
Abstract
The covalent functionalization of exfoliated semiconducting MoS2 by 1,2-dithiolanes bearing an ethylene glycol alkyl chain terminated to a butoxycarbonyl-protected amine and a photoactive pyrene moiety is accomplished. The MoS2-based nanohybrids were fully characterized by complementary spectroscopic, thermal, and microscopy techniques. Markedly, density functional theoretical studies combined with X-ray photoelectron spectroscopy analysis demonstrate preferential edge functionalization, primarily via sulfur addition along partially sulfur saturated zig-zag MoS2 molybdenum-edges, preserving intact the 2D basal structure of functionalized MoS2-based nanohybrids as confirmed by high-resolution transmission electron microscopy and electron energy loss spectroscopy. Furthermore, in the MoS2-pyrene hybrid, appreciable electronic interactions at the excited state between the photoactive pyrene and the semiconducting MoS2 were revealed as inferred by steady-state and time-resolved photoluminescence spectroscopy, implying its high potentiality to function in energy conversion schemes.
Similar content being viewed by others

Introduction
Transition metal dichalcogenides (TMDs) are layered nanomaterials consisting of an atomic plane of a transition metal sandwiched between two atomic planes of chalcogens. Bulk TMDs are semiconductors with an intrinsic band-gap and exhibit noteworthy optical and electronic properties desirable for energy conversion and storage,1,2,3,4,5,6,7 sensing,8,9,10 and photodynamic/photothermal therapy.11,12,13 Analogously to graphite, numerous van der Waals forces keep together the layered sheets of TMDs forming stacks. However, despite their close similarity to graphene, TMDs offer significant advantages related to their band-gap dependent electronic properties,14, 15 which can be tuned by altering the transition metal and/or the chalcogen composition. In addition, although functionalization of graphene is greatly developed, allowing the preparation of a plethora of modified graphene-based hybrid materials, the corresponding field for TMDs is still at its infancy. Particularly, a central aspect of layered TMDs chemistry, which has yet to be fully exploited, concerns their functionality in energy conversion applications. In this context, it is imperative the functionalization of TMDs, permitting their better manipulation and processing in wet media by improving solubility in common organic solvents. At the same time, of particular importance is the incorporation of organic electron donor units by robust and stable chemical bonds with the network of TMDs in order to alter their properties and entirely harness their capabilities.
Diverse methodologies for exfoliating TMDs from the bulk exist, giving access predominantly to the metallic polytype lattice structure with octahedral coordination geometry.16,17,18,19,20,21,22 On the other hand, we recently reported a facile approach for the exfoliation of MoS2 and WS2 polytypes with trigonal prismatic geometry and semiconducting properties with a direct band-gap, by treating the corresponding bulk materials with chlorosulfonic acid.23 The TMD layers are protonated by the superacid and kept apart due to developed electrostatic repulsive interactions, however, without being oxidized. Nevertheless, despite the progress achieved so far targeting the exfoliation of MoS2, its covalent functionalization is still hampered. In fact, the limited functionalization procedures reported to date mainly utilize exfoliated MoS2 by n-butyl lithium (n-BuLi), thus of the metallic polytype 1T-MoS2 in which the negative charges on MoS2 due to charge-transfer from n-BuLi, are quenched by alkyl halides,24 and diazonium salts,22 while thiols are conjugated at sulfur vacancies21, 25,26,27,28 and metal carboxylate salts coordinate to surface sulfur atoms of 2H-MoS2.29 Therefore, it is absolutely desirable and timely to functionalize semiconducting MoS2 species with organic electron donor units.30 Additionally, it is necessary to understand where and how functionalization occurs, as to date the characterization performed is rather vague.
Herein, we report for the first time on the covalent functionalization of exfoliated semiconducting MoS2 by employing 1,2-dithiolane units as parts of organic derivatives bearing either an ethylene glycol alkyl chain terminated to a butoxycarbonyl (BOC)-protected amine 1a or a pyrene 1b (Fig. 1) as an electron donor. Moreover, we disclose the location (“where”) and the manner (“how”) this functionalization occurs based on density functional theoretical studies supported by X-ray photoelectron spectroscopy (XPS) and confirmed by high-resolution transmission electron microscopy (HRTEM) imaging and electron energy loss spectroscopy (EELS). Notably, the 1,2-dithiolane component possesses high binding affinity for Mo atoms, particularly those located at the edges of the corresponding TMDs, where sulfur vacancy sites were naturally introduced after chemical exfoliation from the bulk.31 Such a facile approach for the chemical functionalization of MoS2 offers the advantage of keeping intact the basal plane of MoS2 and preserving to great extent their novel surface properties, as well as their semiconducting character, while also allows the incorporation of a plethora of suitably modified organic derivatives targeting diverse applications. Importantly, the presence of defects at the edges of MoS2, as generated by either chemical exfoliation21, 25,26,27,28 or ion irradiation,32 guarantees the universal utility and versatility of the functionalization route. The newly prepared MoS2-based nanohybrids were fully characterized, while examination of the optical properties revealed the occurrence of significant electronic interactions in the MoS2-pyrene 2b hybrid.

