
Abstract
Oxidative stress induced endothelial dysfunction plays a particularly important role in promoting the development of cardiovascular diseases (CVDs). Salvianolic acid A (SalA) is a water-soluble component of traditional Chinese medicine Salvia miltiorrhiza Bunge with anti-oxidant potency. This study aims to explore the regulatory effect of SalA on oxidative injury using an in vitro model of H2O2-induced injury in human umbilical vein endothelial cells (HUVECs). In the study, we determined cell viability, the activities of Lactate dehydrogenase (LDH) and Superoxide dismutase (SOD), cell proliferation rate and intracellular reactive oxygen species (ROS). Flow cytometry was used to detect cell apoptosis. Western-blotting was used to evaluate the expression of cell senescence, apoptosis, autophagy and pyroptosis protein factors. The expression level of miRNA was determined by qRT-PCR. Compared with H2O2-induced HUVECs, SalA promoted cell viability and cell proliferation rate; decreased LDH and ROS levels; and increased SOD activity. SalA also significantly attenuated endothelial senescence, inhibited cell apoptosis, reversed the increase of LC3 II/I ratio and NLRP3 accumulation. Furthermore, miR-204-5p was regulated by SalA. Importantly, miR-204-5p inhibitor had similar effect to that of SalA on H2O2-induced HUVECs. Our results indicated that SalA could alleviate H2O2-induced oxidative injury by downregulating miR-204-5p in HUVECs.
Similar content being viewed by others

Introduction
Cardiovascular disease (CVD) is a leading cause of morbidity and mortality worldwide, especially among the aging population1,2. Endothelial dysfunction is an early marker of vascular damage linked to the disruption of the vascular homeostasis and represents a key step in the pathogenesis of CVDs. The redox imbalance is the main cause of endothelial dysfunction. Accumulating evidence suggested that oxidative stress occurs throughout the causes of endothelial cell (EC) dysfunction, leading to cellular senescence and death, including apoptosis, autophagic cell death, pyroptosis, necroptosis and ferroptosis3,4,5. It is crucial to rescue ECs from oxidative injury, implementation of effective interventions can alleviate endothelial dysfunction and offer important preventative and therapeutic benefits for patients with CVDs.
The senescence and death of ECs are not only adverse outcomes, but also causal contributors to endothelial dysfunction. The intracellular reactive oxygen species (ROS) when present in excess, result in oxidative stress, EC senescence and even cell death. However senescent ECs can produce high levels of ROS, mainly including superoxide, H2O26. Then, H2O2 or ROS can induce multiple modes of EC death. Moreover, different modes of cell death may compensate for each other. EC autophagy is generally considered to be vasoprotective, which is attributed to its ability to balance the status of cellular redox and bioenergetics. Apoptosis is one of the most widely studied forms of programmed cell death, and excess ROS is a major driver of EC apoptosis7. Necroptosis, pyroptosis, and ferroptosis are three main modes of regulated necrosis, a majority of cell death events in EC dysfunction. Necroptotic inflammation can be enhanced by crosstalk with pyroptosis, NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome is considered as a key step of this crosstalk8. Herein, adequate protection of EC from oxidative stress mediated cellular senescence and death appears to be promising therapeutics.
Danshen, Salvia miltiorrhiza Bunge, is a traditional Chinese medicine widely used for the prevention and treatment of CVDs9. Salvianolic acid is the main active ingredients in Salvia miltiorrhiza Bunge, including Salvianolic acid A (SalA), Salvianolic acid B (SalB) and Salvianolic acid C and so on. The results of pharmacological researches have showed that Salvianolic acid exerts antioxidation, anti-inflammatory and antithrombotic effects10. In our previous work, we revealed that H2O2 could induce oxidative damage to human umbilical vein endothelial cells (HUVECs), which could be reversed by SalB11. However, the therapeutic mechanisms of SalA are not fully understood, and the regulation of signaling transduction pathways remains limited.
