
Abstract
Titanium (IV) sulphide (TiS2) is a layered transition metal dichalcogenide, which we exfoliate using liquid phase exfoliation. TiS2 is a candidate for being part of a range of future technologies. These applications are varied, and include supercapacitor and battery energy storage devices, catalytic substrates and the splitting of water. The driving force behind our interest was as a material for energy storage devices. Here we investigate a potential failure mechanism for such devices, namely oxidation and subsequent loss of sulphur. This degradation is important to understand, since these applications are highly property-dependent, and changes to the chemistry will result in changes in desired properties. Two approaches to study oxidisation were taken: ex situ oxidation by water and oxygen at room temperature and in situ oxidation by a 5% O2/Ar gas at elevated temperatures. Both sources of oxygen resulted in oxidation of the starting TiS2 flakes, with differing morphologies. Water produced amorphous oxide slowly growing in from the edge of the flakes. Oxygen gas at ≥375 °C produced crystalline oxide, with a range of structures due to oxidation initiating from various regions of the observed flakes.
Similar content being viewed by others

Introduction
Titanium (IV) sulphide, TiS2, is a candidate for applications in a range of future technologies, including catalytic substrates and the splitting of water, supercapacitors and energy storage devices. TiS2 is a layered transition metal dichalcogenide (TMD) with hexagonal crystal symmetry and takes the 1T phase and is relatively under explored in two-dimensional form. Each layer consists of three sheets of atoms: the two outer planes are sulphur, coordinated to three titanium atoms with a trigonal pyramidal geometry; the middle plane is titanium and each atom takes octahedral coordination to six sulphur atoms. These atomic sheets are stacked ABC (Supplemental Information Fig. 1), while the layers themselves stack AA in the 1T phase.

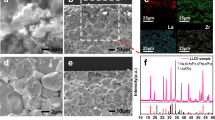
Initial status of TiS2. a TEM overview of TiS2 flake, b HAADF STEM of region from A, from which maps C-E were acquired, c Ti L3 EELS map, d O K EELS map, e S K EDX map
The excellent capacitive properties of bulk TiS2 (high energy-density and power-density1, 2 and excellent electronic conductivity3) have attracted interest since the 1970s, with bulk TiS2 used as a cathode material in lithium-metal/alloy anode batteries, where lithium metal ions were intercalated, transferring charge to reduce titanium 3d orbitals and causing reduction from Ti4+ to Ti3+ (refs. 4, 5). High rates of charging and discharging were demonstrated with near-perfect reversibility. In addition, the material forms a single phase with lithium (Li x TiS2) over the entirety of the range 0 ≤ x ≤ 1, avoiding the need for phase changes upon (de-)lithiation which removes much of the strain induced in cycling, prolonging battery life. However, TMD/Lithium-metal batteries were withdrawn from consumers due to accidents in 1989 where the metallic lithium caught fire as a consequence of short-circuiting due to lithium-dendrite growth.6
Other battery-applications of bulk TiS2 include as an encapsulation material of Li2S cathodes and as part of nanocomposite cathodes in solid-state lithium batteries.7, 8 It has also been used as a hybrid cathode in Li-S batteries, where sulphur is intercalated into a TiS2 foam, and found to have a very high capacity.9 Furthermore, TiS2 has also been used as a substrate for the growth of water-splitting Pt and Au catalysts.10 It has also been proposed that alloying with TiO2 would reduce the bandgap to optimise photon absorption for photocatalytic water splitting.11 The exact bandgap of TiS2 is hard to specify, as a range of values have been put forward across the literature, ranging from −1.5 to 2.5 eV, but it is usually considered to be either a semi-metal (negative bandgap arising from overlapping conduction and valence bands) or a small-gap semiconductor.12,13,14 One possible explanation for this range of values is that the samples of TiS2 being measured had already undergone some small degree of oxidation, or that samples differed stoichiometrically.
Here our interest in the degradation of this material was initialised when, upon opening a dispersion of TiS2, a sulphurous smell was noted. In the presence of water TiS2 could oxidise according to the following reactions:
While in the presence of oxygen gas, the following is proposed:
Cucinotta et al.15 performed density functional theory (DFT) calculations on the above reactions, predicting that in an aqueous environment water would preferentially oxidise a monolayer from the edge or from a point defect in the case that the edge was 50% terminated in sulphur. It is worth noting that other possible mechanisms could result in the formation of different species including SO2, but this was not included in these calculations. This correlated well with work by Han et al.16 who observed similar behaviour in TiS2 nanocrystals in a water/toluene mixture. Here we expand this work to study the oxidation of material produced through liquid phase exfoliation methods. We combine ex situ studies with advanced electron microscopy techniques including in situ oxidation studies to reveal the mechanisms underlying the oxidation of TiS2 nanosheets.
Results
Oxidation by water
We studied the oxidation of TiS2 by two common oxygen-containing species: deionised water (H2O) and gaseous oxygen (O2). We used ex situ ageing experiments initially to study oxidation over the course of many weeks within aqueous and atmospheric environments. We subsequently conducted in situ gas-heating and vacuum-heating experiments to try and observe oxidation in action, but decoupled from purely thermal effects. Oxidation occurs on the nanoscale, so we used transmission electron microscopy (TEM) to acquire both structural and spectroscopic information with high spatial resolution, in combination with first principles calculations to determine likely reaction mechanisms.