Reaction route for obtaining functionalized MoS2-based nanohybrids 2 from exfoliated semiconducting MoS2 flakes
Results
The exfoliated semiconducting MoS2 was prepared from the bulk upon chlorosulfonic acid treatment by following our recently published procedure.23 The general scheme for the functionalization process is illustrated in Fig. 1 (structural characterization data for 1,2-dithiolanes 1a and 1b are presented at the Supplementary Information, Figs. S1–S4). The newly functionalized MoS2-based nanohybrids 2 found to be soluble in N,N-dimethylformamide (DMF), ca. 1.2 mg/mL, while their solubility in dichloromethane, acetone, and THF was limited. The so-derived MoS2-based nanohybrids can be stored in solution for months, without observing precipitation, hence justifying their stability in wet media. Treatment of 2a with gaseous HCl cleaves the BOC-protecting group, yielding the cationic ammonium derivatized MoS2-based material 2c, soluble in protic solvents and aqueous media. The amino-loading of 2c was calculated by performing the Kaiser test and found to be 103 μmol g−1. The latter result allows manipulating and processing such modified MoS2-based materials in biotechnological applications, where handling in physiological media is a prerequisite. The MoS2-based nanohybrids 2a–c were fully characterized by complementary spectroscopic and thermal techniques. Markedly, the functionalization appears to primarily occur via sulfur addition along partially sulfur saturated zig-zag MoS2 molybdenum-edges. Density functional studies combined with XPS analysis demonstrated the preferential edge functionalization, preserving the 2D basal structure of the functionalized MoS2-based nanohybrids as confirmed by HRTEM imaging and EELS. This is an extremely important finding, shedding ample light on the location and the manner the functionalization of exfoliated MoS2 with 1,2-dithiolanes proceeds, hence allowing the design and development of a plethora of interesting hybrid materials for diverse applications. A natural consequence of this finding is that chemical functionalization behavior of exfoliated MoS2 should occur similarly to that for bulk material, where edge functionalization is likely to be predominant. Thus, we would expect the possibility of direct transfer of functionalization strategies from bulk to exfoliated MoS2, for example transition metal doping of edge sites to improve catalytic behavior.33 Particularly in the MoS2-pyrene 2b hybrid material, appreciable electronic interactions at the excited state between the photoactive pyrene and the semiconducting MoS2 nanosheets were revealed as inferred by steady-state and time-resolved photoluminescence spectroscopy studies. An electron donor-acceptor combination, such as the one found in 2b hybrid material, mimics the simple photosynthetic system and imitates charge-transfer processes that ensue in nature. Moreover, such MoS2-based hybrid materials, incorporating photo-active and/or electro-active species, feature novel light harvesting, charge-separation and/or charge-transport properties, hence considered ideal for photovoltaics and optoelectronic devices. Thereby, assays on artificial photosynthetic models capable of undergoing charge-separation phenomena are of primary importance in constructing economic and light-to-electricity converting devices. Currently, we are extending the applicability of the current functionalization methodology to other layered transition metal dichalogenides incorporating diverse photoactive organic electron donors en route the preparation of advanced hybrid materials suitable for energy conversion applications. For example, solar cells convert the energy captured from sunlight into electricity by exploiting the generation of electrons and holes, namely the generation of charge-separation, that move to the corresponding electrodes of the photovoltaic cell. Needless to mention that the development of a long-lived charge-separated state is desirable for achieving high energy conversion efficiency, since it can effectively stream electrons and holes to the respective electrodes. Efforts along those lines are underway in our laboratories.
Discussion
Our functionalization largely deviates from recent studies based on thiol reactions, which were shown to oxidize to the corresponding disulfides which eventually only adsorb onto MoS2 and are not chemically anchored.34 In stark contrast, in our experimental protocol a mixture of exfoliated MoS2 and 1,2-dithiolane derivatives 1a or 1b in DMF was stirred at 70 °C for 36 h. Then, the reaction mixture was filtered over a polytetrafluoroethylene (PTFE) membrane (0.1 μm pore size) and the solid residue collected onto the filter was extensively washed with dichloromethane to remove organic impurities, yielding MoS2-based nanohybrids 2. The strong binding of the organic addends on the MoS2 was proved by examining their electronic absorption profile after extensive washing cycles to remove any loosely bound (physisorbed) organic compound. Evidently, after four consecutive washing cycles, the UV-Vis spectrum of functionalized MoS2-based materials 2 remain unaffected, namely the characteristic absorptions due to pyrene, as well as due to the MoS2 retained their intensity, while also the filtrate fail to show characteristic absorptions owed to the pyrene species.
Attenuated-total-reflectance IR assays gave spectroscopic proof for the success of the functionalization. Briefly, strong C–H stretching and bending modes are discernible in the 2840–2970 cm−1 range, while the ethylene glycol unit gives rise to vibrational modes in the region 1130–1190 cm−1 for 2. Particularly for 2a, two carbonyl moieties are identified at 1650 and 1710 cm−1 owed to the amide and BOC protecting units, respectively—the latter is absent in the IR spectrum of 2c, in which free amine functionalities are present—while for 2b, only the ester carbonyl is identified at 1730 cm−1 (Fig. 2a, b).

ATR-IR spectra for a 1,2-dithiolane derivative 1a (red) as compared with functionalized MoS2-based nanohybrids 2a (black) and 2c (gray), and b 1,2-dithiolane derivative 1b (red) and MoS2-pyrene 2b (black). Raman spectra for c exfoliated MoS2 (gray) as compared with functionalized MoS2-pyrene 2b (black), obtained upon 633 nm excitation. Thermographs for d exfoliated MoS2 (gray) as compared with that due to functionalized MoS2-pyrene 2b (black), obtained under nitrogen atmosphere
Next, Raman spectroscopy was employed as a valuable tool to obtain meaningful insights on the functionalization of MoS2. Upon in-resonance excitation at 633 nm, more rich spectra as compared with measurements conducted under off-resonance conditions at 514 nm were acquired. Upon comparison of the Raman spectra for functionalized MoS2 with those owed to exfoliated MoS2 flakes the following points are deduced: (a) the intensity of the 2LA(M) mode at 447 cm−1 is decreased for the functionalized MoS2 nanohybrids, for spectra normalized at the A1g mode located at 404 cm−1, in accordance with Raman studies performed on differently functionalized MoS2 species,22 and (b) phonon modes directly related to the metallic polytype of MoS2 at 150, 225, and 325 cm−1,35, 36 are totally absent, hence proving the semiconducting behavior of MoS2 in the functionalized nanohybrids 2. A representative Raman spectrum for 2b is shown in Fig. 2c (for 2a, see Supplementary Information, Figure S5).
