
Abstract
This retrospective study investigated the association between the pattern of disease progression and molecular mechanism of acquired resistance in a large cohort of 49 patients with ROS1-rearranged advanced non-small-cell lung cancer treated with first-line crizotinib. We found that treatment-emergent ROS1 point mutations were the major molecular mechanism of crizotinib resistance, particularly for patients who developed extracranial-only disease progression. Our findings highlight the importance of rebiopsy and gene testing for subsequent-line therapeutic management.
Similar content being viewed by others

Gene rearrangements involving the ROS proto-oncogene-1 (ROS1) are actionable therapeutic targets for non-small cell lung cancer (NSCLC). ROS1 fusions occur at a rate of 2% in NSCLC and up to 3.3% in lung adenocarcinoma1,2. Crizotinib is the first tyrosine kinase inhibitor (TKI) to show clinical activity in ROS1-rearranged NSCLC3. Cancer cells that are previously sensitive to TKIs such as crizotinib develop resistance thru on-target and off-target mechanisms, which are the main causes of treatment failure and lead to disease progression4. Brain metastasis is commonly observed in patients with lung cancer and is also one of the primary modes of disease progression in ROS1-positive patients5. So far, few studies have comprehensively analyzed the frequency of ROS1 resistance mutations and the pattern of disease progression after crizotinib therapy of ROS1-rearranged NSCLC6. This retrospective study aimed to explore the molecular mechanism of crizotinib resistance and its relationship with the mode of progression.
Of the 117 patients screened, 49 patients with ROS1-rearranged advanced lung adenocarcinoma were included in our analysis. A total of 30 patients (61.2%, 30/49) were identified with secondary ROS1 mutations using NGS analysis of rebiopsy samples collected at progression from first-line crizotinib. The study cohort comprised of 57.1% female (n = 28), 75.5% patients who had no smoking history (n = 37), and 67.3% of patients without baseline brain metastasis (n = 33). The median age of 50 years (range: 26–66). All patients had lung adenocarcinoma (100%). CD74-ROS1 was observed in 57.1% (28/49) of the cohort and was the most common fusion partner of ROS1. SDC4-ROS1 was detected in ten patients (20.4%, 10/49), EZR-ROS1 in six patients (12.2%, 6/49), two patients each (4,1%, 2/49) with SLC34A2-ROS1 and TPM3-ROS1, and a patient (2%, 2.1/49) had CCDC6-ROS1. Most patients (91.8%, 45/49) had single gene fusion, whereas the remaining four patients (8.2%, 4/49) had multiple gene fusions wherein one gene fusion is a canonical ROS1 gene fusion, and the other is a retained 5′-ROS1 fused to another gene. Table 1 lists the baseline characteristics of the cohort.
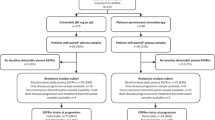
For patterns of disease progression analyses, all 49 patients were included. Patient details are listed in Supplementary Table 1. Extracranial-only progression was confirmed in 33 (67.3%) patients, intracranial-only progression in 11 (22.4%) patients, and both intracranial and extracranial progression in five (10.2%) patients (Fig. 1). The clinical characteristics among the three groups were not statistically different (Supplementary Table 2). The detailed information of the patients with intracranial-only progression and both intracranial and extracranial progression are also summarized in Supplementary Table 3 and Supplementary Table 4, respectively.

Flow chart illustrating the study design.
Treatment-emergent ROS1 point mutations were detected in 61.2% of patients (30/49) after crizotinib progression, with ROS1 G2032R being the most common mutation, detected in 14 patients (28.5%, 14/49). The other secondary ROS1 mutations detected in our cohort were G2032K (8.3%, 4/49), G2026M (6.1%, 3/49), L2086F (6.1%, 3/49), S1986Y (4.1%, 2/49), S1986F (2%, 1/49), L1174F (2%, 1/49), and L2155S (2%, 1/49). The remaining 19 (38.7%, 19/49) patients were not detected with any mutations related to crizotinib resistance (Fig. 2a).

a Distribution of molecular mechanisms of crizotinib resistance of the cohort. b Patients with intracranial-only progression on crizotinib had a significantly lower frequency of treatment-emergent ROS1 point mutations than patients with extracranial-only progression.