Flakes exfoliated by ultra-sonication had thicknesses in the approximate range 10–60 nm as measured by energy filtered transmission electron microscope thickness mapping, taking the electron inelastic mean free path at 300 kV to be 127.4 nm (further information can be found in the supplementary information, Supplementary Fig. 2).17 This range of thickness is typical of flakes exfoliated in this manner without undergoing size-selection, and includes changes in thickness within a single flake.18 Lateral flake dimensions range from several hundred nanometres up to several micrometres.
Figure 1 shows the status of a flake of TiS2 immediately after exfoliation. Figure 1a shows an overview of a flake acquired with TEM, the boxed region was imaged with scanning transmission electron microscopy (STEM) in Fig. 1b. Figure 1c–e shows elemental maps. We mapped sulphur with energy-dispersive X-ray spectroscopy (EDX) due to the weak EELS L2,3 edge (Fig. 1e), which is fairly uniform across the entirety of the mapped region. Titanium L2,3 (Fig. 1c) and oxygen K (Fig. 1d) edges were mapped using electron energy loss spectroscopy (EELS). EDX maps of the Ti K, L and O K peaks are provided in Supplementary Fig. 3, but the overlap between the Ti L and O K peaks due to poorer energy resolution means they are not as useful to map as the associated EELS edges and the low counts makes EDX quantification meaningless. As expected, titanium was found uniformly across the flake, and a narrow boarder (~2 nm) of oxygen was observed at flake/step edges, attributed to oxidation of the starting powder prior to exfoliation. The small bright feature present in Fig. 1c, d is attributed to a particle of titanium oxide on top of the sulphide flake, whereas no feature is present at that location in the S map (Fig. 1e).
Figure 2 maps across a flake stored in H2O for 21 days, during which time the oxidation region grew ~80 nm into the flake. A band of material was mapped across the flake to identify changes over the flake which are not localised to the edge, which is narrow to minimise the effects of sample-drift. Figure 2a is an HAADF (high-angle annular dark-field) STEM image of part of the TiS2 flake, across which EELS and EDX spectra were acquired. Two regions were identified based on contrast levels and morphology—an outer and an inner region. Speckled contrast was observed in the outer region and to a lesser extent across the centre. This is indicative of fragmented pieces, suggesting the flakes are breaking down during the reaction to form a shell. A region of darker HAADF intensity was observed along the border between these regions – implying reduced thickness or mass. It is expected that the thickness of these flakes resulted in the retention of this inner region, due to difficulties in getting water molecules into the middle where the flakes were thickest. Thinner flakes would likely fully oxidise faster due to the relative increase in the number of layers near to the surface. Figure 2b–d shows elemental maps across these regions, consisting of a Ti L2,3 EELS map (2 B), an O K EELS map (2C) and a S K EDX map (2 D). Figure 2b, c shows Ti and O are present across all areas although both seem reduced in the inner region, while S was confined to only the inner region. Mapping the shift in energy of the maximum of the Ti L3 edge (Fig. 2e) shows a shift of 0.8 eV from the sulphide to the oxide region, an expected chemical shift.19 This chemical shift will be expanded upon later in comparison to other samples. There is a ~5 nm wide intermediary region (green in Fig. 2e) which matches with the decay in sulphur intensity in Fig. 2d, in which a mixture of sulphide and oxide is expected.

State of TiS2 after 21 days oxidation in water. a HAADF STEM of a partially oxidised flake of TiS2 from which B-G were acquired in the location indicated by the orange rectangle, b Ti L3 EELS map, c O K EELS map, d S K EDX map, e map of the peak shift on the Ti L3 edge, f: Summed nanoprobe diffraction patterns from sulphide region, g Summed nanoprobe diffraction patterns from oxide region. f and g have been contrast inverted for clarity
A ‘parallel nanoprobe’ beam was employed to compare the crystallinity between these regions. The nanoprobe had a diameter of ~15 nm, and was used to obtain local diffraction patterns from across the flake, providing a sampling region over 130 times smaller than that obtainable with our smallest selected area electron diffraction aperture. The sulphide region (Fig. 2f) was crystalline, matching TiS2 down (001), while the oxide region (Fig. 2g) was amorphous. Figure 2f, g was obtained by summing the diffraction patterns from their respective regions, and contrast was inverted for clarity.
This oxidation process in water, matched the way flakes were observed to degrade within the dispersion, shown in Supplementary Fig. 4.