Additional proof for the success of the covalent functionalization of MoS2 with 1,2-dithiolane derivatives 1a and 1b was delivered by thermogravimetric analysis (TGA). Exfoliated MoS2 is thermally stable up to 250 °C under nitrogen atmosphere. However, in MoS2-based nanohybrid 2b 8.1% weight loss in the temperature range 250–550 °C is observed (Fig. 2d). The latter is attributed to the thermal decomposition of the organic functionalities covalently bonded to Mo at the edges of MoS2, i.e., at sulfur vacancies. Actually, the loading of pyrene onto MoS2 is calculated to be one per every 32 MoS2 units. Similarly, from the TGA for 2a (Supplementary Information, Figure S6), the maximum loading of one organic addend is calculated to be per every 24 MoS2 units. Markedly, the relatively low mass loss observed in the TGA performed for nanohybrids 2 clearly proves that the organic functionalities were covalently anchored onto MoS2, contrasting the case where physisorption occurs, in which the coverage of MoS2 is very high reaching a loading of one organic unit per every two Mo atoms.34
HRTEM images show that the flakes owed to functionalized MoS2-pyrene 2b vary from single-layer to oligo-layer (the exfoliated MoS2 was composed of 6 layers on average according to calculations performed on their extinction spectra),37 with largely damage-free basal plane (Fig. 3a, b). Although the edge structure is difficult to be determined conclusively, there is evidently strong preference for zig-zag edge orientation, consistent with theoretical predictions38, 39 and experimentally observed structures,39,40,41,42,43 suggesting that 50% S and 100% S saturated zig-zag molybdenum-edges should be thermodynamically preferred.43,44,45 The HRTEM images also show the presence of material at the edges, which may correspond to carbonaceous species. In order to investigate this point, we carried out spatially-resolved EELS studies, as the most appropriate technique for such kind of analyses.46, 47 In more detail, Fig. 3c displays the EEL spectrum recorded in a region close to the edge of a flake belonging to functionalized MoS2-pyrene 2b nanohybrid—see also high-angular annular dark field, scanning TEM image displayed as inset of Fig. 3c. Apart from the expected Mo and S contributions, the C–K edge is visible. The fine structure of this C–K signal (shown in Fig. 3d) includes a significant π* peak and other contributions at ~288 eV and in the σ* region (290-310 eV), consistent with attribution to the organic addend incorporated in 2b hybrid material and different to the one of amorphous carbon, further proving the success of functionalization of semiconducting MoS2 with pyrene species.46,47,48,49

a, b HRTEM micrographs of functionalized MoS2-pyrene 2b hybrid. Edges are primarily zig-zag type, with evidence of protruding chain-like carbonaceous material, terraced layers with extensive exposed edges. c EEL spectrum acquired in a flake of functionalized MoS2-pyrene 2b shown in the inset. The S–L2,3, Mo–M4,5, C–K, and Mo–M2,3 edges are highlighted in this spectrum. d C–K edge fine structures (ELNES), extracted from the spectrum of Fig. 3c. Scale bar of all (S)TEM images is 1 nm
We next turn to density functional theory (DFT) modeling to better understand the functionalization mechanism. In order to gain insight into the covalent binding behavior, we first model the interaction of 3-methyl-1,2-dithiolane with the MoS2 edge and basal plane. The former molecule represents the covalent linking segment of 1a–c with the carbon chain attached to neighboring S atoms of MoS2 either in the basal plane or at zig-zag 100% S molybdenum-edge (Fig. 4a–d and Supplementary Information, Figure S7). Preferential binding for the edge compared to the basal plane is found (2.54 eV per molecule for the most stable configuration).

a–d 3-methyl-1,2-dithiolane binding to MoS2 edge: a Schematic representation, b most stable binding configuration found for the edge functionalization, with the aliphatic chain connected to S atoms from the same reconstructed edge S2 pair, c, d less stable configurations with the aliphatic chain connected to S atoms from neighboring S2 pairs. Enthalpy differences (eV) are relative to structure in Fig. 4b. e Calculated structure for 1b binding to MoS2 (upper panel) 100% S zig-zag molybdenum-edge, and (bottom panel) basal plane, yielding functionalized MoS2-pyrene 2b. f Top and side view of MoS2 zig-zag molybdenum-edges 50% S (left panel) and 100% S saturated (right panel). Atom color code: green-blue = Mo, yellow = S, gray = C, white = H, red = O
At the edge, 3-methyl-1,2,-dithiolane is most stable with its carbon chain approximately perpendicular to the edge (Fig. 4b) breaking apart a single S2 pair. The other less stable edge configurations result in two broken S2 pairs, which then reconstruct horizontally (Fig. 4c, d). Neighboring bonds in MoS2 around the covalent anchoring site are also distorted, notably in Fig. 4c and d, the distance between Mo atoms connected to the S atoms bound to the carbon chain of 3-methyl-1,2,-dithiolane is reduced from 3.13 Å for the bulk to around 2.96 Å.
