH3 and 50 bar H2. The autoclave was placed in a preheated aluminium block at 130 °C and the reactions were stirred for the required time. During the reactions, the inside temperature of the autoclave was measured to be 120 °C. After completion of the reactions, the autoclave was cooled to room temperature. The remaining ammonia and hydrogen were then discharged, and the reaction products were removed from the autoclave. Next, 250 µl n-hexadecane was added as standard to the reaction products. The catalyst was separated by filtration and the filtrate containing the reaction products was subjected to GC analysis to determine the yield of benzylamine. The separated catalyst was washed with ethyl acetate, dried under vacuum and used without further purification or reactivation for the next run.
Data availability
All data are available from the authors upon reasonable request.
References
Beller, M. & Bolm, C. Transition Metals for Organic Synthesis (Wiley-VCH, 2008).
Negishi, E.-I. Magical power of transition metals: past, present, and future (Nobel lecture). Angew. Chem. Int. Ed. 50, 6738–6764 (2011).
Hagen, J.Industrial Catalysis: A Practical Approach (Wiley‐VCH, 2006).
Catlow, C. R., Davidson, M., Hardacre, C. & Hutchings, G. J. Catalysis making the world a better place. Philos. Trans. R. Soc. A 374, 20150089 (2016).
Busacca, C. A., Fandrick, D. R., Song, J. J. & Senanayake, C. H. The growing impact of catalysis in the pharmaceutical industry. Adv. Synth. Catal. 353, 1825–1864 (2011).
Rylander, P. N.Catalytic Hydrogenation in Organic Syntheses (Academic Press, 1979).
Alonso, D. A. et al. in Science of Synthesis: Water in Organic Synthesis (ed. Kobayashi, S.) 95–119 (Thieme, 2012); https://doi.org/10.1055/sos-SD-206-00075
Oger, C., Balas, L., Durand, T. & Galano, J.-M. Are alkyne reductions chemo-, regio-, and stereoselective enough to provide pure (Z)-olefins in polyfunctionalized bioactive molecules? Chem. Rev. 113, 1313–1350 (2013).
Massaro, L., Zheng, J., Margarita, C. & Andersson, P. G. Enantioconvergent and enantiodivergent catalytic hydrogenation of isomeric olefins. Chem. Soc. Rev. 49, 2504–2522 (2020).
Chirik, P. J. Iron- and cobalt-catalyzed alkene hydrogenation: catalysis with both redox-active and strong field ligands. Acc. Chem. Res. 48, 1687–1695 (2015).
Magano, J. & Dunetz, J. R. Large-scale carbonyl reductions in the pharmaceutical industry. Org. Process Res. Dev. 16, 1156–1184 (2012).
Zhong, R., Wie, Z., Zhang, W., Liu, S. & Liu, Q. A practical and stereoselective in situ NHC-cobalt catalytic system for hydrogenation of ketones and aldehydes. Chem 5, 1552–1566 (2019).
Formenti, D., Ferretti, F., Scharnagl, F. K. & Beller, M. Reduction of nitro compounds using 3d-non-noble metal catalysts. Chem. Rev. 119, 2611–2680 (2019).
Jagadeesh, R. V. et al. Nanoscale Fe2O3-based catalysts for selective hydrogenation of nitroarenes to anilines. Science 342, 1073–1076 (2013).
Wiesenfeldt, M. P., Nairoukh, Z., Dalton, T. & Glorius, F. Selective arene hydrogenation for direct access to saturated carbo- and heterocycles. Angew. Chem. Int. Ed. 58, 10460–10476 (2019).
Murugesan, K. et al. Cobalt-nanoparticles catalyzed efficient and selective hydrogenation of aromatic hydrocarbons. ACS Catal. 9, 8581–8591 (2019).
Sahoo, B. et al. A robust iron catalyst for the selective hydrogenation of substituted (iso)quinolones. Chem. Sci. 9, 8134–8141 (2018).
Lawrence, S. A. Amines: Synthesis, Properties and Applications (Cambridge Univ. Press, 2004).
Ricci, A. Amino Group Chemistry: From Synthesis to the Life Sciences (Wiley-VCH, 2008).
Qureshi, M. H. Top 200 Pharmaceuticals by Retails Sales in 2020 (University of Arizona, 2020); https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/Top%20200%20Pharmaceuticals%20By%20Retail%20Sales%202020V3.pdf
Murugesan, K. et al. Catalytic reductive aminations using molecular hydrogen for synthesis of different kinds of amines. Chem. Soc. Rev. 49, 6273–6328 (2020).
