
Abstract
Since its first fabrication by exfoliation in 2014, phosphorene has been the focus of rapidly expanding research activities. The number of phosphorene publications has been increasing at a rate exceeding that of other two-dimensional materials. This tremendous level of excitement arises from the unique properties of phosphorene, including its puckered layer structure. With its widely tunable band gap, strong in-plane anisotropy, and high carrier mobility, phosphorene is at the center of numerous fundamental studies and applications spanning from electronic, optoelectronic, and spintronic devices to sensors, actuators, and thermoelectrics to energy conversion, and storage devices. Here, we review the most significant recent studies in the field of phosphorene research and technology. Our focus is on the synthesis and layer number determination, anisotropic properties, tuning of the band gap and related properties, strain engineering, and applications in electronics, thermoelectrics, and energy storage. The current needs and likely future research directions for phosphorene are also discussed.
Similar content being viewed by others
Introduction
Since the discovery of graphene in 2004 (ref. 1), there has been a quest for new two-dimensional (2D) materials aimed at fully exploring new fundamental phenomena stemming from quantum confinement and size effects. This quest has spurred new areas of research with rapid growth from both theoretical and experimental fronts aimed at technological advancements. Among recently discovered 2D materials, phosphorene is one of the most intriguing due to its exotic properties and numerous foreseeable applications.2 This review discusses recent advances in phosphorene research with special emphasis on: (i) fabrication and techniques for rapid identification of the number of layers; (ii) anisotropic behavior; (iii) band gap and property tuning; (iv) strain engineering and mechanical properties; (v) devices and applications; and (vi) future directions.
2D materials composed of a single-atom-thick or a single-polyhedral-thick layer can be grouped into diverse categories.3,4,5 The first is comprised of layered van der Waals solids such as atomically flat graphene, h-BN, phosphorene, SiC, Si2BN, transition metal dichalcogenides (TMDs) (MX2 (X-M-X layer) where M=Ti, Zr, Hf, V, Nb, Ta, Mo, W, and X = S, Se, Te),6 layered metal oxides such as vanadium oxide and Sb2Te3, etc. The second category is comprised of layered ionic solids, which consist of a charged polyhedral layer sandwiched between hydroxide or halide layers by electrostatic forces such as La0.90Eu0.05Nb3O10, KLnNb2O7, Eu(OH)2.5(DS)0.5. Finally, the third category includes surface-assisted nanolayered solids such as silicene, germanene, stanene, etc. In a different approach, Gibaja and co-worker recently isolated few-layered antimonene by both liquid phase exfoliation7 and mechanical cleavage8 methods.
Graphene, the first demonstrated and the most explored 2D material, has a crystalline form of sp2 carbon atoms packed in a single-atom-thick planar honeycomb network. Its superior properties, including high carrier mobility, ultrahigh surface area, excellent thermal conductivity, and quantum confinement effect have been well documented.9 However, the lack of band gap is a serious limitation for the use of graphene in electronic devices. Lately, another unique 2D material consisting of a monolayer, or few layers, of black phosphorus (BP) has been attracting attention. BPs most remarkable properties include thickness-dependent band gap, strong in-plane anisotropy, and high carrier mobility. Recent reports have brought to light the highly encouraging prospects of using this novel 2D material in both nano- and optoelectronics. The key characteristics that make it so promising are high carrier mobility, strong in-plane anisotropy, particularly the anisotropy of electric conductance,10 and highly tunable band gap, which changes with doping, functionalization, and the number of layers from ~1.5 eV for a monolayer to 0.3 eV for bulk BP.11 The on/off ratio and the carrier mobility are also layer-dependent.12 Furthermore, compatibility of phosphorene with other 2D materials in so-called van der Waals heterostructures can offer solutions for important issues, such as surface degradation, doping, or control of surface/interface electronic structure, and can also enable novel functionalities and devices with unique or unprecedented performance. In the last 2 years, since the first experimental demonstration of phosphorene,13 research activities related to this material have rapidly expanded. The timeline reflecting this progress and how it compares to other 2D materials is summarized in Fig. 1a. The rapid take off in research activities on phosphorene is evident by the number of related publications that appeared in the last 2 years.

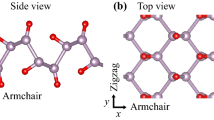
a Comparison of 2D materials in terms of number of published articles during a decade (The vertical axis is in log scale). KEY WORDS: "Graphene", "Hexagonal Boron Nitride", "Phosphorene", "Mxenes", "MoS2 or WS2 or MoSe2 or WSe2 or MoTe2", "Silicene or Germanene". SEARCH ENGINE: Google Scholar. b Crystal structure of phosphorene (side and top view). c Structures of three predicted polymorphs of phosphorene. d Crystal structures of monolayer of MoS2 (left) and graphene (right). Images c adapted with permission from ref. 26 [Nature Publishing Group], and ref. 34 [American Chemical Society]
Phosphorene basic properties
Phosphorene is a single atomic layer of BP, one of four known allotropes of phosphorus that shows semiconducting properties and was first discovered more than a century ago by Bridgman.14 BP, similar to graphite and other layered materials commonly referred to as van der Waals materials, is comprised of 2D sheets that are vertically stacked with respect to one another. However, unlike graphite, in-plane bonding in BP is due to sp3 hybridization.15 Each phosphorus atom is bonded to three adjacent phosphorus atoms; therefore, each “p” orbital retains a lone pair of electrons. The crystalline structure of layered bulk BP is orthorhombic with space group Cmca (#64) (ref. 16). Its structural and other properties have previously been discussed.11, 17 Due to the sp3 hybridization, phosphorene does not form atomically flat sheets like graphene.15 Instead, they form a puckered honeycomb-structured layers18 (Fig. 1b). These layers are stacked together by weak van der Waals forces.19 As a result of the puckered structure, each single layer of the honeycomb network contains two atomic layers where the distance between the two nearest atoms (d 1 = 2.224 Å) and the distance between top and bottom atoms (d 2 = 2.244 Å) are slightly different. The values of d 1 and d 2 are very close to each other because of the covalent bonds between phosphorus 3p orbitals.20 The electronic band gap of phosphorene has been predicted to be 1.5 eV (ref. 11), which is much higher than the 0.3 eV value of bulk BP.21, 22 Theoretical calculations indicate that the change in band gap is due to a loss of interlayer hybridization in few layer systems.12 Main phonon modes of phosphorene are defined as \({{\rm{A}}}_{{\rm{g}}}^{1}\) (out of plane), \({{\rm{B}}}_{2{\rm{g}}}\) and \({{\rm{A}}}_{{\rm{g}}}^{2}\) (in-plane).23 These modes have been predicted to be found at around 368, 433, and 456 cm−1, respectively.23
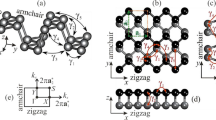
Due to its unique topological structure and differences between the armchair (AM) and zigzag (ZZ) directions, phosphorene displays strong in-plane anisotropy with many properties in these two principal directions drastically different. Anisotropy is strongly reflected in effective masses24 with an eight times increase in hole effective mass along the ZZ direction compared to the AM direction.12, 25 Phosphorene also has strong anisotropic phonon dispersions that lead to much larger group velocities along the ZZ direction than those along the AM direction.26,27,28 This is the reason for a strong orientation-dependent shear modulus29 and thermal transport.30 Strong anisotropic behavior, showing thickness and spectral dependence, is also observed for electron–photon and electron–phonon interactions.31 The anisotropy of phosphorene’s magnetic properties has also been predicted. In particular, for edges terminated along the ZZ direction, a dissipative quantum Hall effect has been calculated.32 Figure 2 lists some of the most significant anisotropic properties of phosphorene. Figure 2a, b show effective masses of the electron and hole in 2D phosphorene as a function of strain applied along the zigzag (x) and armchair (y) directions. It is evident that effective masses of charge carriers depend on the direction of charge transfer and also can change significantly by strain. However, the change of effective masses also depends on the direction of applied strain, which is a further indication of the anisotropic nature of phosphorene. Moreover, as it can be seen, the sharp changes in the effective masses in Fig. 2a, b occur around critical strains for the direct-indirect band gap transitions (e.g., the effective masses in the ZZ direction in Fig. 2a have a sudden change around critical strain ε x~8%). Additionally, based on effective masses in relaxed phosphorene (zero applied strain), one can conclude that AM is preferred direction for charge carriers’ transport as the effective masses are significantly smaller in this direction than ZZ direction.