MicroRNAs (miRNAs) are a class of small non-coding RNAs that regulate gene expression post transcriptionally, and thus participate in various physiopathological processes, including cell proliferation, differentiation, cell death12. SalA was confirmed to protect blood-spinal cord barrier integrity by modulation of miRNA (miR-101a) expression in primary rat brain microvascular EC13. Therefore, miRNA could be a promising therapeutic target for SalA rescue endothelial oxidative injury. miR-204-5p is derived from the 5′ end arm of hsa-miR-204 precursors (pre-miRNAs)14. Growing evidence have proved that miR-204-5p plays crucial role in EC dysfunction15,16. Our previous works had showed that the expression of miR-204-5p was conspicuously elevated in H2O2 induced HUVECs17. Thus, we aimed to find whether SalA could rescue ECs from oxidative injury by targeting regulation of miR-204-5p.
Results
SalA ameliorated H2O2-induced HUVECs injury
First, we used different concentrations of H2O2 to induce HUVECs for 4 h to construct oxidative damage models. The results of CCK-8 (Fig. 1A) showed that cell viability was inhibited to different degrees with increasing concentrations of H2O2 (50–1000 μM), which indicated that cell was damaged by oxidative stress. Among them, when the H2O2 concentration was 200 μM, the cell survival rate was around 50% and statistically different. Therefore, we chose 200 μM H2O2 to stimulate HUVECs in the subsequent experiments.

Effects of SalA on cell viability, cell proliferation and LDH release. (A) The effects of different concentration of H2O2 (50–1000 μM) treated for 4 h on the cell viability of HUVECs. (B) The effects of different treatment time of 200 μM H2O2 on the cell viability of HUVECs. (C–F) HUVECs were treated with SalA for 24 h after 12 h 200 μM H2O2 inducing: (C) The effect of SalA on the cell viability of HUVECs. HUVECs were treated with different concentration of SalA (0.25–16 μM). (D) The effect of SalA on the cell viability in H2O2-induced HUVECs. (E) The effect of SalA on the cell proliferation rate in H2O2-induced HUVECs were determined using a EdU assay. (F) The effect of SalA on LDH release in H2O2-induced HUVECs. Values are expressed as the mean ± SD (n = 3) (*P < 0.05, **P < 0.01, ***P < 0.001 vs. Control, #P < 0.05, ##P < 0.01 vs. H2O2).
Then, we continued to optimal the H2O2 treatment time with the concentration of 200 μM. The results (Fig. 1B) showed that 200 μM H2O2 significantly inhibited the cell viability of HUVECs at 4 h, 6 h, 8 h,12 h treatment time. According to the recent reports and our results18,19,20, the treatment time of 12 h was selected to develop a model of oxidative damage.
To define the optimal concentration of SalA, a dose–response curve was performed within the previously established nontoxicity range from 0 to 16 μΜ, and examined for cell viability. SalA (0.25–8 μM) did not alter cell viability compared to the control group (Fig. 1C), but improved the cell viability compared to H2O2 group (P < 0.05), and SalA (0.25, 0.5 μM) significantly increased the cell viability against H2O2-induced injury (Fig. 1D) (P < 0.01). Therefore, we selected 0.25 μM and 0.5 μM as the optimal concentration of SalA in subsequent experiments.
The Edu assay was used to examine the effect of SalA treatment on cell proliferation. The results (Fig. 1E) showed that compared with the control group, H2O2 induced group was significantly decreased cell proliferation rate (P < 0.01). Compared with H2O2 group, SalA increased the cell proliferation rate, but only 0.5 μM SalA group exhibited a significant change (P < 0.05).
Lactate dehydrogenase (LDH) leakage as a marker for cell degradation was also monitored. LDH release in H2O2 group was highly increased than control group (P < 0.05). After treated with SalA (0.25, 0.5 μM), the LDH release rate was significantly inhibited (P < 0.01) (Fig. 1F).
Thus, SalA is effective in protecting HUVECs against cell injury caused by H2O2.