Oxidation by air
An additional oxidation process explored was the reaction with oxygen gas. Figure 3 shows the result of ageing in normal atmospheric conditions for 46 days. This sample showed more oxidation than the as-exfoliated flake shown in Fig. 1. Figure 3a is a STEM image of the edge-region, and fast fourier transforms (FFT) (green and red insets to Fig. 3a) demonstrate the loss of crystallinity at the edge. Variations in contrast in STEM are due to both differences in average atomic number and sample thickness. Along the right hand edge, the flake has fully oxidised and appears dark relative to the rest of this area due to titanium oxide being of a lower mean atomic number than titanium disulphide. However the bright region towards the bottom of the probed region arises from a TiO x particle on top the TiS2. The oxide region looks very different from that in Fig. 3a, uniform contrast suggesting a different structure was produced without the small nanoflakes observed in the water-oxidised sample. We observed strong enhancement of the oxygen signal around the flake edges (Fig. 3c), similar to the ‘as-exfoliated’ sample in Fig. 1, which had only grown ~5 nm into all layers after 46 days. A line of oxide extending into the flake was observed, suggesting that the oxide can form along boundary defects, but that growth into the bulk of the flake is limited. A shift in the peak of the Ti L3 edge (Fig. 3e) was again observed in the oxide region as in the pristine sample but there was now with a larger shift of about 1.5 eV compared to the sulphide. Again, a 5-nm wide intermediary region was observed.

State of TiS2 after 46 days oxidation in air. a HAADF of TiS2 flake from which b–e were acquired. Inserted are FFTs from the middle (green) and edge (red) showing loss in crystallinity in the oxide, b Ti L3 EELS map, c O K EELS map, d S K EDX map, e Map of the shift of the maximum of the Ti L3 edge. Scale bars on b–e are 20 nm
Overall, ageing in water produced significantly more oxidation despite exposure for only half as long as a sample exposed only to air. A possible explanation for this difference is that water molecules dissociate into H+ and OH− ions, creating a more reactive. This suggests that to prevent oxidation of TiS2 a desiccated environment may be sufficient and a good vacuum or an inert gas environment may not be necessary. An aqueous electrolyte, however, would likely lead to premature failure battery devices.
Oxidation by oxygen + heating
We studied the response of TiS2 flakes to exposure to a 5% oxygen/argon gas mixture at temperatures between 150 and 500 °C. Thermogravimetric analysiss showed rapid conversion of TiS2 to TiO2 at 325 °C.14 This heating induced transformation is of potential importance to alternate battery systems. For example, Na-ion batteries often operate at elevated temperature to compensate for the sluggish movement of the larger ions versus Li ions. In order to better understand this behaviour we used EDX and EELS spectrum imaging and nanoprobe line profiles, in conjunction with a heating/gas in situ TEM holder to study the oxidation of these materials at high spatial resolution. To minimise initial oxidation, the flakes were dispersed in anhydrous isopropyl alcohol (IPA) in the inert atmosphere of a nitrogen glove-box. The sample was put under vacuum within a few hours of being drop-cast, and a flake was initially inspected at room temperature in vacuum (Supplementary Fig. 5), however the flakes bore a strong resemblance to those in Fig. 1, with a thin oxide border around all flake steps. The sample was heated up to 150 °C and allowed to settle before the gas mixture was introduced, which produced no obvious changes to flakes (Fig. 4a). Gas was then introduced at a pressure of 330 mbar, and the temperature increased to 250 °C and then to 375 °C. At 375 °C a rapid and dramatic morphological change occurred in many flakes, at which point the temperature was dropped to 325 °C to prevent further reaction while analysis was performed.

Oxidation at 375 °C. a Initial HAADF of material at 150 °C in vacuum, b HAADF of region from which b–h were acquired after heating to 375 °C in 330 mbar gas, c Ti K EDX map, d O K EDX map, e S K EDX map, f Summation of nanoprobe diffraction patterns from line across edge of flake, contrast inverted for clarity, g Ti L3 EELS map, h O pre-K EELS map, i O K EELS map
Figure 4 demonstrates that the flake undergoes both chemical and morphological changes when heated to 375 °C in this environment. The HAADF contrast (Fig. 4b), formerly uniform across the flake, showed striations running through it parallel to the edges of the flake, and the flake appears to have shrivelled up and possibly delaminated, very different to the pristine starting material. We saw near-complete loss of sulphur throughout (Fig. 4e), apart from a small region top the top left underneath a much thicker flake. Comparison of the Ti and O EDX maps (Fig. 4c and d) suggests that there is an oxide layer which does not contain Ti around the edge of the flake. This is confirmed by EELS analysis in which we can distinguish different local environments for oxygen from the shape of the O K edge. By producing a chemical map using the O pre-K edge (π* and σ*) (Fig. 4h) we find this coincides well with the Ti L2,3 map (Fig. 4g). In contrast the main O K-edge map (Fig. 4i) extends outside the Ti-rich region but agrees with the O EDX map, suggesting the presence of an additional surface oxide layer. A combination of the surface layer and the thickness likely protected the top region from oxidation.