Anisotropic properties of phosphorene. a, b Effective masses of the electron and hole under uniaxial strain “ε” in both zigzag “x” and armchair “y” directions, respectively. c Frequency of Raman modes of phosphorene (\({{\rm{A}}}_{{\rm{g}}}^{1}\), \({{\rm{B}}}_{2{\rm{g}}}\), and \({{\rm{A}}}_{{\rm{g}}}^{2}\)) as a function of uniaxial strain along zigzag and armchair directions. d, e Raman spectra of phosphorene as a function of uniaxial strain along zigzag and armchair directions, respectively. f, g Direction dependence of Young’s modulus and Poisson’s ratio of phosphorene. h Electron mobility of phosphorene along zigzag (μ x) and armchair (μ y) directions as a function of biaxial strain at room temperature. Images a–b, c–e, f, g, and h adapted with permission from ref. 79 [American Physical Society], ref. 23 [American Institute of Physics], ref. 33 [American Institute of Physics], ref. 72 [Royal Society of Chemistry], and ref. 128 [American Chemical Society], respectively
In Fig. 2c, Raman modes show significant shifts as a function of uniaxial strain. However, the rate of frequency shift depends on the direction of applied strained (i.e., ZZ and AM). The Atomic vibration of modes, orientation and amplitude of strain, and consequently variations of bond lengths are parameters that can determine the frequency shift in Fig. 2c.
In Fig. 2d, e, prominent peaks for relaxed phosphorene (\({{\rm{A}}}_{{\rm{g}}}^{1}\), \({{\rm{B}}}_{2{\rm{g}}}\), and \({{\rm{A}}}_{{\rm{g}}}^{2}\)) are located at 368, 433, and 456 cm−1, which are in good agreement with experimental results. These two plots shows that relative Raman peak positions and spacing can give information about the strain condition (i.e., amplitude and direction). As shown in Fig. 2d, all Raman peaks shift monotonically as a function of strain along AM direction. \({{\rm{B}}}_{2{\rm{g}}}\), and \({{\rm{A}}}_{{\rm{g}}}^{2}\) show red shift under stretching and blue shift under compression while \({{\rm{A}}}_{{\rm{g}}}^{1}\) modes shift in the opposite direction in both stretching and compression. In other words, energy spacing between \({{\rm{A}}}_{{\rm{g}}}^{1}\) and \({{\rm{B}}}_{2{\rm{g}}}\) reduces under compression and rises under stretching strain in the AM direction. In the case of strain along the ZZ direction (Fig. 2e) \({{\rm{B}}}_{2{\rm{g}}}\) and \({{\rm{A}}}_{{\rm{g}}}^{2}\) modes shift monotonically, i.e., blue shift under compression and red shift under stretching, which is in contrast with AM strain. However, \({{\rm{A}}}_{{\rm{g}}}^{1}\) modes exhibit redshift under both compression and stretching strain. Interestingly, energy spacing between \({{\rm{A}}}_{{\rm{g}}}^{1}\) and \({{\rm{B}}}_{2{\rm{g}}}\) shows clear potential to justify the direction of the strain. Their energy spacing reduces under stretching and increases under compression strain. Additionally, a significant increase of \({{\rm{B}}}_{3{\rm{g}}}^{2}\) peak intensity under stretching strain indicates the impact of ZZ strain on the intensity of Raman modes. Comparing Fig. 2d, e, one can see Raman modes are more sensitive to ZZ than AM strain. This is due to the phosphorene puckered structure that makes it softer along the AM than the ZZ direction. Calculated orientation dependence of Young’s modulus is illustrated in Fig. 2f. The Young’s modulus varies between a maximum of 166 GPa and a minimum of 44 GPa, which is along the ZZ and AM directions, respectively. Furthermore, the Young modulus has an average value of 94 GPa for all orientations.
Figure 2g presents the direction dependence of the Poisson’s ratio of phosphorene as another strong indication of anisotropic nature of phosphorene. The Poisson’s ratio along ZZ and AM directions is 0.703 and 0.175, respectively. However, it has the minimum value of 0.064 around θ = 47.5°. Figure 2h shows how the applied biaxial strain modifies the calculated electron mobility of phosphorene at room temperature (T = 300 K). Furthermore, under around 3% biaxial strain, the mobilities along ZZ and AM directions undergo the critical transition and, after this transition (switch), the ZZ mobility becomes larger than the AM mobility in contrast with the unstrained state of phosphorene. The AM mobility of the relaxed state at room temperature estimates around 2200 cm2 V-1 s-1, which is much smaller than that at T = 4K (1.6 × 105 cm2 V-1 s-1). However, smaller mobility at room temperature is due to additional effects that we may not need to consider at low temperature, such as electron-phonon scattering and band average.33
Unique properties of phosphorene can be further enhanced in allotrope structures. Based on computational work, a number of new phosphorene polymorphs/allotrope with buckled honeycomb structure or non-honeycomb structure has been predicted (e.g., non-honeycomb: ζ-P and η-P; honeycomb: β-P that is called blue phosphorene and γ-P).34 The prediction of several polymorphs adds more variety to the family of phosphorene-based materials and enables possibilities of potential novel phenomena and applications.34,35,36
Phosphorene fabrication methods
Since bulk BP is classified as a van der Waals material, similar top-down techniques as those employed to produce graphene using graphite as the precursor can also be applied to make phosphorene from BP. In such approach, bulk BP needs to be first obtained. Usual synthesis routes include high pressure,14, 37, 38 high-speed ball milling,39 and short transport reactions.40, 41 Polycrystalline BP can also be grown from red phosphorus under high pressure in an anvil press, as reported recently.42 The successful conversion of red phosphorus to BP has also been demonstrated through sonication alone,43 which further lowers the cost and simplifies the production process.
The interlayer cohesive energy of BP has been predicted at-151 meV per atom,44 which is somewhat larger than that of graphite at about −35 meV per atom.45 However, mechanical13 and liquid exfoliation46 of BP have already shown encouraging results. In 2014, two separate research teams reported the first successful isolations of a single atomic layer of BP using mechanical exfoliation.12, 47 Samples of a few layer-thick phosphorene can also be produced using liquid exfoliation.46, 48 Other techniques such as pulsed laser deposition,49 and plasma thinning50 have also been demonstrated to fabricate few-layer phosphorene using BP as the precursor material.
One of the major issues hindering phosphorene application in modern day devices is its lack of stability under ambient conditions. Due to the two uncoupled pairs of electrons in each phosphorus atom (caused by sp3 hybridization) and the enormously high surface to volume ratio, phosphorene is highly reactive to combinations of oxygen, water, and light.51 Under such conditions, it undergoes degradation where oxidized phosphorus52 and phosphoric acid species are formed.51 The most likely mechanism of the chemical degradation is oxidation followed by exothermic reaction with water.53
While this is still in an early development stage and more efforts are required, several approaches, especially of using protective capping layers,13 have already been evaluated for achieving higher stability phosphorene including encapsulating phosphorene by atomic layer deposition (ALD) of a dielectric layer (e.g., alumina),52 oxidizing top layers of phosphorene by oxygen plasma dry etching to protect remaining phosphorene along with ALD of alumina,54 van der Waals passivation by 2D graphene and/or h-BN,55 and solvent exfoliation with anhydrous organic solvents such as N-methylpyrrolidone (NMP).56 When using NMP, Kang et al. realized that residual NMP protects the phosphorene against fast degradation and postulated that this is potentially caused by NMP encapsulation or intercalation. Very soon after this work, Guo et al.57 reported that using saturated NaOH NMP solution for liquid exfoliation of bulk BP drastically increases the stability of phosphorene. They found that OH− ions were adsorbed onto phosphorene and prevent water from degrading the material. The results of their characterization show a clear improvement in comparison to reports by Kang et al. work. The significant role of solvents to stabilize the liquid-exfoliated phosphorene have been emphasized in other research by Hanlon et al.58 They also suggested that some solvents (e.g., N-cyclohexyl-2-pyrrolidone) can protect BP against oxidation via encapsulating the phosphorene during the liquid exfoliation. Interestingly, after removing the solvent, the sample remained intact under ambient conditions for several days, as the solvation shell still encapsulates it. Therefore, in the liquid exfoliation approach, selecting a solvent that forms the solvation shell in addition to having sufficient surface energy can enhance the quality of the production.