Hahn, G., Kunnas, P., de Jonge, N. & Kempe, R. General synthesis of primary amines via reductive amination employing a reusable nickel catalyst. Nat. Catal. 2, 71–77 (2019).
Sabatier, P. & Senderens, J. B. Application aux nitriles de la methode d’hydrogenation directe par catalyse: synthese d’amines primaires, secondaires et tertiaires. C. R. Hebd. Séances Acad. Sci. 140, 482–484 (1905).
Paal, C. & Gerum, J. Über katalytische Wirkungen kolloidaler Metalle der Platingruppe. VI. Reduktionskatalysen mit kolloidalem Palladium. Ber. Dtsch. Chem. Ges. 42, 1553–1560 (1909).
Braun, V. J., Blessing, G. & Zobel, F. Katalytische hydrierungen unter Druck bei Gegenwart von nickelsalzen. VI.: nitrile. Ber. Dtsch. Chem. Ges. A 56, 1988–2001 (1923).
Raney, M. Method of preparing catalytic material. US patent 1563587A (1925).
Carothers, W. H. & Jones, G. A. The preparation of some primary amines by the catalytic reduction of nitriles. J. Am. Chem. Soc. 47, 3051–3057 (1925).
Aller, B. V. Raney cobalt hydrogenation catalysts. I. The preparation of the catalyst. J. Appl. Chem. 7, 130–134 (2007).
Adkins, H. Reactions of Hydrogen with Organic Compounds Over Copper-Chromium Oxide and Nickel Catalysts 53 (Univ. Wisconsin Press, 1937).
Freifelder, M. A low pressure process for the reduction of nitriles. Use of rhodium catalyst. J. Am. Chem. Soc. 82, 2386–2389 (1960).
Freidlin, L. K. & Sladkova, T. A. Catalytic reduction of dinitriles. Russ. Chem. Rev. 33, 319–330 (1964).
Barnett, C. Hydrogenation of aliphatic nitriles over transition metal borides. Ind. Eng. Chem. Prod. Res. Dev. 8, 145–149 (1969).
de Bellefon, C. & Fouilloux, P. Homogeneous and heterogeneous hydrogenation of nitriles in a liquid phase: chemical, mechanistic, and catalytic aspects. Catal. Rev. 36, 459–506 (1994).
López-De Jesús, Y. M., Johnson, C. E., Monnier, J. R. & Williams, C. T. Selective hydrogenation of benzonitrile by alumina-supported Ir–Pd catalysts. Top. Catal. 53, 1132–1137 (2010).
Lévay, K. & Hegedűs, L. Recent achievements in the hydrogenation of nitriles catalyzed by transitional metals. Curr. Org. Chem. 23, 1881–1900 (2019).
Yoshimura, M. et al. Selective synthesis of primary amines from nitriles under hydrogenation conditions. Adv. Synth. Catal. 360, 1726–1732 (2018).
Liu, Y. et al. Mild palladium-catalysed highly efficient hydrogenation of C≡N, C–NO2, and C=O bonds using H2 of 1 atm in H2O. Green Chem. 21, 830–838 (2019).
Wang, H. et al. Quasi Pd1Ni single-atom surface alloy catalyst enables hydrogenation of nitriles to secondary amines. Nat. Commun. 10, 4998 (2019).
Ryabchuk, P. et al. Intermetallic nickel silicide nanocatalyst—a non-noble metal–based general hydrogenation catalyst. Sci. Adv. 4, eaat0761 (2018).
Murugesan, K. et al. Cobalt-based nanoparticles prepared from MOF–carbon templates as efficient hydrogenation catalysts. Chem. Sci. 9, 8553–8560 (2018).
Mitsudome, T., Sheng, M., Nakata, A., Mizugaki, T. & Jitsukawa, K. A cobalt phosphide catalyst for the hydrogenation of nitriles. Chem. Sci. 11, 6682–6689 (2020).
Enthaler, S., Junge, K. & Beller, M. Sustainable metal catalysis with iron: from rust to a rising star. Angew. Chem. Int. Ed. 47, 3317–3321 (2008).
Plietker, B. Catalysis in Organic Chemistry: Reactions and Applications (Wiley-VCH, 2008).
Bornschein, C. et al. Mild and selective hydrogenation of aromatic and aliphatic (di)nitriles with a well-defined iron pincer complex. Nat. Commun. 5, 4111 (2014).
Liu, L. & Corma, A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 118, 4981–5079 (2018).
Jagadeesh, R. V. et al. MOF-derived cobalt nanoparticles catalyze a general synthesis of amines. Science 358, 326–332 (2017).
Yang, X.-F. et al. Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 46, 1740–1748 (2013).