SalA reduced oxidative stress in H2O2-induced HUVECs
ROS is often used as an index to evaluate oxidation. Results as shown in Fig. 2A, ROS levels in H2O2 group was significantly higher than that in control group (P < 0.01). Compared with H2O2 group, SalA (0.25, 0.5 μM) treatment significantly decreased ROS levels in HUVECs (P < 0.01).

Effect of SalA on Intracellular ROS level and SOD activity in HUVECs induced by H2O2. HUVECs were treated with SalA for 24 h after 12 h 200 μM H2O2 inducing. (A) The effect of SalA on ROS level in H2O2-induced HUVECs were determined using a ROS assay. (B) The effect of SalA on SOD activity in H2O2-induced HUVECs. Values are expressed as the mean ± SD (n = 3) (*P < 0.05, **P < 0.01 vs. Control, #P < 0.05, ##P < 0.01 vs. H2O2).
Superoxide dismutase (SOD) is one of the most important antioxidants. In Fig. 2B, the SOD activity in the H2O2 group was significantly lower than in the control group (P < 0.05), and SalA treatment ameliorated the decrease in SOD activity in H2O2 group, especially in 0.25 μM SalA group (P < 0.05).
SalA attenuated cell senescence in H2O2-induced HUVECs.
Oxidative stress can induce cell senescence, the expression levels of cell cycle-related proteins such as p53, p21, and Cyclin E1 were detected to analyze the cellular senescence with western blot analysis. In Fig. 3A–D, compared with the control group, our results showed that the expression levels of p53 and p21 were increased (P < 0.05) while Cyclin E1 expression level were decreased (P < 0.05) in H2O2 induced HUVECs. SalA treatment decreased the expression of p53 and p21 proteins, and increased Cyclin E1 level, in which 0.25 μM SalA had the most significant effect (P < 0.05, P < 0.01, P < 0.05).

Effect of SalA on cell senescence in HUVECs induced by H2O2. HUVECs were treated with SalA for 24 h after 12 h 200 μM H2O2 inducing. (A) The effect of SalA on the (B) p53, (C) p21, (D) Cyclin E1, were detected by western blotting. Uncropped gel images are provided in the supplementary file. Values are expressed as the mean ± SD (n = 3) (*P < 0.05 vs. Control, #P < 0.05, ##P < 0.01 vs. H2O2).
SalA attenuated programmed cell death in H2O2-induced HUVECs
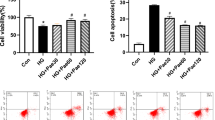
Apoptosis mostly led by oxidative stress, and Bcl-2 and BAX are the main regulatory genes of apoptosis. The ratio of Bcl-2/BAX can show the level of apoptosis. As shown in Fig. 4A–D, compared with control group, Bcl-2 protein expression diminished significantly (P < 0.05), and BAX protein increased substantially (P < 0.05) in H2O2 group. On the contrary, SalA treatment increased Bcl-2 level (P < 0.01) while reduced BAX level significantly (P < 0.01), and 0.25 μM SalA had significant effect by increasing the ratio of Bcl-2/BAX (P < 0.05). The cell flow result also indicated that SalA significantly reduced the apoptosis induced by H2O2 in Fig. 4E–F.

Effect of SalA on cell death in HUVECs induced by H2O2. HUVECs were treated with SalA for 24 h after 12 h 200 μM H2O2 inducing. (A) The effect of SalA on the protein expressions of (B) Bcl-2 and (C) BAX were detected by western blotting. (D) The expression ratio of Bcl-2/BAX. (E,F) Representative images of flow cytometry analysis of the effect of SalA on cell apoptosis rate. (G) The effect of SalA on the (H) LC3 II/I ratio and (I) NLRP3 protein expression were detected by western blotting. Uncropped gel images are provided in the supplementary file. Values are expressed as the mean ± SD (n = 3) (*P < 0.05, ***P < 0.001 vs. Control, #P < 0.05, ##P < 0.01 vs. H2O2).
Autophagy is an evolutionarily conserved cellular process which is essential for cell survival and maintenance of intracellular homeostasis21. The level of LC3 II/I ratio was considered as a biomarker of the changes of autophagy in cells. The increase of LC3 II/I ratio in H2O2 group was significant (P < 0.05), whereas SalA (0.25, 0.5 μM) significantly inhibited LC3 II/I ratio in Fig. 4G–H.