Determination of the number of layers
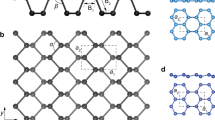
Several methods have been proposed in the literature for determination of the number of layers in phosphorene and BP flakes. However, to prevent unwanted reactions and material degradation, fast and accurate characterization techniques are needed. Atomic force microscopy (AFM) is one of the primary tools allowing measurements of the thickness down to one atomic layer, which for phosphorene is ~0.9 nm (ref. 12). However, the drawback of using AFM is that the analysis requires extended periods of time. An AFM image of a monolayer exfoliated phosphorene on a SiO2-coated silicon wafer with a thickness of ~0.85 nm is shown in Fig. 3a. This measured thickness is slightly larger than the theoretical value of 0.6 nm for a monolayer of phosphorene. However, this slight discrepancy is acceptable as it is observed in other 2D materials with SiO2/Si substrate.12

Determination of phosphorene thickness. a Atomic force microscopy image of a monolayer phosphorene. b Theoretical and PL-based phosphorene band gap as a function of the number of phosphorene layers. c Intensity ratio of \({{\rm{A}}}_{{\rm{g}}}^{1}\) Raman peak of phosphorene and Silicon Raman peak as a function of thickness. d Phase-shifting interferometry (PSI) image of phosphorene flake to determine the number of layers. e Raman spectra of bulk black phosphorus and 2–5 layers thick of exfoliated phosphorene. f \({{\rm{A}}}_{{\rm{g}}}^{2}\) and \({{\rm{B}}}_{2{\rm{g}}}\) Raman modes of phosphorene (right axis) and difference of them (left axis) as a function of number of layers of phosphorene. g Thickness dependence of intensity ratio of Raman modes \({{\rm{A}}}_{{\rm{g}}}^{1}\) and \({{\rm{A}}}_{{\rm{g}}}^{2}\) (ref. 50). h Layering number dependence of electron diffraction intensity ratio of (101) and (200) planes.18 i Optical contrast of 1–5 layers of phosphorene. Images a, b, c, and h, d, and e–g and i adapted with permission from ref. 12 [American Chemical Society], ref. 59 [American Chemical Society], ref. 18 [IOP Publishing], ref. 61 [Nature Publishing Group], and ref. [50] Springer, respectively
Photoluminescence (PL) is another determination method59 and is based on the fact that the band gap, which is represented by the excitonic emission in the PL spectrum, increases with the decrease of the number of layers. However, recent PL excitation spectroscopy suggests a quasiparticle band gap of 2.2 eV, suggesting an exciton binding energy of around as high as 0.9 eV (ref. 60).
Figure 3b illustrates the high dependence of phosphorene band gap on its layer number. Experimental band gap values in Fig. 3b are based on the PL spectroscopy measurements with 532 nm laser for excitation. Additionally, during the PL measurement, a flow of nitrogen gas was used to avoid degradation of few-layer phosphorene. PL peaks were detected at 1.29, 0.98, 0.88, and 0.80 eV for two to five layers, respectively. The band gap drops by an increase in the number of layers, which is in agreement with the trend predicted by theoretical values of the band gap (Fig. 3b).
Several Raman spectroscopy-based calibration methods have also been developed18, 50 as well as a method based on electron diffraction that uses a thickness dependent intensity ratio between the (101) and (200) diffraction spots.18 Thickness of few-layer phosphorene on silicone substrate can be determined by the intensity ratio between the silicon Raman peak and phosphorene \({{\rm{A}}}_{{\rm{g}}}^{1}\) peak as shown in Fig. 3c.
Recently, a fast, low-intensity and less destructive LED light-based phase-shifting interferometry (PSI) method has also been developed.61 Measurement of the layer number of phosphorene by the optical interferometry method is rapid, noninvasive, and accurate (Fig. 3d). In this approach, the optical path length (OPL) can be measured by PSI and evaluate the digitized interference pattern. Ultimately, by knowing the OPL value for each layering step change, the number of layers can be determined. A detailed summary of layer number determination techniques is depicted in Fig. 3. Figure 3 represents the Raman spectra of a few different thicknesses of exfoliated phosphorene. Thickness dependence of \({{\rm{A}}}_{{\rm{g}}}^{2}\) and \({{\rm{B}}}_{2{\rm{g}}}\) modes is shown in Fig. 3f. The frequency difference of these modes varies between 27.7 and 31 cm−1 for bulk and monolayer, respectively. As illustrated in Fig. 3g, the intensity ratio of \({{\rm{A}}}_{{\rm{g}}}^{1}\) to \({{\rm{A}}}_{{\rm{g}}}^{2}\) exhibits a monotonic behavior with respect to change in the number of layers, therefore, can be used as another means to determine the number of layers. As can be seen in Fig. 3i, the optical contrast of the phosphorene drops noticeably as the number of layers decrease. This dependence of optical contrast to the number of layers of phosphorene offers a practical resource of thickness measurement.
Subsequently, each mentioned approach has merits and weaknesses. In particular, AFM and Raman spectroscopy have been routinely used to reliably determine the sample thickness of TMD semiconductors with monolayer precision. However, these two methods are not reliable for the identification of very-few-layer phosphorene (one or two layers) under ambient conditions. The scanning rate of AFM is slow compared to the fast degradation of very-few-layer phosphorene under ambient conditions. AFM can also introduce potential contaminants that might affect further characterizations on the same sample. Unlike in TMD semiconductors, where Raman mode frequency has a monotonic dependence on the layer number, phosphorene has a non-monotonic dependence owing to the complicated Davydov-related effects. Moreover, the relatively high-power laser used in Raman spectroscopy can significantly damage the phosphorene samples. Due to the aforementioned challenges, optical interferometry offers a rapid, noninvasive, and highly accurate approach to determine the layer number.61
Property tuning: heterostructures, doping, surface functionalization, and composites
Figure 4 summarizes phosphorene band gap variation for various methods reported in the literature. As mentioned, the band gap can be increased by reducing the number of layer from the bulk value of 0.3 to ~1.5 eV for a monolayer. However, more research has recently been focused on tuning the band gap and also altering other properties of phosphorene through functionalization and doping. It has been found, for example, that oxygen adsorbs strongly onto the reactive edges of phosphorene and can increase its stability.62, 63 Similarly, covalent diazonium functionalization of phosphorene suppresses its chemical degradation and significantly improves its stability.64 Other studies have found that the addition of Cu adatoms65 or chemical doping with benzyl viologen66 can be used as an effective electron doping strategy to obtain n-type conductivity. It has also been found that doping, even with non-magnetic atoms such as oxygen, sulfur, or selenium, can induce magnetism in phosphorene.63 This is believed to be due to that fact that such dopant atoms only bond to two phosphorus atoms leaving an unpaired electron. Similar effects have been calculated for blue phosphorene.67

Another means of affecting properties of phosphorene is by applying external electric fields which, in some cases can even lead to semiconductor-to-metallic transitions.68 Combining phosphorene with other materials and forming heterostructures or interfaces has also being explored as means of altering material properties. For example, graphene/phosphorene stacked heterostructures are promising systems since the anisotropy of the electric polarization of phosphorene has been calculated to disappear in such structures.68 Of interest are also interfaces between phosphorene and high-k dielectric materials due to their ability to improve the performance of scaled electronics. For example, heterostructures of phosphorene and hafnium dioxide (HfO2) have been found to induce topological changes in phosphorene at elevated temperatures, which could alter device performance.69 Highly effective photovoltaic systems, with exciton recombination times up to 1.08 ps and over 98% quantum efficiency, have also been predicted theoretically in heterostructures of phosphorene and TiO2 (ref. 70).
Effects of stress and mechanical properties
Strain engineering or applications of pressure can also be used to modify properties of BP or phosphorene. At high pressures, BP undergoes crystal structure transitions, first from an orthorhombic to a rhombohedral structure at ~5 GPa and then to a simple cubic structure at ~10 GPa (ref. 71). Lower pressures or strains have also significant impact on electronic and other properties of phosphorene. For example, the application of uniaxial stress can break the symmetry and enhance in-plane anisotropy.72 Other effects include electron–phonon coupling enhancement,73 superconductivity,74 semiconductor to metal transition,75 band gap modification,15 and direct-indirect band gap transition12 (Fig. 5). There have been several theoretical studies on the effect of strain on the electronic structure of single- and/or few-layer phosphorene. density functional theory (DFT) calculations by Ju et al.76 of few-layer (1–5 layers) BP have indicated that compressive strain can result in a semiconductor-metal transition (SMT), whereas the biaxial tensile strain only affects the band gaps and does not affect essentially the electronic properties of the system. Based on the band structure and charge density calculations, they have predicted that the compressive strain leads to the reduction of P–P bond length, downward movement of the conduction band, and increased delocalization of states around the Fermi level, which at the critical compressive strain are essential reasons of SMT. Indeed, the calculations have shown a reverse relation between the critical compressive strain for the SMT and the thickness of few-layer BP. In a similar DFT study of bilayer phosphorene, Manjanath et al.75 found strong interlayer interactions between phosphorpus pz orbitals and a reversible SMT at ~13% of normal compressive strain. They also observed a unique band gap modulation pattern leading to a direct-indirect band gap transition at ~3% of strain (Fig. 5c). Furthermore, the study has suggested high structural integrity at relatively high strains, as demonstrated by the absence of negative frequencies in phonon spectra. Raman active modes (\({{\rm{A}}}_{{\rm{g}}}^{1}\), \({{\rm{B}}}_{2{\rm{g}}}\) ,and \({{\rm{A}}}_{{\rm{g}}}^{2}\)) have been found to increase linearly with normal compressive strain, with the highest increase rate for the out-of-plane \({{\rm{A}}}_{{\rm{g}}}^{1}\) mode due to strong interlayer interaction (Fig. 5a). In another study, Seixas et al.77 have analyzed the effects of uniaxial compression on the optical response of phosphorene and the exciton stability. They have found that under high pressure, the material gradually undergoes the transition into an indirect gap semiconductor and eventually into a semimetal. They have also shown that uniaxial strain affects effective masses and, in consequence, the exciton anisotropy and binding strength. They have calculated the exciton binding energy of 0.87 eV for 5% of strain, which is among the highest value ever reported for 2D materials. Additionally, they have suggested that due to its strong and linear strain dependence, luminescence peak can be used to probe directly the strain state of single layer phosphorene as an alternative to using Raman peaks, which are weakly affected by AM strain.23 However, the use of PL as an experimental probe to study strain in phosphorene may not be reliable due to the lack of proper understanding of the origin of the PL in the material. It is well known that PL response due to the excitons and trions strongly depend on the dielectric environment, defects, surface states etc. Consequently, many important fundamental properties, such as exciton and trion dynamics, remain underexplored.61 The origin of the reported emissions due to highly anisotropic bright excitons and quasi particle band gap in phosphorene are a still topic of debate.60 This makes PL a less reliable technique to directly probe the strain state of single-layers of phosphorene.