Of the 11 patients with intracranial-only progression, the sample types submitted for NGS testing were cerebrospinal fluid (positive cytology, as liquid biopsy sample) and plasma samples (n = 7), plasma samples (n = 2), or brain metastasis tissue sample (n = 2) (Fig. 1). We further compared the frequency of treatment-emergent ROS1 point mutations in patients with either intracranial or extracranial progression. Comparative analysis revealed that patients with extracranial-only progression had a significantly higher frequency of ROS1 point mutations than patients with intracranial-only progression (72.7% vs. 15.2%, p = 0.001, Fig. 2b). No significant difference was observed between the patients with CD74-ROS1 and non-CD74 ROS1 fusions for the frequency of treatment-emergent ROS1 mutations (70.4% vs. 52.9%, p = 0.337, Supplementary Fig. 1) and the rates of intracranial or extracranial progression (57.63% vs. 63.6%, p = 1.000, Supplementary Fig. 2).
Supplementary Table 4 summarizes the clinical information of the five patients with both intracranial and extracranial metastasis at crizotinib progression. These five patients were comprised of two patients harboring CD74-ROS1, two patients with SDC4-ROS1, and a patient with TPM3-ROS1. A patient was detected with ROS1 L1174F, while no mutation was detected in the other four patients.
The molecular mechanisms of acquired resistance to crizotinib have been well-described both in vitro and in patients with ROS1-rearranged lung cancer7,8,9,10. Previous studies have shown that the mechanisms of crizotinib resistance in patients with ROS1 fusion-positive NSCLC are mainly divided into two categories: on-target mechanisms involving mutations in the ROS1 kinase domain and off-target mechanisms involving the activation of other signaling pathways11,12,13,14. However, most of these clinical studies reporting on the inhibitor resistance mechanisms of ROS1-rearranged lung cancer were conducted with only limited samples, and only a few explored the association between treatment-emergent ROS1 point mutations and patterns of disease progression. Hence, our study extends the understanding of this association by performing retrospective analysis of a larger cohort of patients with ROS1-rearranged advanced NSCLC treated with crizotinib.
In our cohort, 57.4% had CD74-ROS1 at diagnosis, which is consistent with previous reports10,15. We also found four patients with multiple fusions, which may pose as a predictive factor for the poor prognosis of patients harboring ROS1 fusions16. Although these 5’-ROS1 fusions are not expressed into RNA and protein, and no study has provided evidence on their oncogenic function, we speculate that the detection of these 5’-end fusions using DNA-NGS reflects a more complex biological scenario17. Future studies are warranted to shed light on the molecular mechanisms associated with the retention of these 5′-ROS1 fusions.
Among the patients in our cohort, ten patients had brain progression. Approximately 63.8% of patients were detected with treatment-emergent ROS1 point mutations after crizotinib progression and were not detected at baseline. ROS1 G2032R was the most common, which is consistent with other published reports1,10,15,18. The secondary ROS1 missense mutations detected from our cohort were all previously reported. These ROS1 missense mutations affect the residues located in the ATP binding pocket of the kinase domain, resulting in steric hindrance and blocking the binding of ROS1 inhibitors at varied levels, which leads to inhibitor resistance1,10,15,18. Our results support that secondary mutations in the ROS1 kinase domain are the major molecular mechanisms of crizotinib resistance in patients with ROS1-rearranged NSCLC, particularly those with extracranial progression. After crizotinib progression, these patients with secondary mutations could benefit from next-generation ROS1 inhibitors such as entrectinib, lorlatinib, or cabozantinib, depending on the sensitivity profile of the ROS1 point mutation15. This raises the need to perform rebiopsy and elucidate the mutational profile after developing disease progression to understand the resistance mechanism and plan optimal therapeutic strategies to improve the survival outcomes of these patients. In addition, no resistance-related mutation was detected in some patients, particularly those with intracranial progression, despite the use of their cerebrospinal fluid or brain tissue samples for molecular analysis, suggesting that unknown resistance mechanism needs further exploration. As previously discussed by Gainor et al., patients with intracranial progression might reflect pharmacokinetic failure due to the inherent limitation of crizotinib in penetrating the blood-brain barrier, rather than true biological resistance10. Moreover, the comparable modes of disease progression and rates of secondary ROS1 point mutations in patients with either CD74-ROS1 or non-CD74-ROS1 suggest that the ROS1 gene fusion partners did not contribute to the differences in either the site of disease progression or the mechanisms of acquired resistance.