We next perform calculations on covalently attaching the full 1,2-dithiolane-based derivative 1b to both basal plane and edge sites of MoS2 (Fig. 4e and Supplementary Information Figure S8). The edge functionalization configuration is constructed based on the 3-methyl-1,2,-dithiolane configuration in Fig. 4c, since this maintains the same conformation of the aliphatic chain and pyrene moiety as in the basal case. For MoS2 basal plane functionalization we assume direct substitution of S into neighboring S vacancies on the basal plane, resulting in neighboring carbon atoms bound directly to S atoms in the basal plane. One of these carbon atoms sits directly above the lattice S, while the second is displaced, pulling its S neighbor 0.32 Å out of the plane and distorting neighboring Mo–S bonds.
In all cases, the pyrene and aliphatic tail preferentially stack parallel to the MoS2 basal plane. The pyrene moiety and chain stack at approximately their van der Waals radii distance above the MoS2 surface (3.16 Å above the S plane, 4.73 Å above the Mo plane; Fig. 4e and Supplementary Information Figure S8). In the basal plane case this adds 2.0 eV binding enthalpy per molecule compared to alternative non-interacting configurations, of which ~ 1.1 eV comes from the pyrene moiety (Supplementary Information, Figure S9).
Once again edge binding is preferred, with the 1b molecule 1.96 eV more stable at the edge than on the basal plane. This value is very close to that for 3-methyl-1,2-dithiolane in the equivalent binding configuration (2.03 eV). In addition, the distances between pyrene and the S atoms from the top plane of MoS2 is the same as for the basal functionalization model (3.16 Å). This suggests that the preference for the edge in this model is mainly due to the changes produced by the covalent linkage between the molecule and MoS2, while the non-covalent interaction with the basal plane, due to pyrene and the aliphatic chain, is similar in both basal and edge functionalization models. Moreover, structures 1a, 1b, and 1c all connect to MoS2 through the same functional group, and the 3-methyl-1,2-dithiolane molecular fragment is a valid model for all three. While the non-covalent binding of 1a and 1c may not behave similarly to that of 1b, even in the unlikely case that the non-covalent interactions show a preference for basal plane binding, such a difference would have to be larger than 2.54 eV to give net favorable basal plane binding. Thus, the calculations suggest the edge binding preference of 1b will be shared by 1a and 1c.
Complete energy cycles are given in Supplementary Information, including either vacancy formation followed by molecular insertion into the vacancy sites, or S2 removal from the molecule followed by direct addition to either the basal plane (which is endothermic in this case) or the edge. The relative energy between basal and edge functionalization is independent of the energy cycle chosen, i.e., while the sulfur reference state energy will depend on the sulfur chemical potential, this is a ‘constant’ in both cases. Actual energy cycles could involve other processes such as creation of monovacancies and vacancy migration, and in practice the experimental cycle followed is likely to be considerably more complex (e.g., dealing with oxygen impurities). We note also that even if the basal plane is successfully functionalized, there is then a 2.54 eV driving force for the functional group to migrate to the sheet edge.
Hence, we suggest that edge binding is strong energetically favored, and functionalization will likely progress via physisorption to the basal plane followed by migration and chemisorption to the edge. This leads us to propose a new model for covalent functionalization of semiconducting MoS2 with the 1,2-dithiolanes. There is evidence that edges play an important role in chemical processes like catalysis, in contrast to the less reactive basal plane.50, 51 Literature studies of edge configurations give the 50% S and 100% S saturated zig-zag molybdenum-edges as the most thermodynamically favored (Fig. 4f), with the 50% S edge more stable over a wide range of S chemical potential.40,41,42,43,44 These edges have been both observed experimentally and it has been shown that the edge configuration depends on the synthesis conditions.40,41,42,43 Although further studies need to be done in order to fully unveil the processes occurring at these edges, some theoretical studies have already shown the possibility of their modification through adsorption and desorption processes.52,53,54
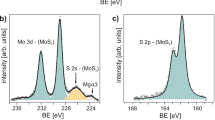
Starting with the most stable 50% S zig-zag edges, 1,2-dithiolanes can directly bind to the exposed Mo at this edge, saturating and resulting in 100% S coverage with covalently attached species, preferentially in a binding configuration perpendicular to the edge (cf. Fig. 4b). This converts the sulfur bonding at the extensive edge sites to a configuration much closer to the bulk, and can also be viewed as filling ‘vacant sulfur’ edge sites. The model is supported by XPS studies of the semiconducting MoS2 samples before and after functionalization with 1b (i.e., MoS2-pyrene 2b hybrid material) as depicted in Fig. 5. Before functionalization, the S 2p signal shows a superposition of the conventional semiconducting MoS2 signal, the split peak pair at 163.4 and 162.2 eV, with a second pair of peaks at 163.1 and 161.9 eV, typically associated with “vacancy” or damaged material,55 which we associate here with the unsaturated sulfur edges. After functionalization, these additional peaks are removed as the edges all become fully sulfur saturated. At the same time, there is very little change in the Mo 3d signal, the slight peak shifts consistent with small charge transfer from π-stacked pyrene found in our calculations. We note that the XPS signal shows no secondary peaks corresponding to oxidized S and Mo proving the high purity of functionalized MoS2-pyrene 2b hybrid material. The same was also confirmed by the EELS measurements (cf. Fig. 3c).

XPS a–b S 2p and c–d Mo 3d signal from a, c exfoliated semiconducting MoS2 and b, d functionalized MoS2-pyrene 2b hybrid material. Components in maroon show the presence of MoS2, while in gray are due to vacancies or damaged material
The UV-Vis spectrum for 2a in DMF exhibits the characteristic features due to the semiconducting polytype of MoS2.
Discernible are the excitonic transitions at 400, 500, 630, and 690 nm (Supplementary Information, Figure S10), justifying the semiconducting nature of the functionalized MoS2-based species.56 For the pyrene modified MoS2-based nanohybrid 2b, additional bands directly derived from the optical absorption of the pyrene are identified at 315, 330, and 345 nm (Fig. 6a), hence, directly implying the success of the functionalization reaction. Overall, the UV-Vis spectrum of MoS2-pyrene nanohybrid 2b is a simple superimposition of the spectra owed to the semiconducting polytype of MoS2 and to the pyrene derivative 1b, implying the absence of appreciable electronic interactions between the two species in the ground state.