a Raman frequencies as a function of normal compressive (NC) strain for three Raman active modes. b The stress dependence of strain for a monolayer phosphorene under isotropic strain. c Band gap change as a function of strain for Perdew Burke-Ernzerhof (PBE), HeydScuseria-Ernzerhof (HSE) and G0W0 methods. d Band gap of the monolayer phosphorene (MLP) as a function of the strain. Images a, b, c, and d adapted with permission from ref. 75 [IOP Publishing], ref. 134 [IOP Publishing], ref. 132 [American Physical Society], and ref. 135 [IOP Publishing], respectively
Phosphorene shows unique mechanical properties, which make it a promising novel material for nanoelectromechanical systems and devices.78 It can sustain tensile strains of up to 30% (refs 33, 79), which is a higher value of those reported for graphene or single layer MoS2 (ref. 33). Phosphorene has a negative Poisson’s ratio that is attributed to its unique puckered structure.80 It shows higher ductility for the AM direction compared to the ZZ direction and good mechanical flexibility, as indicated by its Young’s modulus value lower that those reported for graphene and single layer MoS2 (ref. 81).
Based on the remarkable mechanical properties, phosphorene has a potential as an efficient filler to reinforce polymers and composites. For example, incorporation of phosphorene (0.3 vol%) into polyvinylchloride (PVC) substantially enhanced its mechanical properties. The remarkable improvement of all mechanical parameters PVC after phosphorene reinforcement such as Yang modulus, composite strength, and tensile toughness make it a great competitor for graphene reinforcement.58
Applications of phosphorene
Electronic devices and sensors
Some of the first studies of phosphorene-based devices involved studying the basic properties of field effect transistors (FETs).12 The hole mobility in phosphorene is over 1000 cm2 V−1 s−1 (ref. 10), whereas phosphorene 2D electron gas shows exceptional Hall mobility of up to 6000 cm2 V−1 s−1 and quantum Hall effect in high-quality van der Walls heterostructures.82 Recent studies have also found that trions and excitons in phosphorene can be tuned by using a transistor topography set-up.83 It is noted that trions can be used in spintronic-based devices due to their non-zero spin.83 By having more control over hole–electron interactions, efficient optoelectronic and spin-based devices can be fabricated. In high-quality phosphorene-based heterostructures with negative compressibility, a many-body effects strong correlations lead to an enhanced capacitance in 2D electronic systems, suggesting potential for low-power nanoelectronics and optoelectronics.82 The anisotropy of transport in phosphorene can also be incorporated into devices. FET device performance and speed may be increased by orientating the conducting channel along the AM direction.25 The AM direction has lower effective mass, which increases ON current.25 Transistors based on heterostructures with high-k dielectrics have shown ~8 times increase of the ON current when utilizing phosphorene’s anisotropy.84 In addition to several phosphorene-based transistors based on in-plane transport geometry, first high performance structures with a vertical FET geometry, such as a graphene/phosphorene van der Walls heterostructure,85 have also been realized recently. Figure 6a, illustrate the application of phosphorene in a tunable photoinduced-carrier-transported FET.86

Thermoelectric (a–c), electronic (d–f), and sensor (g–i) applications of Phosphorene. a A schematic illustration of an electric-double-layer transistor (EDLT) for thermoelectric measurements. b Tunable thermoelectric power of a BP-EDLT as a function of gate voltage at T = 210 K (inset: the optical microscope image of the real thermoelectric device of a BP flake with a thickness of 40 nm). c Theoretical calculations of thermoelectric power S x and S y for single-layer, bilayer, and five-layer phosphorene and bulk BP (from top to bottom) as a function of volumetric carrier density dependence at T = 210 K. d top: Schematic image of a BP/TiO x transistor under illumination (λ = 365 nm, power = 3.5 × 10–4 μW μm−2); bottom: Mechanism of photoinduced charge transfer at the TiO x /BP interface. e Evolution of conductivity vs. gate-voltage curves for the TiO x /BP FET with increased irradiation time. The inset is the one for a pristine BP under the same illumination condition. f Recovery process of photoinduced doping by applying a positive bias. Log-scale curve of the device after photodoping was given in the inset. g Black phosphorus nanosheets current–voltage (I–V) characteristics at different relative humidity (RH), h sensitivity vs. RH plot (inset top: schematic diagram of the typical transistor device; bottom: optical photograph of the real transistor device), i typical current-time plot for three cycles of humidity switching between 32 and 97%. Images a–c, d–f, and g–i adapted with permission from ref. 136 [American Chemical Society], ref. 86 [American Chemical Society], and ref. 137 [American Chemical Society], respectively
The essential factors of materials for transistor applications include high charge carrier mobility, high conductivity, high on/off ratio and low off-state conductance. Comparing all these 2D materials, the carrier mobility of graphene can reach to 200,000 cm2 V−1 s−1 (ref. 87), but it difficult to switch on/off due to its zero band gap and semimetallic nature, which limits its use as a digital logic device.88 TMDs layers such as MoS2 usually have band gap around 1.5 eV can provide high on/off ratio, the low carrier mobility (200 cm2 V−1 s−1, even lower) limits the performance on transistor applications. Phosphorene seems to satisfy all the key factors for a transistor material. The band gap of 0.3–2 eV ensures a large on/off ratio and its carrier mobility has been found over 1000 cm2 V−1 s−1 (ref. 47), which can produces reasonably fast operations.
2D Mott’s variable range hopping is a dominant in-plane electrical and thermoelectric transport mechanism.89 For the out-of-plane electronic transport in hetereostructures, thermionic emission over the Schottky barrier determines charge transport at high temperatures, whereas hopping transport dominates only at low temperatures.85
Phosphorene-based device structures show high photo-sensitivity and high-performance broad spectral range photodetectors have been demonstrated.90 Solar cells have also been suggested, as energy conversion efficiencies as high as 20% have been predicted for heterojunctions involving phosphorene with hydrogen and fluorine.91 Experimentaly, first in-plane p-p junctions have recently been fabricated using chemical doping and its considerable performance has been shown.66
Due to the lone electron pair on each phosphorus atom in phosphorene, it is also a very promising material for gas sensing applications.92 Organic molecules such as tetracyano-p-quinodimethane (TCNQ) have been predicted to act as acceptors once absorbed onto phosphorene.93 TCNQ provides shallow states near the valence band, which enhances p-type conductivity. On the contrary, the doping with electron donor tetrathiafulvalene does not enhance n-type conductivity, as the introduced donor states are too deep into the band gap. Only by applying out of plane electric fields or in-plane tensile strain n-type conductivity can be enhanced in this case.
First principal studies have found that for gas species containing nitrogen-based molecules, transport properties in phosphorene may be altered by as few as one molecule.94 Single molecule detection would render phosphorene as one of the most sensitive gas sensors ever created. Charge transfer between physisorbed gas molecule and phosphorene can be enhanced by introducing structural ripples, as the surface curvature increases the interaction between the molecule and phosphorene atoms.95 The introduction of dopants into phosphorene is another method of increasing gas-sensing abilities. For example, it has been calculated that for Ca doped or Ca-decorated phosphorene, the binding affinity towards gas molecules increases.96 Ultrahigh sensitivity and layer-dependent sensing performance of phosphorene-based gas sensors have been reported by performing the testing in an air-tight chamber.97 But this poses many practical problems for it to be implemented a real gas sensor. However, few-layer BP nanosheets, produced by liquid phase exfoliation under ambient conditions in solvents such as N-cyclohexyl-2-pyrrolidone (CHP) have been found to be surprisingly stable probably due to the solvation shell protecting the nanosheets from reacting with water or oxygen.58 The reactions are believed to occur only at the nanosheet edge. Biological agents have also been shown to have an impact on the electrical resistance of phosphorene.98 For example, a lower detection limit of about ten ng ml−1 for human immunoglobulin G, which is one of the main antibodies found in humans, has been reported,98 suggesting that phosphorene-based sensors may be used to detect immune system disorders in humans.
Thermoelectric applications
All electronic devices generate heat during operation. Therefore, it is advantageous to utilize the thermoelectric materials as the FET channel to find a high efficient chip-cooling method by converting the waste dissipation heat to electrical energy and also to protect the device against performance degradation and breakdown. It has been shown that direct conversion between thermal and electrical energy using thermoelectric properties can play an important role to answer future energy and environmental concerns.