NLRP3 is a key marker of inflammatory death induced by oxidative stress22. As result in Fig. 4I, compared with the control group, the protein expressions of NLRP3 were obviously upregulated (P < 0.05) in H2O2-induced HUVECs, while 0.25 μM and 0.5 μM SalA treatments reversed these aberrant changes (P < 0.05).
These findings indicated that SalA could attenuate H2O2-induced programmed cell death in HUVECs.
SalA decreased miR-204-5p expression in H2O2-induced HUVECs.
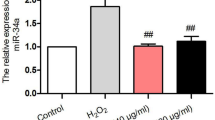
To determine whether the effect of SalA on the expression of these miR-204-5p was in accordance with our hypothesis, miRNA expression was measured by qRT–PCR (Fig. 5A). Compared with control group, miR-204-5p was up-regulated by H2O2 inducement. SalA treatment significantly reduced the expression of miR-204-5p (P < 0.01) in H2O2 induced HUVECs.

Effects of SalA on miR-204-5p expression of HUVECs. HUVECs were first transfected with miR-204-5p mimic/inhibitor for 6 h, and then cells were induced by 200 μM H2O2 for 12 h before 24 h SalA treatment. (A) The effect of SalA on the miR-204-5p expression of HUVECs induced by H2O2. (B,C) The effect of SalA on the miR-204-5p expression of H2O2-induced HUVECs with miR-204-5p mimic/inhibitor. Values are expressed as the mean ± SD (n = 3) (*P < 0.05, **P < 0.01 vs. Control, ##P < 0.01 vs. H2O2, &&P < 0.01 vs. H2O2 + SalA).
To further confirm the interaction of SalA and miR-204-5p, miR-204-5p mimic or miR-204-5p inhibitor were transferred into H2O2-induced HUVECs. Consistent with expected, the high expression of miR-204-5p induced by miR-204-5p mimic transfection reversed the decrease effect of SalA treatment (Fig. 5B); The transfection of miR-204-5p inhibitor downregulated the expression of miR-204-5p (P < 0.01) compared with the H2O2 group, which was similar as the effect of SalA, and miR-204-5p inhibitor enhanced the inhibition effect of SalA on miR-204-5p (P < 0.01) (Fig. 5C).
SalA alleviated H2O2-induced HUVECs apoptosis though miR-204-5p/p53 pathway
miR-204-5p was found to promote apoptosis by downregulating Bcl-2 in prostate cancer cells23. It is thought that p53 causes apoptosis, which is governed by the presence of several apoptotic genes, including Bcl-2 and BAX24. Therefore, we were interested in determining the role of miR-204-5p/p53 pathway. The transfection of miR-204-5p inhibitor and SalA has a similar effect on p53, and the combined effect was stronger than on their alone (Fig. 6). Meanwhile, miR-204-5p inhibitor enhanced the effect of SalA on p53 (P < 0.05), Bcl-2 (P < 0.05), BAX (P < 0.05) and Bcl-2/BAX ratio (P < 0.05).

Effects of SalA on expression of apoptosis-related protein in H2O2-induced HUVECs by inhibiting miR-204-5p. HUVECs were first transfected with miR-204-5p inhibitor for 6 h, and then cells were induced by 200 μM H2O2 for 12 h before 24 h SalA treatment. (A) The effect of SalA on the protein expressions of (B) p53, (C) Bcl-2 and (D) BAX were detected by western blotting. (E) The expression ratio of Bcl-2/BAX. Uncropped gel images are provided in the supplementary file. Values are expressed as the mean ± SD (n = 3) (**P < 0.01 vs. Control, #P < 0.05, ##P < 0.01 vs. H2O2, $P < 0.05 vs. H2O2 + miR-204-5p inhibitor).