a UV-Vis electronic absorption, and b photoluminescence spectra (excitation at 340 nm) for 1,2-dithiolane-based pyrene derivative 1b (red) as compared with functionalized MoS2-pyrene 2b hybrid material (black), obtained in DMF
Next, focusing at the excited states, strong electronic communication between MoS2 and pyrene within 2b is identified. More precisely, based on steady-state photoluminescence assays, the characteristic emission bands of pyrene in 1b, located at 376, 396, and 418 nm upon photoexcitation at 340 nm, were quenched in 2b (Fig. 6b), for samples possessing equal absorptions at the excitation wavelength. Notably, a blank sample prepared by mixing exfoliated MoS2 with pyrene derivative 1b failed to show photoluminescence quenching of the pyrene emission (Supplementary Information, Figure S11). Hence, intrahybrid transduction of electron and/or energy from the pyrene singlet excited state to MoS2 for 2b occurs, implying that MoS2 acts as electron/energy acceptor. Moving further, based on the time-correlated-single-photon-counting method, the fluorescence lifetime profiles for 1b were obtained and analyzed. The evaluation of the time profile of the fluorescence decay at 396 nm for the singlet-excited state of free pyrene in 1b was monoexponentially fitted with a lifetime of 3.79 ns. Conversely, two decay components in MoS2-based nanohybrid 2b were identified. Biexponential fitting for the fluorescence decay gives rise to a slower component with 5.01 ns, attributed to non-interacting pyrene and a faster one with 280 ps, corresponding to the fluorescence quenching of the emission intensity of the singlet excited state of pyrene in MoS2-pyrene 2b. Then, based on Eq. (1) the quenching rate constant k S q for the singlet excited state of pyrene in MoS2-pyrene 2b was evaluated as 3.3 × 109 s−1. Furthermore, by employing the Eq. (2), the quantum yield Φ S q for 2b was determined to be 0.85.
where T f refers to the lifetime of the fast-decaying component in 2b, and T o refers to the lifetime of free pyrene in 1b.
In conclusion, we demonstrate here that covalent chemical functionalization of exfoliated MoS2 with 1,2-dithiolane derivatives occurs preferentially at the sheet edges, with functionalization likely progressing via physisorption to the basal plane followed by migration and chemisorption to the edge. The presence of the 1,2-dithiolane end-group in our attach-ing species facilitates incorporation at the available edge sites. This is mediated through easy variation in sulfur saturation (50–100%) along the edge sites. Preferential edge reactivity is highly reminiscent of the chemistry of non-exfoliated MoS2, known for its strong catalytic capability, and suggests that other bulk MoS2 functionalization and treatment strategies may be transferable to the 2D system. Moreover, covalent incorporation of pyrene modified 1,2-dithiolane 1b on exfoliated MoS2 furnished donor-acceptor 2b hybrid material featuring charge-transfer interactions potentially suitable for managing energy conversion schemes. Similar edge-functionalization behavior is likely to also hold true for other 2D TMDs such as WS2, while we are currently extending our investigations with the incorporation of diverse photoactive molecules to yield novel functional materials.
Methods
General procedure for the synthesis of 1,2-dithiolanes 1a and 1b
In a round bottom flask, α- lipoic acid (1.6 mmol), tert-butyl (2-(2-(2-aminoethoxy)ethoxy)ethyl)carbamate (2.00 mmol, 1.25 equiv.) for 1a or 1-pyrenebutanol (1.6 mmol, 1 equiv.) for 1b, EDCI (4.8 mmol, 3 equiv.), and DMAP (4.8 mmol, 3 equiv.) were added in dry dicloromethane (100 mL). The reaction mixture was stirred under nitrogen at room temperature for 18 h. Then, the organic phase was extracted with H2O (5 × 100 mL), dried over MgSO4 and purified by column chromatography (petroleum ether/ethyl acetate 50%). The experimental procedure was replicated five times in our laboratories. Spectroscopic data for 1a: 1H NMR (300 MHz, CDCl3) δ: 6.07 (s, 1H), 5.01 (s, 1H), 3.58 (m, 8H), 3.44 (d, J = 4.6 Hz, 2H), 3.30 (s, 2H), 3.11 (m, 2H), 2.44 (m, 1H), 2.18 (t, J = 7.3 Hz, 3H), 1.89 (m, 1H), 1.66 (s, 4H), 1.43 (s, 9H); 13C NMR (75 MHz, CDCl3) δ: 172.80 (1C), 155.97 (1C), 79.31 (1C), 70.24 (1C), 70.15 (1C), 56.40 (1C), 40.32 (1C), 40.19 (1C), 39.14 (2C), 38.42 (1C), 36.20 (1C), 34.61 (2C), 28.86 (1C), 28.39 (3C), 25.37 (1C). ESI-HRMS calcd for C19H36N2O5S2: 436.2066, found: m/z 437.2138 [M + H]+. Spectroscopic data for 1b: 1H NMR (300 MHz, CDCl3) δ: 8.17 (d, J = 9.3 Hz, 1H), 7.97 (m, 7H), 7.77 (d, J = 7.8 Hz, 1H), 4.06 (t, J = 6.4 Hz, 2H), 3.39 (m, 1H), 3.28 (t, J = 7.5 Hz, 2H), 2.98 (m, 2H), 2.23 (m, 3H), 1.77 (m, 5H), 1.51 (m, 4H), 1.32 (m, 2H). 13C NMR (75 MHz, CDCl3) δ: 173.54, 136.31, 131.42, 130.87, 129.86, 128.59, 127.49, 127.27, 127.22, 126.64, 125.83, 125.09, 125.01, 124.90, 124.81, 124.72, 123.27, 64.18, 56.30, 40.15, 38.44, 34.54, 34.10, 33.04, 28.73, 28.66, 28.13, 24.71. ESI-HRMS calcd for C28H30O2S2: 462.6660, found: m/z 485.1579 [M + Na]+. A total of six NMR spectroscopy data and two ESI-HRMS were taken.