Zhang, J. et al. Tuning polarity of Cu-O bond in heterogeneous Cu catalyst to promote additive-free hydroboration of alkynes. Chem 6, 725–737 (2020).
Bukur, D. B. et al. Binder/support effects on the activity and selectivity of iron catalysts in the Fischer-Tropsch synthesis. Ind. Eng. Chem. Res. 29, 1588–1599 (1990).
Suo, H. et al. Chemical and structural effects of silica in iron-based Fischer–Tropsch synthesis catalysts. J. Catal. 286, 111–123 (2012).
Xu, C. et al. Interfacing with silica boosts the catalysis of copper. Nat. Commun. 9, 3367 (2018).
Dhavale, V. M., Singh, S. K., Nadeema, A., Gaikwad, S. S. & Kurungot, S. Nanocrystalline Fe–Fe2O3 particle-deposited N-doped graphene as an activity-modulated Pt-free electrocatalyst for oxygen reduction reaction. Nanoscale 7, 20117–20125 (2015).
Qafoku, O. et al. Fayalite dissolution and siderite formation in water-saturated supercritical CO2. Chem. Geol. 332–333, 124–135 (2012).
Machala, L., Zboril, R. & Gedanken, A. Amorphous iron(III) oxide—a review. J. Phys. Chem. B 111, 4003–4018 (2007).
Hinzmann, A. & Gröger, H. Selective hydrogenation of fatty nitriles to primary fatty amines: catalyst evaluation and optimization starting from octanenitrile. Eur. J. Lipid Sci. Technol. 122, 1900163 (2020).
X’Pert High Score Plus version 4.8 (4.8.0.25518) (Malvern Panalytical B.V., 2018).
PDF-4+ database (ICDD, 2021).
ICSD database version 4.5.0 (build 20200929-1047) (FIZ Karlsruhe GmbH, 2021).
Acknowledgements
We acknowledge the European Research Council (EU project 670986-NoNaCat) and the State of Mecklenburg-Vorpommern for financial and general support. We thank the analytical team of the Leibniz-Institut für Katalyse for their excellent service. We thank O. Tomanec, M. Petr, I. Medřík and G. Zoppellaro (all from Palacky University Olomouc) for TEM, XPS and EPR measurements and control sample synthesis. The authors from Palacky University acknowledge the support of the Operational Program Research, Development and Education - European Regional Development Fund (project no. CZ.02.1.01/0.0/0.0/16_019/0000754) and the ERDF project ‘Development of pre-applied research in nanotechnology and biotechnology’ (project no. CZ.02.1.01/0.0/0.0/17_048/0007323) of the Ministry of Education, Youth and Sports of the Czech Republic. R.Z. acknowledges support from the Czech Science Foundation (project no. 19-27454X).
Author information
Authors and Affiliations
Contributions
R.V.J. and M.B. supervised the project. V.G.C., R.V.J. and M.B. planned and developed the project. V.G.C. developed and prepared the catalytic materials and performed the catalytic experiments. T.S. assisted in the catalyst preparation and catalytic experiments and also reproduced the results. R.G.K., J.K., O.M., M.B.G. and R.Z. characterized the catalysts. R.V.J., V.G.C. and M.B. wrote the paper with contributions from M.B.G., R.G.K. and R.Z.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Catalysis thanks Bhalchandra Bhanage, Hironao Sajiki and Dingsheng Wang for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Methods, Tables 1–7, Figs. 1–19, Notes 1–3, NMR data and spectra, and references.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chandrashekhar, V.G., Senthamarai, T., Kadam, R.G. et al. Silica-supported Fe/Fe–O nanoparticles for the catalytic hydrogenation of nitriles to amines in the presence of aluminium additives. Nat Catal 5, 20–29 (2022). https://doi.org/10.1038/s41929-021-00722-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41929-021-00722-x
This article is cited by
-
Constructing multiple active sites in iron oxide catalysts for improving carbonylation reactions
Nature Communications (2023)
-
Iron phosphide nanocrystals as an air-stable heterogeneous catalyst for liquid-phase nitrile hydrogenation
Nature Communications (2023)
-
In Situ Mineralization of Akageneite (β-FeOOH) on Cotton for Water Purification via Fenton-Like Reaction
Fibers and Polymers (2023)
-
Tunable hydrogen coverage on electron-deficient platinum nanoparticles for efficient hydrogenation reactions
Nano Research (2023)
-
One-pot H/D exchange and low-coordinated iron electrocatalyzed deuteration of nitriles in D2O to α,β-deuterio aryl ethylamines
Nature Communications (2022)