Discussion
Endothelial cells are directly exposed to endogenous danger signals and metabolites in the circulatory system. Endothelial senescence and death associated EC dysfunction are widely recognized to be associated with vascular diseases such as atherosclerosis, hypertension and hyperlipidaemia and is a common early event in CVD25,26. Thus, research on EC senescence and death could accelerate the discovery of more effective drugs for the treatment of vascular diseases. In this study, our results demonstrated that SalA reversed the effects of H2O2-induced on endothelial oxidative injury, EC senescence, apoptosis, autophagy and NLRP3 expression.
The oxidative pathway is one of the important mechanisms associated with cell senescence, and the role of oxidative stress in the initiation and progression of vascular endothelial cell senescence has been widely reported. The excessive production of ROS is one of the prime responses of oxidative stress, result in cell dysfunction, senescence, and even cell death. SOD is an important component of cellular defense against free radical-mediated cellular oxidative damage27. In our study, SalA significantly improved cell viability and increased SOD activity after H2O2-induction, as well as reduced intracellular ROS levels. Consistent with the previous studies28,29, SalA reversed the oxidative injury on HUVECs.
Peroxidation products activate transcription factors (including p53) associated with senescence at high levels of oxidative stress30,31. It has been found that exposure of cells to oxidative stress leads to senescence by activating p53 through the initiation of the DNA damage response pathway and activating the downstream factor p21. Activated p21 blocks the G1 cell cycle and inhibits the activity of cell cycle proteins, such as Cyclin E132. In our results SalA inhibited p53, p21 and promoted Cyclin E1 expression levels, these results indicated that SalA rescued H2O2-induced cellular senescence.
Each mode of cell death is associated with the specific cellular signaling cascades. Autophagy is involved in the antioxidant defense mechanism, but excessive or autophagy can be detrimental to cells. LC3 protein is one of the key components of autophagosomes, which regulates the formation and recycling of autophagic vesicles and plays an important role in the initiation and expansion of autophagy. When autophagy is active, LC3 II/I ratio was up-regulated. Pyroptosis is a proinflammatory cell death characterized by the formation of inflammasomes. NLRP3 is a component of the inflammasome, which is activated by specific signals to trigger a series of inflammatory responses and contribute to pyroptosis, thus participating in inflammatory regulation and cell death mechanisms. Apoptosis is one of the most widely studied forms of programmed cell death. There are three classical apoptosis signaling pathways: the extrinsic (death receptor) pathway, the intrinsic (mitochondrion) pathway and the endoplasmic reticulum stress pathway (including ischemia, oxidative stress, hypoxia, etc.). All of which can modulate the expression of the Bcl-2 family of proteins and alter the permeability of the mitochondrial membrane, thereby inducing apoptosis. Our study found that SalA reversed the H2O2-induced increase in the LC3 II/I ratio and NLRP3 expression, and inhibited HUVECs apoptosis. These results indicating that SalA could alleviate the oxidative stress induced endothelial cell death.
The molecular mechanisms underlying EC senescence or death are not fully understood. In recent years an increasing number of studies on miRNAs as modulators of endothelial dysfunction have provided new insights into endothelial dysfunction, leading to potential new therapeutic approaches. miR-204-5p was reported to regulate apoptosis, autophagy, and cellular senescence by inhibiting target genes in multiple cell types33,34,35. Apoptosis is one of the most common phenotypes regulated by miR-204-5p and its target genes. Bcl-2 is the most common target of miR-204-5p and a key apoptosis regulator by interacting with proapoptotic members of the Bcl-2 family36,37. Our data showed that SalA inhibited the expression of miR-204-5p in H2O2-induced HUVECs, and prompted the expression of Bcl-2.
Notably, when cells are injured, p53 protein acts as a major transcription factor, which is involved in the activation of various factors of the cell cycle, in addition to controlling multiple pathways such as cell senescence, apoptosis, autophagy, etc., and determining the multiple fates of cells under stress conditions38,39. Bcl-2 has been confirmed to prevent p53-mediated apoptosis. In addition, p53 has a dual regulatory effect on autophagy40,41. It has also been demonstrated that Bcl-2 complex has a key role in the link between autophagy and apoptosis responses42. From this it is clear that p53 are important in cellular senescence and cell fate decision. Our results suggested that SalA may target miR-204-5p to regulate the expression of p53.