General procedure for the preparation of functionalized MoS2-based nanohybrids
In a round bottom flask, exfoliated MoS2 (20 mg) and 1,2-dithiolane derivative (1a or 1b; 10 mg) in DMF (10 mL) were stirred at 70 °C for 36 h. After that period, the reaction mixture was filtered through a PTFE membrane (0.1 mm pore size), the solid residue was extensively washed with DMF and dichloromethane and then collected as a dispersion in dichloromethane. The experimental procedure was replicated ten times in our laboratories.
Computational methodology
DFT calculations were performed using the local density approximation (LDA) as implemented in the density functional code AIMPRO.57,58,59 The spin-averaged charge density is fitted to plane waves with an energy cut-off of 100 Ha for the pristine MoS2 models and the S2 molecule model,150 Ha for the 3-methyl-1,2-dithiolane molecule model and the MoS2 models functionalized with it and 200 Ha for the MoS2 models functionalized with the pyrene derivative. Relativistic pseudopotentials generated by Hartwigsen, Goedecker and Hutter60 were used. Independent Gaussian-based polynomial functions up to angular momentum l = 3 (‘f’) were used to construct the basis set for the different atom species. The number of independent functions for each species were 50 for Mo (l ≤ 3), 28 for S, 38 for C, 40 for O (l ≤ 2), and 12 for H (l = 0), giving a highly versatile and transferable basis description. A finite electron temperature of 0.04 eV was used to aid self-consistent convergence. Absolute energies were converged in the self-consistency cycle to better than 10−5 Ha. Atomic positions and lattice parameters were geometrically optimized until the maximum atomic position change in a given iteration dropped below 10−4 a0 (a0: Bohr radius) and/or energy change less than 10−5 Ha.
While the LDA does not include explicit van der Waals interaction terms, its known over-binding ‘mimics’ these interactions. Additionally the focus of these calculations is on the relative covalent bonding behavior of the species, which is largely independent of the physisorbed pyrene moiety where van der Waals effects are most important. This can be seen in the similar relative energies between the basal and edge sites for the complete 1b molecule (1.96 eV) and the truncated molecule (3-methyl-1,2-dithiolane) at 2.03 eV, the same species as 1b without the pyrene moiety present.
The continuous monolayer MoS2 models use a hexagonal Mo64S128 repeating unit cell (8 × 8 supercell) with optimized lattice constant a = 25.02 Å, and a single k-point. Edge calculations use a Mo32S64 (Mo32S60) ribbon for the 100% (50%) S saturated molybdenum-edge, with a ribbon width of 8 MoS2 units. The supercell length in the direction of the slab is 4 MoS2 units, with cell lattice parameters of a = 63.50 Å, b = 12.51 Å and an angle between lattice vectors in the slab plane of 120°, with a 1 × 2 k-point grid. Edge calculations for the functionalization of 1,2-dithiolane-based derivative 1b used a doubled cell (Mo64S128) with single k-point. Unit cell parameters are fixed at the infinite monolayer value and all atoms are relaxed, with the exception in the edge calculations where the MoS2 units at the opposing edge are held fixed.
Large cells were used to avoid self-interaction, with a fixed interlayer distance of 31.1 Å (giving a vacuum spacing between MoS2 monolayers of 27.9 Å), and an in-plane vacuum spacing between edges of 34.1 Å. This ensures that the stable functionalized structures are all more than 23 Å from atoms in neighboring slabs.
Code availability
Further details of the code AIMPRO are available from the website, http://aimpro.ncl.ac.uk/. Calculations presented here used version 3.21. The code can be accessed by arrangement with the code authors Patrick Briddon and Mark Rayson at Newcastle University, UK, who should be contacted directly (contact details available through the website).
References
Voiry, D. et al. Enhanced catalytic activity in strained chemically exfoliated WS2 nanosheets for hydrogen evolution. Nat. Mater. 12, 850–855 (2013).
Wang, K. et al. Ultrafast saturable absorption of two-dimensional MoS2 nanosheets. ACS Nano 7, 9260–9267 (2013).
Huang, X., Zenga, Z. & Zhang, H. Metal dichalcogenide nanosheets: preparation, properties and applications. Chem. Soc. Rev. 42, 1934–1946 (2013).
Pospischil, A., Furchi, M. M. & Mueller, T. Solar-energy conversion and light emission in an atomic monolayer p–n diode. Nat. Nanotechnol. 9, 257–261 (2014).
Ross, J. S. et al. Electrically tunable excitonic light-emitting diodes based on monolayer WSe2 p–n junctions. Nat. Nanotechnol. 9, 268–272 (2014).
Pumera, M., Soferb, Z. & Ambrosi, A. Layered transition metal dichalcogenides for electrochemical energy generation and storage. J. Mater. Chem. A 2, 8981–8987 (2014).
Stephenson, T., Li, Z., Olsen, B. & Mitlin, D. Lithium ion battery applications of molybdenum disulfide (MoS2) nanocomposites. Energy Environ. Sci. 7, 209–231 (2014).
Radisavljevic, B., Radenovic, A., Brivio, J., Giacometti, V. & Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 6, 147–150 (2011).