Efficient conversion between thermal and electrical energy can be realized by low thermal and high electrical conductivity at the same time. Subsequently, phosphorene, with much lower thermal conductivity than graphene, has great potential for novel thermoelectric applications.99 It also shows strong thermal anisotropy and the orientation-dependent figure of merit ZT value that is larger along the AM than the ZZ direction. However, the Seebeck coefficient is practically isotropic28, 100,101,102 (Fig. 6a). Preferred electrical and thermal transport are orthogonal in phosphorene and large ratios of electrical over thermal conductivity can be achieved.101 Several theoretical studies revealed that strain engineering has significant influence on the ZT value, therefore, optimized conditions and strain engineering make it possible to reach very large ZT values.101,102,103,104 Theoretical calculations indicate that photocurrent response can be used to measure thermoelectric properties of BP indirectly. As calculated by Hong et al.105, the generated electron–hole pairs contribute to photocurrent, which leads to the voltage difference and the Seebeck coefficient difference between BP and the electrode. The study by Low et al.106 has demonstrated that the photocurrent is dominated by thermally driven thermoelectric and bolometric processes rather than the photovoltaic effect. Recently, Flores et al.107 have been able to measure thermoelectric properties directly. Figure 6 shows three recent examples of phosphorene devices.
Energy storage
BP has a high theoretical Li-storage capacity of 2596 mAh g−1 (ref. 108, 109), but suffers from the large volume change of ~300% and rapid capacity loss during charging-discharging cycling when used as an anode material in Li-ion batteries.110 Phosphorene does not show such problems and can also provide additional advantages thanks to its 2D structure properties. It shows the reversible capacity of ~433 mA h g−1, low open circuit voltage, small volume change, and good electrical conductivity.111 The diffusion of Li in phosphorene is ultrafast and anisotropic112 (1.6 × 109 times faster along the ZZ direction than along the AM direction, and 260 times faster than in graphite). Energy barriers for the Li diffusions are very low (~0.09 eV)112, 113 suggesting excellent rate capabilities. Due to significant energy barriers difference (Fig. 6a), Li diffusion takes place mostly between grooves.11
Relatively weak (~1.9 eV) binding energy of Li in phosphorene can be significantly increased (even by ~1 eV) by the introduction of intrinsic point defects.11 However, defects can also cause a significant increase of Li diffusion barriers, like in the case of Stone-Wales defects, which increase the diffusion barrier for the ZZ direction from −0.17 to 0.49 eV. Defects, however, can also lead to new diffusion channels and effectively enhance the Li diffusion. For example, recent DFT study has shown that vacancy can allow for the Li atoms to diffuse between two adjacent grooves with a low energy barrier of 0.13 eV (ref. 11). The binding energy of Li can also be enhanced in heterostructured composites. DFT calculations for phosphorene/graphene composite indicate a significant increase in binding energy to 2.6 eV without affecting the high mobility of Li within the layers.11
Electrochemical performance of phosphorene is predicted to be further improved in nanoribbons especially, in ZZ nanoribbons, where a moderate working voltage (0.504–0.021 V), high capacity (541 mA h g−1), and fast charge/discharge rate is calculated.114 Compared to phosphorene (1.98 eV), there is a moderate enhancement of the Li binding energy for AM (2.18–2.50 eV) and ZZ (2.07–2.52 eV) phosphorene nanoribbons (PNRs) due to the presence of unique edge states. However, a strong anisotropy of the Li diffusion, with a much lower barrier for the diffusion within two neighboring grooves (0.09 eV) than across a grove (0.65 eV), is also calculated for the nanoribbon morphology.115, 116 In addition, similarly, to pristine phosphorene, room temperature diffusion constants inside the groove in AM and ZZ nanoribbons are about 109–1011 times larger than between two adjacent grooves.
In addition to LIBs, phosphorene has also been considered for other electrochemical energy storage applications, such as all-solid-state supercapacitors117 and Na ion batteries (NaIBs),118 which are considered safer than LIBs and use more abundant Na ions. The promise of using phosphorene for NaIB anode materials comes from the fact that, unlike the traditional, carbon-based anode materials for LIBs, it can accommodate Na ions, which are significantly larger than Li ions (2.04 vs. 1.52 Å). Because of this, BP with its larger interlayer channel size (3.08 vs. 1.86 Å for graphite) is a highly promising NaIB anode material with a theoretical specific capacity of ~2600 mAh g−1, which dwarfs that of any other present materials. Recent DFT study of the intercalation of Na in BP119 and few-layer phosphorene118 has demonstrated a fast (energy barrier of 0.04 eV) and anisotropic (Fig. 6b) Na diffusion and, additionally, that phosphorene undergoes SMT at high Na intercalation concentration.118 The preferred Na ion transport along the ZZ edge compared to that along the AM edge has also been shown recently by in-situ aberration-corrected TEM study.120 Furthermore, high performance NaIB anode in the form of BP/Ketjenblack-multiwalled carbon nanotubes composite has also been recently obtained using ball milling.121 While most of the reports on the use of phosphorene for electrochemical energy storage applications are theoretical DFT-based calculations,11, 111, 112, 122 some first experimental studies have also been reported,123, 124 including the work of Cui et al.97, on the mechanism of sodiation in BP and hybrid material of few-layer phosphorene sandwiched between graphene (P/G) investigated using in-situ TEM and ex-situ XRD methods.123 This study showed a two-step mechanism of intercalation of sodium along the x-axis followed by alloying and formation of Na3P. The strong anisotropic volume expansion (estimated at 0, 92, and ~160%, along the x, y, and z-axis, respectively) observed in few-layer phosphorene samples was found to be suppressed in P/G nanocoposite electrodes. This was due to graphene layers, which accommodated the volumetric change and provided high electrical conductivity pathways. This resulted in high specific capacity of ~2440 mAh g−1 (calculated using the mass of phosphorus only) at a current density of 0.05 A g−1 and good capacity retention of 83% after 100 cycles between 0 and 1.5 V (Fig. 6c). In another in-situ TEM study, Xu et al.124 investigated the thickness-dependent lithiation/delithiation mechanims and have found significant capacity and electrical conductivity losses in thick multilayer phosphorene due to structural decomposition and irreversibility of lithiated Li3P phase during delithiation processes. However, few-layer phosphorene samples have shown much better lithiation/delithiation cycling performance. Interestingly, in-situ lithiation of an individual phosphorene nanoflake have shown nearly the same rate of expansion of ~35% for both longitudinal and lateral directions, suggesting the same diffusion rate of Li+ ions in the stark contrast to highly anisotropic Li+ diffusion predicted by several DFT studies (Fig. 7).

a Energy barrier and diffusion paths of Li atom inside and between two adjacent grooves of monolayer phosphorene. b Diffusion paths and energy barriers for the Na diffusion on monolayer phosphorene. c Schematic of structural evolution of the sandwiched P/G structure during sodiation (top), TEM images and Raman spectra of P/G hybrid (bottom-left), and reversible desodiation capacities of various phosphorene–graphene electrodes with different carbon–phosphorus mole ratios (C/P) between 0.02 and 1.5 V at a current density of 0.05 A g−1. Images a, b, and c adapted with permission from ref. 11 [Royal Society of Chemistry], ref. 118 [Royal Society of Chemistry], and ref. 123 [Nature Publishing Group], respectively
One of the reasons for discrepancies between experiments and DFT-predicted properties may stem from deficiencies of DFT in capturing the nature of interlayer interactions between phosphorene layers, as shown recently using quantum Monte Carlo calculations by Shulenburger et al.125 According to this study, the interlayer interaction is associated with a significant charge redistribution caused by changes in the nonlocal correlation of electrons in adjacent layers, which cannot be captured by DFT methods based on purely dispersive forces and van der Waals (vdW) corrected functionals. DFT with semilocal vdW correction terms does not adequately capture weakly bonded systems including BP and few layer phosphorene, which are not strictly van der Waals solids.19
As mentioned, intercalating Li or Na atoms between phosphorene layers leads to SMT.126 DFT calculations indicate also that intercalating Li atoms between bilayer phosphorene can cause a transformation from a direct-gap semiconductor to a BCS (Bardeen, Cooper, and Schrieffer) superconductor.127
Future research
For practical applications, fabricating large-scale phosphorene two-dimensional (2D) materials for future electronic devices and/or flexible devices needs to be addressed. In addition, there is much experimental research still needed in areas such as thermoelectric studies, magnetism, band structure, and optical properties. The main reason for scare experimental result in these areas is due to the aforementioned lack of large area synthesis methods. Chemical vapor deposition (CVD) of phosphorene may be a possible route for large scale synthesis. While CVD development of wafer-scale synthesis methods of graphene and TMDs has enabled large-scale device fabrication, to date, investigations of chemical synthesis of large-area and uniform phosphorene has not been reported. The significant barriers to direct chemical growth strategies of phosphorene are probably due to its chemically active surface that is fragile when exposed to air, as well as the absence of a suitable substrate for its CVD growth.