The major limitation of this study is all the experiments were performed based on the cell line and without proper in vivo results. On the other hand, as confirmed by our results, SalA rescues endothelial cell injury by regulating senescence, autophagy, and apoptosis, respectively, but whether there is a correlation between the occurrence of these events still needs to be further explored (Fig. 7). Our future attempts will be to correlate all events using p53 and to clarify the experimental evidence of SalA in vivo.

SalA could alleviate H2O2-induced oxidative injury by downregulating miR-204-5p in HUVECs.
Materials and methods
Materials and chemicals
HUVECs were purchased from Cell Bank of Type Culture collection of Chinese Academy of Sciences (Shanghai, China). Salvianolic acid A (C26H22O10, HPLC ≥ 98%) was purchased from Aladdin Bio-Chem Technology Company (Shanghai, China). has-miR-204-5p mimic/inhibitor were purchased from Guangzhou Ribo Biotechnology Company (Guangzhou, China).
Cell culture and treatment
HUVECs were cultured in RPMI 1640 medium (Hyclone, USA) supplemented with 10% fetal bovine serum (Biological Industries, USA), 1% Penicillin–Streptomycin at 37 °C in a humidified atmosphere of 5% CO2. 200 μM H2O2 was chosen as the modeling concentration to induce oxidative damage in HUVECs. Cells were exposed to 200 μM H2O2 for 12 h in serum-free medium following 24 h of normal culture. For SalA treatment, cells were treated with different concentrations of SalA for 24 h after H2O2 inducing. Cells at passages 3 to 8 were used in all experiments.
Cell viability assay
Cells (1 × 104 cells/mL, 100 μL/well) were seeded in the 96-well plates with five replicate wells. After treatment, 20 μL Cell Counting Kit-8 reagent (CCK-8, Beyotime, China) was added into each well and incubated at 37 °C for 2 h. The absorbance was measured at 450 nm. Cell viability was defined as a percentage of the control group.
Cell proliferation detection
After treatments, cell proliferation rate was detected by BeyoClick™ EdU Cell Proliferation Kit with Alexa Fluor 488 (Beyotime, China). In short, cells were incubated with EdU solution at 37 °C for 2 h and fixed with 4% paraformaldehyde at room temperature for 15 min. Then, cells were permeabilized with 0.3% Triton X-100 (prepared with PBS) for 15 min and incubated with the Click reaction solution for 30 min in dark. After, cells were incubated with 1 × Hoechst 33,342 solution at room temperature for 10 min in dark. The ECLIPSE Ti-DH fluorescence microscope (Nikon, Japan) was used to count the numbers of proliferative cells (EdU-positive) in three random fields of view per slide.
Lactate dehydrogenase (LDH) leakage assay
A LDH assay kit (Beyotime, China) was used to determine the level of extracellular LDH for assessment of cell cytotoxicity. After treatment, the culture medium was collected and centrifugated at 1000 rpm/min for 5 min, the supernatant was employed to add 60 µL LDH working solution following the manufacturer's instructions. The relative LDH release rate was calculated using absorbance at 490 nm.
Intracellular reactive oxygen species (ROS) assay
A ROS assay kit (Beyotime, China) was used to determine the ROS levels of cells. Cells were incubated with DCFH-DA solution (1:1000, diluted in serum-free RPMI 1640 culture medium) at 37 °C for 20 min. Then cells were washed with serum-free RPMI 1640 medium thrice, and the relative fluorescence intensity of cells was detected within 30 min under fluorescence microscope.
Superoxide dismutase (SOD) assay
Following the manufacturer’s instructions (Beyotime, China), after treatments, cells were lysed with 50 μL SOD sample preparation solution per well at 4 °C for 15 min and centrifuged at 12,000 rpm/min for 5 min. In a 96-well plate, the supernatant was added to the working solution and incubated at 37 °C for 30 min. The absorbance was determined at 450 nm.