Eda, G. et al. MoS2 Nanosheet phototransistors with thickness-modulated optical energy gap. Nano Lett. 12, 3695–3700 (2012).
Ou, J. Z. et al. Ion-driven photoluminescence modulation of quasi-two-dimensional MoS2 nanoflakes for applications in biological systems. Nano Lett. 14, 857–863 (2014).
Yin, W. et al. High-throughput synthesis of single-layer MoS2 nanosheets as a near-infrared photothermal-triggered drug delivery for effective cancer therapy. ACS Nano 8, 6922–6933 (2014).
Liu, T. et al. Drug delivery with PEGylated MoS2 nano-sheets for combined photothermal and chemotherapy of cancer. Adv. Mater. 26, 3433–3440 (2014).
Anbazhagan, R., Wang, H.-J., Tsai, H.-C. & Jeng, R.-J. Highly concentrated MoS2 nanosheets in water achieved by thioglycolic acid as stabilizer and used as biomarkers. RSC Adv. 4, 42936–42941 (2014).
Matte, H. S. S. R. et al. MoS2 and WS2 analogues of graphene. Angew. Chem. Int. Ed. 49, 4059–4062 (2010).
Mak, K. F., Lee, C., Hone, J., Shan, J. & Heinz, T. F. Atomically thin MoS2: a new direct-gap semiconductor. Phys. Rev. Lett. 105, 136805 (2010).
Zeng, Z. et al. An effective method for the fabrication of few-layer-thick inorganic nanosheets. Angew. Chem. Int. Ed. 51, 9052–9056 (2012).
Conley, H. J. et al. Bandgap engineering of strained monolayer and bilayer MoS2. Nano Lett. 13, 3626–3630 (2013).
Castellanos-Gomez, A. et al. Local strain engineering in atomically thin MoS2. Nano Lett. 13, 5361–5366 (2013).
Coleman, J. N. et al. Two-dimensional nanosheets produced by liquid exfoliation of layered materials. Science 331, 568–571 (2011).
Tang, Z., Weia, Q. & Guo, B. A generic solvent exchange method to disperse MoS2 in organic solvents to ease the solution process. Chem. Commun. 50, 3934–3937 (2014).
Chou, S. S. Ligand conjugation of chemically exfoliated MoS2. J. Am. Chem. Soc. 135, 4584–4587 (2013).
Knirsch, K. C. et al. Basal-plane functionalization of chemically exfoliated molybdenum disulfide by diazonium salts. ACS Nano 9, 6018–6030 (2015).
Pagona, G., Bittencourt, C., Arenal, R. & Tagmatarchis, N. Exfoliated semiconducting pure 2H-MoS2 and 2H-WS2 assisted by chlorosulfonic acid. Chem. Commun. 51, 12950–12953 (2015).
Voiry, D. et al. Covalent functionalization of monolayered transition metal dichalcogenides by phase engineering. Nat. Chem. 7, 45–49 (2015).
Liu, W., Yang, X., Zhang, Y. & Chen, M. X. H. Ultra-stable two-dimensional MoS2 solution for highly efficient organic solar cells. RSC Adv. 4, 32744–32748 (2014).
Liu, T. et al. Combined photothermal and photodynamic therapy delivered by PEGylated MoS2 nanosheets. Nanoscale 6, 11219–11225 (2014).
Nguyen, E. et al. Electronic tuning of 2D MoS2 through surface functionalization. Adv. Mater. 27, 6225–6229 (2015).
Kim, J.-S., Yoo, H.-W., Choi, H. O. & Jung, H.-T. Tunable volatile organic compounds sensor by using thiolated ligand conjugation on MoS2. Nano Lett. 14, 5941–5947 (2014).
Bakes, C. et al. Functionalization of liquid-exfoliated two-dimensional 2H-MoS2. Angew. Chem. Int. Ed. 54, 2638–2642 (2015).
Presolski, S. & Pumera, M. Covalent functionalization of MoS2. Mater. Today 19, 140–145 (2016).
Liu, T. et al. Drug delivery with PEGylated MoS2 nano-sheets for combined photothermal and chemotherapy of cancer. Adv. Mater. 26, 3433–3440 (2014).
Makarova, M., Okawa, Y. & Aono, M. Selective adsorption of thiol molecules at sulfur vacancies on MoS2 (0001), followed by vacancy repair via S–C dissociation. J. Phys. Chem. C 116, 22411–22416 (2012).
Wang, H. et al. Transition-metal doped edge sites in vertically aligned MoS2 catalysts for enhanced hydrogen evolution. Nano Res 8, 566–575 (2015).
Chen, X., Berner, N. C., Backes, C., Duesberg, G. S. & McDonald, A. R. Functionalization of two-dimensional MoS2: On the reaction between MoS2 and organic thiols. Angew. Chem. Int. Ed. 55, 5803–5808 (2016).
Jimenez-Sandoval, S. J., Yang, D., Frindt, R. F. & Irwin, J. C. Raman study and lattice dynamics of single molecular layers of MoS2. Phys. Rev. B 44, 3955–3962 (1991).
Nayak, A. P. et al. Pressure-dependent optical and vibrational properties of monolayer molybdenum disulfide. Nano Lett. 15, 346–353 (2015).
Backes, C. et al. Edge and confinement effects allow in situ measurement of size and thickness of liquid-exfoliated nanosheets. Nat. Comm. 5, 4576 (2014).
Li, Y., Zhou, Z., Zhang, S. & Chen, Z. MoS2 Nanoribbons: high stability and unusual electronic and magnetic properties. J. Am. Chem. Soc. 130, 16739–16744 (2008).