Nanoprobe electron diffraction patterns along the line indicated in Fig. 4b were acquired to study the crystallinity. Figure 4f is the sum of diffraction patterns from 12 positions over a distance of 120 nm from the surface into the flake. The apparent superposition of single-crystal patterns show that oxide is polycrystalline, while the perseverance of the dominant {110} reflections across several steps suggests one grain to be at least 50 nm wide. Of the three main TiO2 polytypes (rutile, anatase and brookite), the reflections observed most closely match to rutile observed along the [001] direction, with traces of anatase. At this temperature, anatase would be the expected phase of TiO2, as the anatase-rutile transformation is not expected until 620 °C.20 Both anatase and rutile nanosheets have been observed,21,22,23,24 and 4-layer thick rutile nanosheets are predicted to be stable.25 In situ heating in vacuum showed little change to the flake, expanded upon in Supplementary Fig. 6, but shows it is the presence of O2 that is critical to this transformation.
Analysis of elemental environment from electron energy-loss spectra
Maps of the Ti L3 peak shift relative to its energy in the TiS2 are shown in Figs. 2e and 3e. A shift up in energy is expected upon replacing a Ti–S bond with a Ti–O bond. Oxygen is more electronegative than sulphur so oxygen will form a more ionic bond, increasing the bond energy. Mapping the Ti L3 peak offers a useful tool to distinguish between regions of the sample that have actually oxidised versus those with a residual surface layer containing oxygen. It also offers some insight into the degree to which a sulphurous-region has been oxidised, based on the magnitude of the peak shift. The maximum peak shift observed was 1.5 eV in Fig. 3e; however this peak-shift is reduced in Fig. 2e as a result of some surface oxidation also seen in the O EELS map (Fig. 2c).
Figure 5 compares EELS spectra from flakes oxidised in water, air and the in situ heating, showing the Ti L2,3, L1 (minor) and O K edges. These spectra have been normalised to the same integrated intensity for ease of comparison, and split into the Ti L2,3 and O K/Ti L1 regions. It can be seen that while the intensity is relatively flat after the Ti L2,3 edge in spectra A–D post background removal, it increases in spectra E from the in situ experiments. This is due to increased plural scattering from the SiN windows encasing the gas in the in situ holder adding another 80 nm to the overall specimen thickness. Adding this to the flake thickness approaches the inelastic mean free path of TiS2—128 nm at 300 kV. This worsens the energy resolution, spreading the zero-loss peak to a full width half-max of 1.3 eV, so we were not able to resolve most of the fine detail around the various edges.

Comparison of Ti and O EELS edges from across different sample of oxidised TiS2. a From the sulphide region of the water oxidised flake in Fig. 2, b from the oxide region of the water oxidised flake in Fig. 2, c from the sulphide region of the air oxidised flake in Fig. 3, d from the oxide region of the air oxidised flake in Fig. 3, e from the oxide region of the in situ oxidised flake in Fig. 4. Inset concentration of Ti4+ within oxide spectra
The energy loss near edge structure can act as a fingerprint for specific polymorphs of a given structure, such as the various phases of TiO2—anatase, rutile and brookite, and can also differentiate other oxides of varying stoichiometry.26,27,28 Stoyanov et al.28 proposed the following empirical equation to determine the concentration of Ti4+ within a titanium oxide:
In this equation, x is the Ti4+ concentration (Ti4+/ΣTi4+) and a is the white line intensity ratio I(L2)/I(L3) based on the intensity within two 1 eV energy windows—an L3 window centred around the first white line in the L3 edge in a pure Ti3+ sample (455.8 to 456.8 eV), and an L2 window centred around the second white line in the L2 edge in a pure Ti4+ sample (465.25–466.25 eV).
This analysis was performed on the three oxides in Fig. 5 (extracted from the corresponding oxide regions in Figs. 2–4), produced after exposure to water (Fig. 5b), atmosphere (Fig. 5d), and oxygen at 375 °C (Fig. 5e). It showed that in all cases we have a mixture of Ti3+ and Ti4+ ions present within the oxides. This suggests that oxidation of TiS2 does not form pure TiO2 but instead, we appear to have the formation of an intermediary or non-stoichiometric oxide. This could explain the disordered structures observed in both imaging and diffraction (either amorphous or nanocrystalline).
Standard EELS quantification within Digital Micrograph of the oxides in Fig. 5b (water) and Fig. 5d (air) returned Ti:O ratios of 47.2:52.8 and 43.3:56.7 implying formation of an oxide with a stoichiometry close to Ti3O4. While there is no known oxide with this formula,29 two close oxides are TiO and Ti2O3.28 The spectra show agreement with Ti2O3 with reasonable match in the position of the L3,2 maxima, while the lack of crystallinity would likely cause broadening of the peaks making it hard to resolve the expected splitting.28 However, this stoichiometry would suggest the valence of Ti to be a mixture of Ti3+ and Ti2+, TiO would not be expected to be found at room temperature, typically requiring temperatures of nearly 1000 °C to form, and would oxidise almost immediately upon exposure to air to a TiO2.30, 31 Figure 5e was not considered for quantification due to the rising background resulting from the plural scattering.