However, the recent development on growth of single crystals of orthorhombic BP from red phosphorus with Sn/SnI4 provides a new possibility. Sample sizes of several millimeters can be realized with high crystal quality and purity, making it possible for large-area preparation of single- or multi-layer phosphorene. Interestingly, one major benefit is the possible precursor red phosphorus is very abundant and relatively inexpensive. Using pre-formed bulk black phosphors as a CVD precursor may not be a viable route due to its current expense. Furthermore, the successfully synthesis of monolayer silicene, germanene and stanene with strong chemically active surface on the substrates (for instance silicene grown on Ag substrate) provides a good example for possible CVD growth of phosphorene.
References
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Eswaraiah, V., Zeng, Q., Long, Y. & Liu, Z. Black phosphorus nanosheets: synthesis, characterization and applications. Small 12, 3480–3502 (2016).
Butler, S. Z. et al. Progress, challenges, and opportunities in two-dimensional materials beyond graphene. ACS Nano 7, 2898–2926 (2013).
Bhimanapati, G. R. et al. Recent advances in two-dimensional materials beyond graphene. ACS Nano 9, 11509–11539 (2015).
Xu, M., Liang, T., Shi, M. & Chen, H. Graphene-like two-dimensional materials. Chem. Rev. 113, 3766–3798 (2013).
Duan, X., Wang, C., Pan, A., Yu, R. & Duan, X. Two-dimensional transition metal dichalcogenides as atomically thin semiconductors: opportunities and challenges. Chem. Soc. Rev. 44, 8859–8876 (2015).
Gibaja, C. et al. Few-layer antimonene by liquid-phase exfoliation. Angew. Chem. Int. Ed. doi:10.1002/anie.201605298 (2016).
Ares, P. et al. Mechanical isolation of highly stable antimonene under ambient conditions. Adv. Mater. 28, 6332–6336 (2016).
Ferrari, A. C. et al. Science and technology roadmap for graphene, related two-dimensional crystals, and hybrid systems. Nanoscale 7, 4598–4810 (2015).
Xia, F., Wang, H. & Jia, Y. Rediscovering black phosphorus as an anisotropic layered material for optoelectronics and electronics. Nat. Commun.5 (2014).
Guo, G.-C., Wei, X.-L., Wang, D., Luo, Y. & Liu, L.-M. Pristine and defect-containing phosphorene as promising anode materials for rechargeable Li batteries. J. Mater. Chem. A 3, 11246–11252 (2015).
Liu, H. et al. Phosphorene: an unexplored 2D semiconductor with a high hole mobility. ACS Nano 8, 4033–4041 (2014).
Liu, H., Neal, A. T., Si, M., Du, Y. & Ye, P. D. The effect of dielectric capping on few-layer phosphorene transistors: tuning the schottky barrier heights. IEEE Electron Device Lett. 35, 795–797 (2014).
Bridgman, P. W. Two new modifications of phosphorus. J. Am. Chem. Soc. 36, 1344–1363 (1914).
Rodin, A. S., Carvalho, A. & Castro Neto, A. H. Strain-induced gap modification in black phosphorus. Phys. Rev. Lett. 112, 176801 (2014).
Brown, A. & Rundqvist, S. Refinement of the crystal structure of black phosphorus. Acta Crystallogr. 19, 684–685 (1965).
Du, H. W., Lin, X., Xu, Z. M. & Chu, D. W. Recent developments in black phosphorus transistors. J. Mater. Chem. C 3, 8760–8775 (2015).
Castellanos-Gomez, A. et al. Isolation and characterization of few-layer black phosphorus. 2D Materials 1, doi:10.1088/2053-1583/1/2/025001 (2014).
Appalakondaiah, S., Vaitheeswaran, G., Lebegue, S., Christensen, N. E. & Svane, A. Effect of van der Waals interactions on the structural and elastic properties of black phosphorus. Phys. Rev. B 86, doi:10.1103/PhysRevB.86.035105 (2012).
Takao, Y., Asahina, H. & Morita, A. Electronic structure of black phosphorus in tight binding approach. J. Phys. Soc. Jpn. 50, 3362–3369 (1981).
Warschauer, D. Electrical and optical properties of crystalline black phosphorus. J. Appl. Phys. 34, 1853–1860 (1963).
Akahama, Y., Endo, S. & Narita, S. Electrical-properties of black phosphorus single-crystals. J. Phys. Soc. Jpn. 52, 2148–2155 (1983).
Fei, R. & Yang, L. Lattice vibrational modes and Raman scattering spectra of strained phosphorene. Appl. Phys. Lett. 105, 083120 (2014).
Lam, K.-T., Dong, Z. & Guo, J. Performance limits projection of black phosphorous field-effect transistors. IEEE Electron Device Lett. 35, 963–965 (2014).
Ameen, T. A., Ilatikhameneh, H., Klimeck, G. & Rahman, R. Few-layer phosphorene: an Ideal 2D material for tunnel transistors. arXiv preprint arXiv:1512.05021 (2015).
Jain, A. & McGaughey, A. J. H. Strongly anisotropic in-plane thermal transport in single-layer black phosphorene. Sci. Rep. 5, doi:10.1038/srep08501 (2015).
Zhu, L., Zhang, G. & Li, B. Coexistence of size-dependent and size-independent thermal conductivities in phosphorene. Phys. Rev. B 90, doi:10.1103/PhysRevB.90.214302 (2014).
Ong, Z.-Y., Cai, Y., Zhang, G. & Zhang, Y.-W. Strong thermal transport anisotropy and strain modulation in single-layer phosphorene. J. Phys. Chem. C 118, 25272–25277 (2014).
Cai, Y. et al. Giant phononic anisotropy and unusual anharmonicity of phosphorene: interlayer coupling and strain engineering. Adv. Funct. Mater. 25, 2230–2236 (2015).
Jang, H., Wood, J. D., Ryder, C. R., Hersam, M. C. & Cahill, D. G. Anisotropic thermal conductivity of exfoliated black phosphorus. Adv. Mater. 27, 8017–8022 (2015).
Ling, X. et al. Anisotropic electron-photon and electron-phonon interactions in black phosphorus. Nano. Lett. 16, 2260–2267 (2016).
Ostahie, B. & Aldea, A. Phosphorene confined systems in magnetic field, quantum transport, and supperradiance in the quasi-flat band. arXiv preprint arXiv:1512.04793 (2015).
Wei, Q. & Peng, X. Superior mechanical flexibility of phosphorene and few-layer black phosphorus. Appl. Phys. Lett. 104, 251915, doi:10.1063/1.4885215 (2014).
Wu, M., Fu, H., Zhou, L., Yao, K. & Zeng, X. C. Nine new phosphorene polymorphs with non-honeycomb structures: a much extended family. Nano. Lett. 15, 3557–3562 (2015).
Guan, J., Zhu, Z. & Tomanek, D. Phase coexistence and metal-insulator transition in few-layer phosphorene: a computational study. Phys. Rev. Lett.113, doi:10.1103/PhysRevLett.113.046804 (2014).
Zhu, Z. & Tománek, D. Semiconducting layered blue phosphorus: a computational study. Phys. Rev. Lett. 112, 176802 (2014).
Bridgman, P. W. Effects of high shearing stress combined with high hydrostatic pressure. Phys. Rev. 48, 825–847 (1935).
Shoichi, E., Yuichi, A., Shin-ichi, T. & Shin-ichiro, N. Growth of large single crystals of black phosphorus under high pressure. Jpn. J. Appl. Phys. 21, L482 (1982).
Park, C. M. & Sohn, H. J. Black phosphorus and its composite for lithium rechargeable batteries. Adv. Mater. 19, 2465–2468 (2007).
Köpf, M. et al. Access and in situ growth of phosphorene-precursor black phosphorus. J. Cryst. Growth 405, 6–10 (2014).
Nilges, T., Kersting, M. & Pfeifer, T. A fast low-pressure transport route to large black phosphorus single crystals. J. Solid State Chem. 181, 1707–1711 (2008).
Xuesong, L. et al. Synthesis of thin-film black phosphorus on a flexible substrate. 2D Mater. 2, 031002 (2015).
Moran, S., Khakoo, U., Barron, M., Nayak, A., Akinwande, D. Effect Of Ultrasonic Irradiation On The Allotropes Of Phosphorus, Proceedings of The National Conference On Undergraduate Research (NCUR) 2015 16–18 (Eastern Washington University, Cheney, WA, 2015).
Sansone, G., Maschio, L., Usvyat, D., Schütz, M. & Karttunen, A. J. Towards an accurate estimate of the exfoliation energy of black phosphorus: a periodic quantum chemical approach. J. Phys. Chem. Lett. 7, 131–136 (2016).
Benedict, L. X. et al. Microscopic determination of the interlayer binding energy in graphite. Chem. Phys. Lett. 286, 490–496 (1998).
Brent, J. R. et al. Production of few-layer phosphorene by liquid exfoliation of black phosphorus. Chem. Commun. 50, 13338–13341 (2014).
Li, L. et al. Black phosphorus field-effect transistors. Nat. Nanotechnol. 9, 372–377 (2014).