Wang, Z. et al. Mixed low-dimensional nanomaterial: 2D ultra narrow MoS2 inorganic nanoribbons encapsulated in quasi-1D carbon nanotubes. J. Am. Chem. Soc. 132, 13840–13847 (2010).
Lauritsen, J. V. et al. Size-dependent structure of MoS2 nanocrystals. Nat. Nanotechnol. 2, 53–58 (2007).
Hansen, L. P. et al. Atomic-scale edge structures on industrial-style MoS2 nanocatalysts. Angew. Chem. Int. Ed. 50, 10153–10156 (2011).
Lauritsen, J. V. et al. Hydrodesulfurization reaction pathways on MoS2 nanoclusters revealed by scanning tunneling microscopy. J. Catal. 224, 94–106 (2004).
Lauritsen, J. V. et al. Atomic-scale insight into structure and morphology changes of MoS2 nanoclusters in hydrotreating catalysts. J. Catal. 221, 510–522 (2004).
Schweiger, H., Raybaud, P., Kresse, G. & Toulhoat, H. Shape and edge sites modifications of MoS2 catalytic nanoparticles induced by working conditions: a theoretical study. J. Catal. 207, 76–87 (2002).
Bollinger, M. V., Jacobsen, K. W. & Nørskov, J. K. Atomic and electronic structure of MoS2 nanoparticles. Phys. Rev. B 67, 085410 (2003).
Arenal, R. & Stephan, O. in Advanced Transmission Electron Microscopy: Applications to Nanomaterials (eds Francis, L., Mayoral, A. & Arenal, R.) Ch. 5 (Springer, 2015).
Arenal, R. et al. Identification of electron donor states in N-doped carbon nanotubes. Nano Lett. 14, 5509–5516 (2014).
Suenaga, K. & Koshino, M. Atom-by-atom spectroscopy at graphene edge. Nature 468, 1088–1090 (2012).
Arenal, R. et al. Spatially-resolved EELS analysis of antibody distribution on biofunctionalized magnetic nanoparticles. ACS Nano 7, 4006–4013 (2013).
Jaramillo, T. F. et al. Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts. Science 317, 100–102 (2007).
Wang, H. et al. High electrochemical selectivity of edge versus terrace sites in two-dimensional layered MoS2 materials. Nano Lett. 14, 7138–7144 (2014).
Prodhomme, P.-Y., Raybaud, P. & Toulhoat, H. Free-energy profiles along reduction pathways of MoS2 M-edge and S-edge by dihydrogen: a first-principles study. J. Catal. 280, 178–195 (2011).
Dinter, N. et al. Temperature-programed reduction of unpromoted MoS2-based hydrodesulfurization catalysts: experiments and kinetic modeling from first principles. J. Catal. 267, 67–77 (2009).
Galea, N. M., Kadantsev, E. S. & Ziegler, T. et al. Modeling hydrogen sulfide adsorption on Mo-edge MoS2 surfaces under solid oxide fuel cell conditions. J. Phys. Chem. C 113, 193–203 (2009).
Deokar, G., Vignaud, D., Arenal, R., Louette, P. & Colomer, J.-F. Synthesis and characterization of MoS2 nanosheets. Nanotechnology 27, 075604 (2016).
Eda, G. et al. Photoluminescence from chemically exfoliated MoS2. Nano Lett. 11, 5111–5116 (2011).
Briddon, P. R. & Jones, R. LDA calculations using a basis of Gaussian orbitals. Phys. Status Solidi B 217, 131–171 (2000).
Rayson, M. J. & Briddon, P. R. Highly efficient method for Kohn-Sham density functional calculations of 500-10000 atom systems. Phys. Rev. B 80, 205104 (2009).
Briddon, P. R. & Rayson, M. J. Accurate Kohn–Sham DFT with the speed of tight binding: current techniques and future directions in materials modelling. Phys. Status Solidi B 248, 1309–1318 (2011).
Hartwigsen, C., Goedecker, S. & Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 58, 3641–3662 (1998).
Acknowledgements
This project has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie Grant agreement No 642742. C.B. is a research associate of FRS-FNRS. TEM studies were conducted at the Laboratorio de Microscopías Avanzadas, Instituto de Nanociencia de Aragón, Universidad de Zaragoza, Spain. Some of the research leading to these results has received funding from the European Union FP7 under Grant agreements 312483- ESTEEM2 (Integrated Infrastructure Initiative—I3). The Government of Aragon, and the European Social Fund are gratefully acknowledged. R.A. gratefully acknowledges the project “Construyendo Europa desde Aragon” 2014-2020 (grant number E/26) and the support from the Spanish Ministry of Economy and Competitiveness (MINECO) through project grants FIS2013-46159-C3-3-P and MAT2016-79776-P (AEI/FEDER, UE), as well as from the European Union H2020 program “Graphene Flagship”, Grant Agreement 696656.
Author information
Authors and Affiliations
Contributions
R.C.V. performed the synthesis of 1,2-dithiolanes, the exfoliation and functionalization of MoS2 and the characterization of the exfoliated and modified MoS2 hybrids. Y.S.A.B. performed and analyzed the theoretical calculations. M.P.F. imaged the exfoliated and functionalized MoS2 hybrids. R.A. guided the imaging and performed the spectroscopic analysis for the imaged samples. C.B. performed the XPS analysis. C.P.E. and N.T. designed, guided, supervised the study (theory and experiment, respectively), and co-wrote the manuscript. All authors revised and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Canton-Vitoria, R., Sayed-Ahmad-Baraza, Y., Pelaez-Fernandez, M. et al. Functionalization of MoS2 with 1,2-dithiolanes: toward donor-acceptor nanohybrids for energy conversion. npj 2D Mater Appl 1, 13 (2017). https://doi.org/10.1038/s41699-017-0012-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41699-017-0012-8