Thermodynamics of reaction with oxygen gas
To investigate the difference in reactivity between O2 and H2O, we performed calculations to determine the thermodynamics of oxidation of TiS2 by oxygen gas. In the first step of this reaction, the oxygen molecule should be physisorb on TiS2 edge, which is endothermic (−0.28 eV). By contrast physisorption of a water molecule on a TiS2 edge is quite exothermic ( + 0.76 eV). Oxygen could then either substitute for sulphur ( + 2.00 eV at the edge and + 1.98 eV at the centre of a nanoflake) or fill a pre-existing sulphur vacancy (the most exothermic at + 3.39 eV), both significantly more exothermic than the corresponding reaction with water (0.12, 0.11 and 0.21 eV, respectively). Generation of a sulphur vacancy is strongly endothermic however (−1.4 eV) and under the proposed mechanism, filling one would not result in the formation of a new vacancy (in contrast to the chaining reaction with water).15 Filling a vacancy would also result in the formation of a lone oxygen atom that would then either need a second vacancy to enter the structure, or combination with another lone oxygen atom to form a new O2 molecule (we did not investigate this step). The low reactivity of O2 at room temperature is therefore believed to be due to the relatively large barrier in bringing O2 to the surface and then removing a sulphur atom, which is easier to overcome as thermal energy becomes more plentiful upon heating.
It might be possible to have some degree of control over the electronic properties of a sulphide-oxide hybrid material, by engineering the band gap. Calculations have shown the band gap can be fine-tuned between the extremes of pure TiS2 and TiO2 via oxygen substitution,15 which might also explain why such a range of values exist for the measured band-gap. Aging in water would provide one method of controlling the formation of oxide, although it would be isolated to the edges. Flash heating with oxygen might provide an alternative, if a flake could be controllably damaged such that oxygen vacancies pocket the surface, possibly through ion- or electron-irradiation. Small regions of oxide dispersed among the sulphide could therefore potentially be formed.
Discussion
Possible oxidation mechanism
The mechanism by which water oxidises TiS2 could be envisaged as having two stages. During stage 1, sulphur in the outermost surface regions is exchanged with oxygen to produce the hydrogen sulphide smelled upon opening vials of the dispersion. This has previously been demonstrated theoretically,15 and the mechanism reproduced in Supplementary Fig. 7. Later, titanium in non-surface layers seems to diffuse out to react with oxygen, leading to titanium depletion within the core. The depletion region appears dark in both the HAADF intensity (Fig. 2a) and Ti EDX maps (Fig. 2b), and is rich in sulphur relative to titanium. This would be consistent with the nanoscale Kirkendall effect, whereby atoms diffuse via vacancy exchange, with preferential diffusion of cations outwards over anions inwards.32,33,34 Reaction with water at the flake edge disturbs the crystallinity, generating Ti4+ vacancies. These vacancies diffuse away from the edge as fresh Ti4+ moves towards the edge to react with O2−, leaving sulphur to recombine and leave afterwards. The formation of the oxide layer hinders both the removal of sulphur and inward-diffusion of oxygen, leading to the formation of the depletion region observed. The apparent increase in thickness, observed from the increase in intensity in Fig. 2b, would likely be due to formation a less dense amorphous structure. This swelling of layers as they oxidise would serve to inhibit transport to the interface resulting in the slow rate of degradation of layers away from the surface, which would have the greatest impact on thicker flakes. A schematic representing this mechanism is shown in Supplementary Fig. 8.
We observed edge oxidation to be strongly preferential versus bulk suggesting that diffusion between layers is much faster than through layers. The fragmented nature of the oxidised region could arise during the removal of sulphur, if the sulphurous species act to isolate nucleation of tiny oxide domains.
This contrasts with the oxide morphology seen in the oxygen-gas-heating experiments. There oxidation seems to result in the shrinking and crinkling up of the flake, albeit still with a polycrystalline structure. This could be due to large amounts of strain being rapidly induced in the flakes by the reaction. The elevated temperature makes all species more mobile, which is likely the reason behind both the higher rate of oxidation and the higher rate of sulphur recombination and removal. An alternative explanation for the striped appearance of the oxide is that oxide nucleates and starts to grow, dragging titanium towards it while expelling sulphur away. The sulphur-rich regions eventually combine into gaseous species lost into the atmosphere in the holder, leaving behind regions with much less material than those around the original nucleation sites.
Conclusion
In conclusion, we studied the oxidation of TiS2 in a range of environments using a range of advanced electron microscopy techniques. The kinetic barrier for oxygen gas oxidation appears to be higher than that for oxidation via water. Water was observed to slowly oxidise TiS2 flakes, with oxidation starting at the flake edge and moving inwards, forming an amorphous oxide of Ti3+ and Ti4+. In contrast, the flakes oxidise slowly in atmosphere or vacuum conditions at high temperatures, suggesting moisture to be the critical element to remove from their environment in order to ensure long term stability. TiS2 was fully oxidised when heated above 375 °C in oxygen gas, using an in situ set-up, whereupon flakes oxidised immediately to form poly-crystalline material.
This work demonstrates the potential capabilities of cutting-edge in situ TEM holders for detailed characterisation of two-dimensional nanomaterials degradation behaviour within relevant environmental conditions not commonly available within a transmission electron microscope. In particular simultaneous EDX and EELS spectral imaging has been demonstrated in a gas-cell operating at a pressure of 330 mbar, despite the challenges presented by a combination of 80 nm of electron-transparent window material and a gaseous environment. Mapping of the shift in the Ti L3 in particular offers a valuable method of distinguishing regions of oxide, sulphide, or a mixture of the two species. This is particularly useful when surface oxygen species would otherwise skew elemental mapping.