Apoptosis detection with flow cytometry
Flow cytometry was used to detect cell apoptosis. According to the instructions of Annexin V-FITC/PI Apoptosis Kit (Multi Sciences, China), cells were collected and resuspended by adding 500 μL of 1 × Binding Buffer, and then 5 μL of Annexin V-FITC and 10 μL of PI were added in dark conditions. The apoptotic cells were analyzed by flow cytometry (CytoFlex S, Beckman Coulter, USA).
miRNA transfection
Cells in the exponential phase of growth were plated in six-well plates at 2 × 105 cells/plate and cultured for 24 h. miR-204-5p mimic/inhibitor (100 nM) and negative controls were transfected to cells for 6 h using Lipofectamine RNAimax transfection reagent (Invitrogen, USA) according to the manufacturer’s protocols. Cells were induced with H2O2, 24 h after the end of miR-204-5p mimic/inhibitor transfection.
Western blotting
After treatments, cells were washed with PBS twice and lysed with RIPA lysis buffer (Beyotime, China) on ice, and the protein concentration was determined by BCA Protein assay kit (Beyotime, China). We separated the protein (40 μg/lane) from each sample by SDS-PAGE gel electrophoresis and transferred it to a PVDF membrane (MilliporeSigma, Germany). The membranes were blocked with 5% non‑fat milk blocking buffer for 2 h at room temperature, washed thrice with TBST. Primary antibodies were incubated overnight on PVDF membranes at 4 °C for 12 h: anti-BAX (1:1000; Proteintech, USA); anti-Bcl-2 (1:1000; Proteintech, USA); anti-Cyclin E1 (1:1000; ImmunoWay, USA); anti-p53(1:2000; Proteintech, USA); anti-p21 (1:500; Beyotime, China); anti-LC3 (1:2000; Cell Signaling Technology, USA); anti-NLRP3 (1:1,000; Cell Signaling Technology, USA); and anti-GAPDH (1:2000; ImmunoWay, USA). And then incubated with HRP‑conjugated goat anti‑rabbit (abs20040ss, 1:30,000; Absin, China) or goat anti-mouse antibody (DW0990, 1:50,000; Dawen Biotec., China) at room temperature for 2 h. The ChemiScope 6200 and ChemiScope 3300 detection systems were used to scan immunoblot bands and capture images. Each experiment was repeated three times using the same procedure to obtain an average value. ImageJ software (version 1.53, https://imagej.net) was used to analyze band intensity. GAPDH was used as the loading control.
Reverse transcription and real-time qPCR
RNA was extracted by Trizol method. After treatment, the cells were washed twice with pre-cooled PBS, and split on ice for 30 min with 1 mL of RNAiso Plus reagent (TAKARA, Japan) added to each well. After adding 200 μL of chloroform, the mixture was centrifuged at 12,000 rpm/min for 15 min at 4 °C. The 400 μL of supernatant was aspirated, and an equal volume of isopropanol was added, and placed at room temperature for 20 min before centrifugation at 12,000 rpm/min and 4 °C for 15 min. Then 1 mL of 75% ethanol was added to each tube, followed by centrifugation at 12,000 rpm/min for 5 min at 4 °C. After discarding the supernatant and drying at room temperature, 50 μL DEPC water was added to dissolve the RNA.
A Mir-XTM miRNA First-Strand Synthesis Kit (TAKARA, Japan) was used for reverse transcription. Real-time PCR was performed using SYBR® Prime Ex Taq TM II (Tli RNase H Plus) (TAKARA, Japan). Each qPCR reaction contained 12.5 μL 2X TB Green Premix Ex Taq II, 0.5 μL primers (10 µM), 0.5 μL 50X ROX Reference Dye, 2 μL cDNA template, and 9 μL ddH2O to obtain a final volume of 25 μL. The Step One Plus™ Real-Time PCR System (Applied Biosystems, USA) was used for cDNA amplification and detection. The thermal cycler conditions were as follows: Hold for 10 s at 95 °C, followed by 40 cycles of a two‑step PCR consisting of a 95 °C step for 10 s and a 60 °C step for 20 s. U6 was utilized as a housekeeping gene for normalizing miRNA expression levels. The data were analyzed according to the 2–ΔΔCt method.