Our study is limited by its retrospective nature. Some data were not available for analysis and our study only included a small cohort of patients treated in our institution, which may introduce sample bias. Since the cohort with survival outcomes was based on the patients who submitted rebiopsy samples for NGS, inherent sampling bias might exist. Our study only included the analysis of treatment-emergent ROS1 point mutations as the acquired resistance mechanism. Other ROS1-independent mechanisms of resistance should be investigated further. Multi-omics analysis would also be an interesting avenue to comprehensively understand transcriptomics, proteomics, or metabolomics-related changes at crizotinib progression.
In conclusion, our study provides real-world clinical evidence of the association between the molecular mechanisms of resistance and patterns of disease progression in patients with ROS1-rearranged advanced NSCLC who received first-line crizotinib. Our findings revealed that treatment-emergent ROS1 point mutations were the main mechanism of crizotinib resistance, particularly in patients with extracranial progression. These findings raise the need to develop effective treatment strategies for overcoming crizotinib resistance in patients with ROS1-rearranged advanced NSCLC.
Methods
Patients
We retrospectively analyzed the NGS data of 117 patients with ROS1-rearranged NSCLC. The patients were grouped according to the pattern of disease progression. Group 1 included the patients with intracranial-only disease progression (n = 11); group 2 included the patients with extracranial-only disease progression (n = 33), and group 3 included the patients with both extracranial and intracranial disease progression (n = 5) (Fig. 1). Disease progression is evaluated based on radiographic assessment using computed tomography scanning and magnetic resonance imaging for all patients. All patients provided written informed consent to take part in the study. Approval was obtained from the Hunan Cancer Hospital Institutional Review Board Committee (2017YYQ-SSB-026). All patients provided written informed consent to take part in the study.
NGS
Patient samples from baseline and at confirmation of disease progression were submitted for NGS-based multi-gene panel mutation analysis to Burning Rock Biotech, a College of American Pathologists (CAP)-accredited, Clinical Laboratory Improvement Amendments-certified clinical laboratory. All the gene panels used in our study interrogated whole exons and critical introns of at least the eight classic NSCLC oncogenic drivers, including EGFR, ALK, BRAF, ERBB2, KRAS, MET, RET, and ROS1. The sequencing analyses were performed using optimized bioinformatics pipeline for somatic variant calling that involved accurate identification of base substitutions, small insertions-deletions, copy number variations, and genomic rearrangements as described previously19.
Statistical analysis
We analyzed the NGS results and patterns of disease progression for each group. We compared the frequency of treatment-emergent ROS1 point mutations according to progression pattern and type of ROS1 fusion using chi-square test. P < 0.05 indicated statistical significance. All statistical analyses were performed as two-sided tests using SPSS software (version 22) or GraphPad Prism (version 8).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All the data analyzed for this study are included as Supplementary Table 1.
Code availability
No custom code or scripts were generated or used in our study.
References
Drilon, A. et al. ROS1-dependent cancers - biology, diagnostics and therapeutics. Nat. Rev. Clin. Oncol. 18, 35–55 (2021).
Lin, J. J. & Shaw, A. T. Recent Advances in Targeting ROS1 in Lung Cancer. J. Thorac. Oncol. 12, 1611–1625 (2017).
Shaw, A. T. et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): updated results, including overall survival, from PROFILE 1001. Ann. Oncol. 30, 1121–1126 (2019).
D’Angelo, A. et al. Focus on ROS1-Positive Non-Small Cell Lung Cancer (NSCLC): Crizotinib, Resistance Mechanisms and the Newer Generation of Targeted Therapies. Cancers (Basel) 12, https://doi.org/10.3390/cancers12113293 (2020).
Patil, T. et al. The Incidence of Brain Metastases in Stage IV ROS1-Rearranged Non-Small Cell Lung Cancer and Rate of Central Nervous System Progression on Crizotinib. J. Thorac. Oncol. 13, 1717–1726 (2018).
Lin, J. J. et al. Spectrum of Mechanisms of Resistance to Crizotinib and Lorlatinib in ROS1 Fusion-Positive Lung Cancer. Clin. Cancer Res. 27, 2899–2909 (2021).
Zou, H. Y. et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc. Natl Acad. Sci. USA 112, 3493–3498 (2015).
Awad, M. M., Engelman, J. A. & Shaw, A. T. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N. Engl. J. Med. 369, 1173 (2013).
Facchinetti, F. et al. Crizotinib-Resistant ROS1 Mutations Reveal a Predictive Kinase Inhibitor Sensitivity Model for ROS1- and ALK-Rearranged Lung Cancers. Clin. Cancer Res. 22, 5983–5991 (2016).