DFT calculations found the oxidation of TiS2 by both water and oxygen gas to be thermodynamically favourable, with an edge site being favourable to a vacancy site due to the lack of chain reaction to progress the reaction at the vacancy.
Methods
Commercial TiS2 powder was acquired from Sigma Aldrich (99.9% Lot#333492) and stored in an argon filled glove box with both oxygen and moisture readings <1 ppm. Five milligrams of TiS2 were added to 50 ml anhydrous IPA (99.5% Sigma Aldrich Lot#278475). The unexfoliated dispersion was then exfoliated using a Fisherbrand ultrasonic disintegrator operating at 20 kHz and 34 W. The temperature was kept at a constant 10 °C using a circular IsoUK 4100 R20 refrigeration bath. The dispersion was sonicated for 3 h and quickly placed inside a centrifuge tube. The dispersion was centrifuged at 1000 RPM for 1 h and the supernatant was decanted and stored in the Argon-filled glove box.
For the ex situ ageing experiments, the dispersion was drop-cast onto gold TEM grids, while for the in situ experiments it was drop-casted onto specialised Si-Si3N4 windowed environmental cell chips. The later are used with the in situ gas TEM holder and allow the sample to be viewed at high resolution while being exposed to elevated temperatures (up to 900 °C) and pressures up to 1 bar. The upper environmental cell chips consist of a heating element surrounding 30-nm thick Si3N4 windows through which the electron beam can pass and the sample can be viewed. Material was drop-casted onto these windows for heating and imaging.
The microscope used for Figs. 1 and 2 was an FEI Titan 80–300 S/TEM, operated mainly in STEM mode at 300 kV, with an EDAX EDX detector and a Gatan imaging filter (GIF) for acquiring EEL spectra. The convergence angle was 10 mrad, and HAADF collection angles were 39–200 mrad while EEL spectra were acquired with a collection angle of 21 mrad and an energy dispersion of 0.1 eV.
Figure 3 was acquired on a NION UltraSTEM operated at 200 kV with a Brucker EDX detector and Enfinia EELS spectrometer. The convergence angle was 27 mrad, collection angle of the HAADF detector 99–200 mrad, and EEL spectra were acquired with a collection angle of 15 mrad and an energy dispersion of 0.1 eV.
The procedure for ex situ hydration of TiS2 was to image flakes immediately after dispersion, then store the grid in deionised water between imaging sessions. The grid was dried in an Across International drying oven (V0-16020C3-Prong) at ~5 × 10−2 mbar for an hour prior to imaging to remove as much excess water from the grid as possible, and then returned to DI water immediately afterwards. Attempts were made to re-image the same flakes, but build-up of carbonaceous contamination would coat imaged areas, retarding or fully preventing further oxidation. However, the range of flakes studied was broadly similar in both size and thickness, so reasonable comparisons could be drawn between them.
In situ oxidation experiments have been performed using a Protochips Atmosphere system and a FEI Titan 80–200 ChemiSTEM equipped with probe-side aberration correction, anX-FEG electron source and four windowless SDD detectors. The experiment was performed using an acceleration voltage of 200 kV, a beam current of 600 pA and a convergence angle of 21 mrad. EDX spectrum imaging was performed with the specimen tilted at 25° to prevent shadowing of the X-rays emitted from the specimen by the specimen holder. In this configuration, two of the four (Super-X) EDX detectors were used for the acquisition, providing a solid angle of about 0.4 sr. EEL spectra were acquired using a GIF Quantum with an energy dispersion of 0.1 eV and a collection angle of 62 mrad, providing an effective energy resolution of 1.3 eV. In standard imaging mode, the HAADF collection angles were 62–142 mrad, while in spectroscopy imaging mode the HAADF collection angles were 60–190 mrad. The gas environmental cell holder was run with an oxygen/argon mix at 290–350 mbar operated between 150–500 °C. The holder had been modified previously, similar to the modifications reported for the liquid-cell holder,35, 36 in order to significantly reduce the shadowing of the holder for two of the four available EDX detectors. The internal environment of the holder was shielded from the vacuum of the microscope by two silicon nitride windows, 30 and 50-nm thick.
Selecting an appropriate sample size is a crucial step in designing a successful study. Electron microscopy studies were hereby designed to be representative of the sample. We based this study on multiple samples prepared exactly in the same way. Samples were individually screened at the microscope to make sure our findings were representative. Sheets thickness dependence is discussed in the manuscript.
Computational approach
DFT37, 38 calculations were performed with the CP2K package using the PBE (Perdew–Becke–Ernzerhof) exchange and correlation (XC) functional.39 CP2K uses Goedecker-type pseudopotentials and expands Kohn–Sham orbitals using a combination of a mixed Gaussian-type and plane-wave basis set.40 The atomic orbitals of the atoms involved (O, H, S, and Ti) were expanded in DZVP and DZV (double-zeta valence polarised and double-zeta valence) Gaussian-type basis sets, while charge density was expanded in plane-waves with a density cut-off of 800 Ry.