Yasaei, P. et al. High-quality black phosphorus atomic layers by liquid-phase exfoliation. Adv. Mater. 27, 1887, doi:10.1002/adma.201405150 (2015).
Yang, Z. et al. Field-effect transistors based on amorphous black phosphorus ultrathin films by pulsed laser deposition. Adv. Mater. 27, 3748–3754 (2015).
Lu, W. et al. Plasma-assisted fabrication of monolayer phosphorene and its Raman characterization. Nano. Res. 7, 853–859 (2014).
Favron, A. et al. Photooxidation and quantum confinement effects in exfoliated black phosphorus. Nat. Mater. 14, 826–832 (2015).
Wood, J. D. et al. Effective passivation of exfoliated black phosphorus transistors against ambient degradation. Nano Lett. 14, 6964–6970 (2014).
Gaoxue, W., William, J. S., Ravindra, P. & Shashi, P. K. Degradation of phosphorene in air: understanding at atomic level. 2D Mater. 3, 025011 (2016).
Pei, J. et al. Producing air-stable monolayers of phosphorene and their defect engineering. Nat. Commun. 7, 10450 (2016).
Doganov, R. A. et al. Transport properties of pristine few-layer black phosphorus by van der Waals passivation in an inert atmosphere. Nat. Commun. 6, 6647 (2015).
Kang, J. et al. Solvent exfoliation of electronic-grade, two-dimensional black phosphorus. ACS Nano 9, 3596–3604 (2015).
Guo, Z. N. et al. From black phosphorus to phosphorene: basic solvent exfoliation, evolution of raman scattering, and applications to ultrafast photonics. Adv. Funct. Mater. 25, 6996–7002 (2015).
Hanlon, D. et al. Liquid exfoliation of solvent-stabilized few-layer black phosphorus for applications beyond electronics. Nat. Commun. 6, 8563, doi:10.1038/ncomms9563 (2015).
Zhang, S. et al. Extraordinary photoluminescence and strong temperature/angle-dependent raman responses in few-layer phosphorene. ACS Nano 8, 9590–9596 (2014).
Wang, X. et al. Highly anisotropic and robust excitons in monolayer black phosphorus. Nat Nano. 10, 517–521 (2015).
Yang, J. et al. Optical tuning of exciton and trion emissions in monolayer phosphorene. Light-Sci. Appl. 4, doi:10.1038/lsa.2015.85 (2015).
Ding, B., Chen, W., Tang, Z. & Zhang, J. Tuning phosphorene nanoribbon electronic structure through edge oxidization. J. Phys. Chem. C 120, 2149–2158 (2016).
Yang, L., Mi, W. & Wang, X. Tailoring magnetism of black phosphorene doped with B, C, N, O, F, S and Se atom: a DFT calculation. J. Alloys Comp. 662, 528–533 (2016).
Ryder, C. R. et al. Covalent functionalization and passivation of exfoliated black phosphorus via aryl diazonium chemistry. Nat. Chem. 8, 597–602 (2016).
Koenig, S. P. et al. Electron doping of ultrathin black phosphorus with Cu Adatoms. Nano Lett. 16, 2145–2151 (2016).
Yu, X., Zhang, S., Zeng, H. & Wang, Q. J. Lateral black phosphorene P-N junctions formed via chemical doping for high performance near-infrared photodetector. Nano Energy 25, 34–41 (2016).
Sun, M. L. et al. A first-principles study of light non-metallic atom substituted blue phosphorene. Appl. Surf. Sci. 356, 110–114 (2015).
Hashmi, A., Farooq, U. & Hong, J. Graphene/phosphorene bilayer: High electron speed, optical property and semiconductor-metal transition with electric field. Curr. Appl. Phys. 16, 318–323 (2016).
Ling, Z.-P. & Ang, K.-W. Thermal effects on the Raman phonon of few-layer phosphorene. APL Mater. 3, 126104 (2015).
Zhou, L. et al. Phosphorene and doped monolayers interfaced TiO2 with type-II band alignments: novel excitonic solar cells. arXiv preprint arXiv:1512.01675 (2015).
Morita, A. Semiconducting blackphosphorus. Appl. Phys.A-Mater. Sci. Process 39, 227–242 (1986).
Wang, L., Kutana, A., Zou, X. & Yakobson, B. I. Electro-mechanical anisotropy of phosphorene. Nanoscale 7, 9746–9751 (2015).
Ge, Y., Wan, W., Yang, F. & Yao, Y. The strain effect on superconductivity in phosphorene: a first-principles prediction. New J. Phys. 17, doi:10.1088/1367-2630/17/3/035008 (2015).
Shao, D. F., Lu, W. J., Lv, H. Y. & Sun, Y. P. Electron-doped phosphorene: A potential monolayer superconductor. EPL 108, doi:10.1209/0295-5075/108/67004 (2014).
Manjanath, A., Samanta, A., Pandey, T. & Singh, A. K. Semiconductor to metal transition in bilayer phosphorene under normal compressive strain. Nanotechnology 26, doi:10.1088/0957-4484/26/7/075701 (2015).
Ju, W., Li, T., Wang, H., Yong, Y. & Sun, J. Strain-induced semiconductor to metal transition in few-layer black phosphorus from first principles. Chem. Phys. Lett. 622, 109–114 (2015).
Seixas, L., Rodin, A. S., Carvalho, A. & Castro Neto, A. H. Exciton binding energies and luminescence of phosphorene under pressure. Phys. Rev. B 91, (2015).
Wang, Z. et al. Black phosphorus nanoelectromechanical resonators vibrating at very high frequencies. Nanoscale 7, 877–884 (2015).
Peng, X., Wei, Q. & Copple, A. Strain-engineered direct-indirect band gap transition and its mechanism in two-dimensional phosphorene. Phys. Rev. B 90, 085402 (2014).
Jiang, J.-W. & Park, H. S. Negative poisson’s ratio in single-layer black phosphorus. Nat. Commun. 5, 4727 (2014).
Jiang, J. -W. & Park, H. S. Mechanical properties of single-layer black phosphorus. J. Phys. D-Appl. Phys. 47, doi:10.1088/0022-3727/47/38/385304 (2014).
Wu, Y. et al. Negative compressibility in graphene-terminated black phosphorus heterostructures. Phys. Rev. B 93, doi:10.1103/PhysRevB.93.035455 (2016).
Zhang, S. et al. Extraordinarily bound quasi-one-dimensional trions in two-dimensional phosphorene atomic semiconductors. arXiv preprint arXiv:1411.6124 (2014).
Mukhopadhyay, A., Banerjee, L., Sengupta, A. & Rahaman, H. Effect of stacking order on device performance of bilayer black phosphorene-field-effect transistor. J. Appl. Phys. 118, 224501 (2015).
Kang, J. et al. Probing out-of-plane charge transport in black phosphorus with graphene-contacted vertical field-effect transistors. Nano Lett. 16, 2580–2585 (2016).
Ho, P.-H. et al. Tunable photoinduced carrier transport of a black phosphorus transistor with extended stability using a light-sensitized encapsulated layer. ACS Photonics 3, 1102–1108 (2016).
Bolotin, K. I. et al. Ultrahigh electron mobility in suspended graphene. Solid State Commun. 146, 351–355 (2008).
Das, A. et al. Monitoring dopants by Raman scattering in an electrochemically top-gated graphene transistor. Nat. Nanotechnol. 3, 210–215 (2008).
Choi, S. J. et al. Electrical and thermoelectric transport by variable range hopping in thin black phosphorus devices. Nano Lett. 16, 3969–3975 (2016).
Huang, M. et al. Broadband black-phosphorus photodetectors with high responsivity. Adv. Mater. 28, 3481, doi:10.1002/adma.201506352 (2016).
Hu, W., Lin, L., Yang, C., Dai, J. & Yang, J. Edge-modified phosphorene nanoflake heterojunctions as highly efficient solar cells. Nano Lett., doi:10.1021/acs.nanolett.5b04593 (2016).
Donarelli, M. et al. Exfoliated black phosphorus gas sensing properties at room temperature. 2D Materials 3, doi:10.1088/2053-1583/3/2/025002 (2016).
Zhang, R., Li, B. & Yang, J. A first-principles study on electron donor and acceptor molecules adsorbed on phosphorene. J. Phys. Chem. C 119, 2871–2878 (2015).
Ray, S. J. First-principles study of MoS2, phosphorene and graphene based single electron transistor for gas sensing applications. Sens. Actuators B: Chem. 222, 492–498 (2016).
Kistanov, A. A., Cai, Y., Zhou, K., Dmitriev, S. V. & Zhang, Y.-W. Large electronic anisotropy and enhanced chemical activity of highly rippled phosphorene. J. Phys. Chem. C 120, 6876–6884 (2016).
Lalitha, M., Nataraj, Y. & Lakshmipathi, S. Calcium decorated and doped phosphorene for gas adsorption. Appl. Surf. Sci. 377, 311–323 (2016).