Gainor, J. F. et al. Patterns of Metastatic Spread and Mechanisms of Resistance to Crizotinib in ROS1-Positive Non-Small-Cell Lung Cancer. JCO Precis Oncol 2017, https://doi.org/10.1200/PO.17.00063 (2017).
McCoach, C. E. et al. Resistance Mechanisms to Targeted Therapies in ROS1(+) and ALK(+) Non-small Cell Lung Cancer. Clin. Cancer Res. 24, 3334–3347 (2018).
Sato, H. et al. MAPK Pathway Alterations Correlate with Poor Survival and Drive Resistance to Therapy in Patients with Lung Cancers Driven by ROS1 Fusions. Clin. Cancer Res. 26, 2932–2945 (2020).
Cargnelutti, M. et al. Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget 6, 5182–5194 (2015).
Watanabe, J., Furuya, N. & Fujiwara, Y. Appearance of a BRAF Mutation Conferring Resistance to Crizotinib in Non-Small Cell Lung Cancer Harboring Oncogenic ROS1 Fusion. J. Thorac. Oncol. 13, e66–e69 (2018).
Lin, J. J. et al. Spectrum of Mechanisms of Resistance to Crizotinib and Lorlatinib in ROS1 Fusion-Positive Lung Cancer. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.CCR-21-0032 (2021).
Zhang, Y. et al. Clinical and molecular factors that impact the efficacy of first-line crizotinib in ROS1-rearranged non-small-cell lung cancer: a large multicenter retrospective study. BMC Med. 19, 206 (2021).
Zhang, Y. et al. Detection of Nonreciprocal/Reciprocal ALK Translocation as Poor Predictive Marker in Patients With First-Line Crizotinib-Treated ALK-Rearranged NSCLC. J. Thorac. Oncol. 15, 1027–1036 (2020).
Dagogo-Jack, I. et al. Molecular Analysis of Plasma From Patients With ROS1-Positive NSCLC. J. Thorac. Oncol. 14, 816–824 (2019).
Li, Y. S. et al. Unique genetic profiles from cerebrospinal fluid cell-free DNA in leptomeningeal metastases of EGFR-mutant non-small-cell lung cancer: a new medium of liquid biopsy. Ann. Oncol. 29, 945–952 (2018).
Acknowledgements
This work received financial support from the Natural Science Foundation of Hunan Province (2020SK2031, kq1801102, 2020SK2030, 2018RS3106, 2019-TJ-N04, 2019SK4010, and 2020JJ3025), and CAS “Light of West China Program”. The funding agencies had no role in the study design, data collection, analysis, interpretation, paper writing, and decision to submit the paper for publication.
Author information
Authors and Affiliations
Contributions
Y.Z. and N.Y.: Conceptualization, Organization, Data collection, Auditing, Supervision, Project administration, Funding acquisition, Writing- Reviewing, and Editing. Z.H., L.Z., Y.L. and X.Z.: Data curation, Methodology, Formal analysis, Writing-Original Draft Preparation, Writing- Reviewing, and Editing. Q.X., H.Y.: Software, Validation, Writing- Reviewing, and Editing. J.L.: Formal analysis and Visualization, Writing- Reviewing, and Editing. C.X. and Z.S.: Critical comments and Suggestions, Writing- Reviewing and Editing. A.L. and S-H.I.O.: Formal analysis, Writing- Reviewing, and Editing. All authors approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
A.L. is an employee of Burning Rock Biotech and she declares no non-financial interest. All the other authors declare no competing financial or non-financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Huang, Z., Zeng, L. et al. Disease progression patterns and molecular resistance mechanisms to crizotinib of lung adenocarcinoma harboring ROS1 rearrangements. npj Precis. Onc. 6, 20 (2022). https://doi.org/10.1038/s41698-022-00264-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-022-00264-w
This article is cited by
-
Therapeutic advances of targeting receptor tyrosine kinases in cancer
Signal Transduction and Targeted Therapy (2024)
-
Rutin attenuates ensartinib-induced hepatotoxicity by non-transcriptional regulation of TXNIP
Cell Biology and Toxicology (2024)
-
Lorlatinib and capmatinib in a ROS1-rearranged NSCLC with MET-driven resistance: tumor response and evolution
npj Precision Oncology (2023)
-
Protein tyrosine kinase inhibitor resistance in malignant tumors: molecular mechanisms and future perspective
Signal Transduction and Targeted Therapy (2022)