An ideal TiS2 monolayer was modelled using a hexagonal TiS2 (1,1,1) 8 × 8 supercell containing 192 atoms, in which layers were periodically separated in the z-direction by a vacuum region 10.7 Å wide. A 4 × 8 supercell of TiS2 was created to model a nanoflake, continuous in the y-direction and truncated in the x-direction, such that periodically repeated flakes in the x- (z-direction) direction were separated by 15.37 (10.73) Å. This supercell was created to be orthorhombic, replicating a (√3,1) rectangular unit cell over the primitive hexagonal unit cell. This nanoflake was created to study structural and oxidation properties at the edges, which were modelled with 50% S-passivation.15 The structure of this edge was a zig-zag structure, with S atoms lying across the Ti terminal row such that the edge appears tilted. This has the effect of leaving the Ti atoms accessible to the oxidising molecules—gaseous oxygen and water.
We determined the cell to be sufficiently large to be a good representation of the Brillouin zone at Γ, by comparison of edge geometry and oxidation energies between a 4 × 8 and a 4 × 20 supercell. Substitution of an O atom into the middle of the nanoflake was also performed to check that the same reaction energy was obtained for an infinite surface.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Winter, M., Besenhard, J. O., Spahr, M. E. & Novák, P. Insertion electrode materials for rechargeable lithium batteries. Adv. Mater. 10, 725–763 (1998).
Whittingham, M. S. & Jacobson, A. J. High energy density plural chalcogenide cathode-containing cell. US Patent 4,233,375 (1979).
Whittingham, M. & Jacobson, A. J. A mixed rate cathode for lithium batteries. J. Electrochem. Soc. 128, 485–486 (1981).
Whittingham, M. Lithium batteries and cathode materials. Chem. Rev. 104, 4271–4301 (2004).
Kanno, R., Takeda, Y., Imura, M. & Yamamoto, O. Rechargeable solid electrolyte cells with a copper ion conductor, Rb4Cu16I7−δCl13+δ, and a titanium disulphide cathode. J. Appl. Electrochem. 12, 681–685 (1982).
Brandt, K. Historical development of secondary lithium batteries. Solid State Ion. 69, 173–183 (1994).
Seh, Z. W. et al. Two-dimensional layered transition metal disulphides for effective encapsulation of high-capacity lithium sulphide cathodes. Nat. Commun. 5, 5017 (2014).
Trevey, J. E., Stoldt, C. R. & Lee, S.-H. High power nanocomposite TiS2 cathodes for all-solid-state lithium batteries. J. Electrochem. Soc. 158, A1282–A1289 (2011).
Ma, L. et al. Hybrid cathode architectures for lithium batteries based on TiS2 and sulfur. J. Mater. Chem. A 3, 19857–19866 (2015).
Zeng, Z., Tan, C., Huang, X., Bao, S. & Zhang, H. Growth of noble metal nanoparticles on single-layer TiS2 and TaS2 nanosheets for hydrogen evolution reaction. Energy Environ. Sci. 7, 797–803 (2014).
Yang, C., Hirose, Y., Nakao, S., Hoang, N. L. H. & Hasegawa, T. Metal-induced solid-phase crystallization of amorphous TiO2 thin films. Appl. Phys. Lett. 101, 52101 (2012).
Myron, H. & Freeman, A. Electronic structure and optical properties of layered dichalcogenides: TiS2 and TiSe2. Phys. Rev. B 9, 481–486 (1974).
Greenaway, D. L. & Nitsche, R. Preparation and optical properties of group IV–VI2 chalcogenides having the CdI2 structure. J. Phys. Chem. Solids 26, 1445–1458 (1965).
McKelvy, M. J. & Glaunsinger, W. S. Synthesis and characterization of nearly stoichiometric titanium disulfide. J. Solid State Chem. 66, 181–188 (1987).
Cucinotta, C. S. et al. Electronic properties and chemical reactivity of TiS2 nanoflakes. J. Phys. Chem. C 119, 15707–15715 (2015).
Han, J. H. et al. Unveiling chemical reactivity and structural transformation of Two-dimensional layered nanocrystals. J. Am. Chem. Soc. 135, 3736–3739 (2013).
Malis, T., Cheng, S. C. & Egerton, R. F. EELS log-ratio technique for specimen-thickness measurement in the TEM. J. Electron. Microsc. Tech. 8, 193–200 (1988).
Lotya, M. et al. Liquid phase production of graphene by exfoliation of graphite in surfactant/water solutions. J. Am. Chem. Soc. 131, 3611–3620 (2009).
Egerton, R. F. Electron energy-loss spectroscopy in the TEM. Rep. Prog. Phys. 72, 16502 (2009).
Mckelvy, M. J., Claunsinger, W. S. & Ouvrard, G. Titanium Disulfide 6 (Wiley, 2007).
Yu, J., Fan, J. & Lv, K. Anatase TiO2 nanosheets with exposed (001) facets: improved photoelectric conversion efficiency in dye-sensitized solar cells. Nanoscale 2, 2144–2149 (2010).