Cui, S. et al. Ultrahigh sensitivity and layer-dependent sensing performance of phosphorene-based gas sensors. Nat. Commun. 6, 8632 (2015).
Chen, Y. et al. Field-effect transistor biosensors with two-dimensional black phosphorus nanosheets. Biosens. Bioelectron. 89(Part1), 505–510 (2017).
Slack, G. A. Thermal conductivity of elements with complex lattices: B, P, S. Phys. Rev. 139, A507–A515 (1965).
Jiang, J. -W. Thermal conduction in single-layer black phosphorus: highly anisotropic? Nanotechnology 26, doi:10.1088/0957-4484/26/5/055701 (2015).
Fei, R. et al. Enhanced thermoelectric efficiency via orthogonal electrical and thermal conductances in phosphorene. Nano Lett. 14, 6393–6399 (2014).
Lv, H. Y., Lu, W. J., Shao, D. F. & Sun, Y. P. Large thermoelectric power factors in black phosphorus and phosphorene. arXiv:1404.5171 (2014).
Zhang, J. et al. Phosphorene nanoribbon as a promising candidate for thermoelectric applications. Sci. Rep. 4, doi:10.1038/srep06452 (2014).
Qin, G. et al. Hinge-like structure induced unusual properties of black phosphorus and new strategies to improve the thermoelectric performance. Sci. Rep. 4, doi:10.1038/srep06946 (2014).
Hong, T. et al. Polarized photocurrent response in black phosphorus field-effect transistors. Nanoscale 6, 8978–8983 (2014).
Low, T., Engel, M., Steiner, M. & Avouris, P. Origin of photoresponse in black phosphorus phototransistors. Phys. Rev. B 90, doi:10.1103/PhysRevB.90.081408 (2014).
Flores, E. et al. Thermoelectric power of bulk black-phosphorus. Appl. Phys. Lett. 106, doi:10.1063/1.4905636 (2015).
Stan, M. C., Zamory, Jv, Passerini, S., Nilges, T. & Winter, M. Puzzling out the origin of the electrochemical activity of black P as a negative electrode material for lithium-ion batteries. J. Mater. Chem. A 1, 5293–5300 (2013).
Li, W.-J., Chou, S.-L., Wang, J.-Z., Liu, H.-K. & Dou, S.-X. Simply mixed commercial red phosphorus and carbon nanotube composite with exceptionally reversible sodium-ion storage. Nano Lett. 13, 5480–5484 (2013).
Sun, J. et al. Formation of stable phosphorus–carbon bond for enhanced performance in black phosphorus nanoparticle–graphite composite battery anodes. Nano Lett. 14, 4573–4580 (2014).
Zhao, S. J., Kang, W. & Xue, J. M. The potential application of phosphorene as an anode material in Li-ion batteries. J. Mater. Chem. A 2, 19046–19052 (2014).
Li, W., Yang, Y., Zhang, G. & Zhang, Y.-W. Ultrafast and directional diffusion of lithium in phosphorene for high-performance lithium-ion battery. Nano Lett. 15, 1691–1697 (2015).
Yao, Q. S. et al. Theoretical prediction of phosphorene and nanoribbons as fast-charging Li ion battery anode materials. J. Phys. Chem. C 119, 6923–6928 (2015).
Wang, D., Guo, G.-C., Wei, X.-L., Liu, L.-M. & Zhao, S.-J. Phosphorene ribbons as anode materials with superhigh rate and large capacity for Li-ion batteries. J. Power Sources 302, 215–222 (2016).
Luo, J. et al. Crumpled graphene-encapsulated Si nanoparticles for lithium ion battery anodes. J. Phys. Chem. Lett. 3, 1824–1829 (2012).
Yu, Z. et al. Phosphorus-graphene nanosheet hybrids as lithium-ion anode with exceptional high-temperature cycling stability. Adv. Sci. 2, 1400020, doi:10.1002/advs.201400020 (2015).
Hao, C. et al. Flexible all-solid-state supercapacitors based on liquid-exfoliated black-phosphorus nanoflakes. Adv. Mater. 28, 3194–3201 (2016).
Kulish, V. V., Malyi, O. I., Persson, C. & Wu, P. Phosphorene as an anode material for Na-ion batteries: a first-principles study. Phys. Chem. Chem. Phys. 17, 13921–13928 (2015).
Hembram, K. P. S. S. et al. Unraveling the atomistic sodiation mechanism of black phosphorus for sodium ion batteries by first-principles calculations. J. Phys. Chem. C 119, 15041–15046 (2015).
Nie, A. et al. Selective ionic transport pathways in phosphorene. Nano Lett. 16, 2240–2247 (2016).
Xu, G.-L. et al. Nanostructured black phosphorus/ketjenblack multiwalled carbon nanotubes composite as high performance anode material for sodium-ion batteries. Nano Lett. 16, 3955–3965 (2016).
Li, Q.-F., Duan, C.-G., Wan, X. G. & Kuo, J.-L. Theoretical prediction of anode materials in Li-ion batteries on layered black and blue phosphorus. J. Phys. Chem. C 119, 8662–8670 (2015).
Sun, J. et al. A phosphorene–graphene hybrid material as a high-capacity anode for sodium-ion batteries. Nat. Nanotechnol. 10, 980–985 (2015).
Xu F., B. Ge., Chen J., Huo L., Ma H. et al. Shear-exfoliated phosphorene for rechargeable nanoscale battery. arXiv preprint arXiv:1411.6124 (2015).
Shulenburger, L., Baczewski, A. D., Zhu, Z., Guan, J. & Tománek, D. The nature of the interlayer interaction in bulk and few-layer phosphorus. Nano Lett. 15, 8170–8175, doi:10.1021/acs.nanolett.5b03615 (2015).
Zhang, R., Wu, X. & Yang, J. Blockage of ultrafast and directional diffusion of Li atoms on phosphorene with intrinsic defects. Nanoscale 8, 4001–4006 (2016).
Huang, G. Q., Xing, Z. W. & Xing, D. Y. Prediction of superconductivity in Li-intercalated bilayer phosphorene. Appl. Phys. Lett. 106, 5, doi:10.1063/1.4916100 (2015).
Fei, R. & Yang, L. Strain-engineering the anisotropic electrical conductance of few-layer black phosphorus. Nano Lett. 14, 2884–2889 (2014).
Cai, Y., Zhang, G. & Zhang, Y.-W. Electronic properties of phosphorene/graphene and phosphorene/hexagonal boron nitride heterostructures. J. Phys. Chem. C 119, 13929–13936 (2015).
Guo, H. Y., Lu, N., Dai, J., Wu, X. J. & Zeng, X. C. Phosphorene nanoribbons, phosphorus nanotubes, and van der Waals multilayers. J. Phys. Chem. C 118, 14051–14059 (2014).
Peng, X. H., Copple, A. & Wei, Q. Edge effects on the electronic properties of phosphorene nanoribbons. J. Appl. Phys. 116, 144301 (2014).
Tran, V., Soklaski, R., Liang, Y. & Yang, L. Layer-controlled band gap and anisotropic excitons in few-layer black phosphorus. Phys. Rev. B 89, 235319 (2014).
Tran, V. & Yang, L. Scaling laws for the band gap and optical response of phosphorene nanoribbons. Phys. Rev. B 89 144301 (2014).
Ting, H., Yang, H. & Jinming, D. Mechanical and electronic properties of monolayer and bilayer phosphorene under uniaxial and isotropic strains. Nanotechnology 25, 455703 (2014).
Huang, G. Q. & Xing, Z. W. Band-gap tunability and dynamical instability in strained monolayer and bilayer phosphorenes. J. Phys.: Condens. Matter. 27, 175006 (2015).
Saito, Y. et al. gate-tuned thermoelectric power in black phosphorus. Nano Lett. 16, 4819–4824 (2016).
Erande, M. B., Pawar, M. S. & Late, D. J. Humidity sensing and photodetection behavior of electrochemically exfoliated atomically thin-layered black phosphorus nanosheets. ACS Appl. Mater. Interfaces 8, 11548–11556 (2016).
Acknowledgements
We would like to thank Dr Congyan Zhang for preparing the model of phosphorene crystal structure used in Fig. 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Akhtar, M., Anderson, G., Zhao, R. et al. Recent advances in synthesis, properties, and applications of phosphorene. npj 2D Mater Appl 1, 5 (2017). https://doi.org/10.1038/s41699-017-0007-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41699-017-0007-5
This article is cited by
-
Theoretical Investigation of 2D M3C2S2 (M = Cr, Zr) as Potential Electrode Materials for Li-Ion Batteries
Journal of Electronic Materials (2024)
-
Electronic structure and optical properties of nitrogen-doped antimonene under biaxial strain: first-principles study
Journal of Molecular Modeling (2024)
-
Straintronics in phosphorene via tensile vs shear strains and their combinations for manipulating the band gap
Scientific Reports (2023)
-
Vortex structure in Wigner molecules
Scientific Reports (2023)
-
Gas Sensing Properties of Black Phosphorene-Like InP3 Monolayer: A First-Principles Study
Journal of Electronic Materials (2023)