Sheng, L., Liao, T., Kou, L. & Sun, Z. Single-crystalline ultrathin 2D TiO2 nanosheets: a bridge towards superior photovoltaic devices. Mater. Today Energy 3, 32–39 (2017).
Peng, C. W. et al. Interconversion of rutile TiO2 and layered ranisdellite-like titanates: New route to elongated mesoporous rutile nanoplates. Cryst. Growth Des. 8, 3555–3559 (2008).
Zhang, Y. et al. Raman study of 2D anatase TiO2 nanosheets. Phys. Chem. Chem. Phys. 18, 32178–32184 (2016).
He, T. et al. Layered titanium oxide nanosheet and ultrathin nanotubes: A first-principles prediction. J. Phys. Chem. C 113, 13610–13615 (2009).
Brydson, R. et al. Electron energy loss and X-ray absorption spectroscopy of rutile and anatase: a test of structural sensitivity. J. Phys. Condens. Matter. 1, 797–812 (1989).
Brydson, R., Sauer, H., Engel, W. & Hofer, F. Electron energy-loss near-edge structures at the oxygen Kedges of titanium (IV) oxygen compounds. J. Phys. Condens. Matter. 4, 3429–3427 (1992).
Stoyanov, E., Langenhorst, F. & Steinle-Neumann, G. The effect of valence state and site geometry on Ti L3,2 and O K electron energy-loss spectra of TixOy phases. Am. Minerol. 92, 577–586 (2007).
Pearson, A. D. Studies on the lower oxides of titanium. J. Phys. Chem. Solids 5, 316–327 (1958).
Watanabé, D., Castles, J. R., Jostsons, A. & Malin, A. S. The ordered structure of TiO. Acta Crystallogr. 23, 307–313 (1967).
Bartkowski, S. et al. Electronic structure of titanium monoxide. Phys. Rev. B 56, 10656–10667 (1997).
Yin, Y. et al. Formation of hollow nanocrystals through the nanoscale Kirkendall effect. Science 304, 711–714 (2004).
Li, X., Li, M., Liang, J., Wang, X. & Yu, K. Growth mechanism of hollow TiO2(B) nanocrystals as powerful application in lithium-ion batteries. J. Alloys Compd. 681, 471–476 (2016).
Liang, J. et al. Fabrication of TiO2 hollow nanocrystals through the nanoscale Kirkendall effect for lithium-ion batteries and photocatalysis. New J. Chem. 39, 3145–3149 (2015).
Lewis, E. A. et al. Real-time imaging and local elemental analysis of nanostructures in liquids. Chem. Commun. 50, 10019–10022 (2014).
Zaluzec, N. J., Burke, M. G., Haigh, S. J. & Kulzick, M. A. X-ray energy-dispersive spectrometry during in situ liquid cell studies using an analytical electron microscope. Microsc. Microanal. 20, 323–329 (2014).
Hohenberg, P. & Kohn, W. Inhomgenous electron gas. Phys. Rev. B 136, B864–B871 (1964).
Kohn, W. & Sham, L. J. Self consistent equations including exchange and correlation effects. Phys. Rev. 385, A1133–A1138 (1965).
Vandevondele, J. et al. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 167, 103–128 (2005).
Goedecker, S., Teter, M. & Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 54, 1703–1710 (1996).
Acknowledgements
E.L., S.O.′B. and V.N. wish to thank the support of the SFI PIYRA and AMBER grants, the European Research Council (2DNanoCaps and 3D2D Print projects) and the EU ITN MoWSeS project. The Advanced Microscopy Laboratory (AML) and its staff (in particular CD) are thanked for their assistance in electron microscopy in Dublin. S.S. and C.S.C. have been supported by the European Research Council (Quest-project). All calculations were performed on the Parsons cluster maintained by the Trinity Centre for High Performance Computing, under project id: HPC_12_0722. This cluster was funded through grants from Science Foundation Ireland. SJH, EP and EAL thank the Defence Threat Reduction Agency under grant HDTRA1-12-1-0013. The Titan at Manchester was funded with support from HM Government (UK) and is associated with research capability of the Nuclear Advanced Manufacturing Research Centre.
Author information
Authors and Affiliations
Contributions
E.L. wrote the first draft of the paper, and performed the ex situ experiments. E.A.L., E.P., S.J.H., C.D. and E.L. performed the in situ experiments. C.S.C. and S.S. performed simulations of the oxidation. S.O.′B prepared the dispersions. V.N. and E.L. planned the experiments. All authors contributed to the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Long, E., O’Brien, S., Lewis, E.A. et al. An in situ and ex situ TEM study into the oxidation of titanium (IV) sulphide. npj 2D Mater Appl 1, 22 (2017). https://doi.org/10.1038/s41699-017-0024-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41699-017-0024-4
This article is cited by
-
Oxidation kinetics and non-Marcusian charge transfer in dimensionally confined semiconductors
Nature Communications (2023)
-
An Innovative Process for Production of Ti Metal Powder via TiSx from TiN
Metallurgical and Materials Transactions B (2020)