The primers were as follows: miR-204-5p: 5′-GCGAGCACAGAATTAATACGC-3′ (forward) and 5′-TCAGTGCACTACAGAACTTTGT-3′ (reverse); U6: 5′-CTCGCTTCGGCAGCACA-3′ (forward) and 5′-AACGCTTCACGAATTTGCGT-3′ (reverse).
Statistical analysis
If not stated otherwise, results are based on at least three independent experiments (n ≥ 3), and data are presented as mean ± standard deviation (SD) by using GraphPad Prism 8 software (https://www.graphpad.com). For multiple comparisons, the measurement data were subjected to one-way analysis of variance (ANOVA) followed by Tukey post hoc test. P < 0.05 and P < 0.01 was considered a significant difference.
Data availability
All data generated during this study are included in this published article and its supplementary file.
References
Albany, C. J., Trevelin, S. C., Giganti, G., Lombardi, G. & Scottà, C. Getting to the heart of the matter: The role of regulatory T-cells (Tregs) in cardiovascular disease (CVD) and atherosclerosis. Front. Immunol. 10, 2795. https://doi.org/10.3389/fimmu.2019.02795 (2019).
Zhao, D., Liu, J., Wang, M., Zhang, X. & Zhou, M. Epidemiology of cardiovascular disease in China: Current features and implications. Nat. Rev. Cardiol. 16(4), 203–212. https://doi.org/10.1038/s41569-018-0119-4 (2019).
Moretti, R. et al. Common shared pathogenic aspects of small vessels in heart and brain disease. Biomedicines. 10(5), 1009. https://doi.org/10.3390/biomedicines10051009 (2022).
Ambrosino, P. et al. Mechanisms and clinical implications of endothelial dysfunction in arterial hypertension. J. Cardiovasc. Dev. Dis. 9(5), 136. https://doi.org/10.3390/jcdd9050136 (2022).
Raghunandan, S. et al. Heme oxygenase-1 at the nexus of endothelial cell fate decision under oxidative stress. Front. Cell. Dev. Biol. 9, 702974. https://doi.org/10.3389/fcell.2021.702974 (2021).
Manoharan, R. R., Prasad, A., Pospíšil, P. & Kzhyshkowska, J. ROS signaling in innate immunity via oxidative protein modifications. Front. Immunol. 15, 1359600. https://doi.org/10.3389/fimmu.2024.1359600 (2024).
Lin, X. et al. Focus on ferroptosis, pyroptosis, apoptosis and autophagy of vascular endothelial cells to the strategic targets for the treatment of atherosclerosis. Arch. Biochem. Biophys. 715, 109098. https://doi.org/10.1016/j.abb.2021.109098 (2022).
Bai, B. et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 11(9), 776. https://doi.org/10.1038/s41419-020-02985-x (2020).
Xie, X. et al. The protective effect of an extract of Salvia miltiorrhiza Bunge (Danshen) on cerebral ischemic injury in animal models: A systematic review and meta-analysis. J. Ethnopharmacol. 317, 116772. https://doi.org/10.1016/j.jep.2023.116772 (2023).
Duan, S. et al. Integrated proteomics and phosphoproteomics profiling reveals the cardioprotective mechanism of bioactive compounds derived from Salvia miltiorrhiza Burge. Phytomedicine. 117, 154897. https://doi.org/10.1016/j.phymed.2023.154897 (2023).
Guo, Y. et al. Effect of main ingredients of Danhong Injection against oxidative stress induced autophagy injury via miR-19a/SIRT1 pathway in endothelial cells. Phytomedicine 83, 153480. https://doi.org/10.1016/j.phymed.2021.153480 (2021).
Zhou, Y. et al. The miR-204-5p/FOXC1/GDF7 axis regulates the osteogenic differentiation of human adipose-derived stem cells via the AKT and p38 signalling pathways. Stem. Cell. Res. Ther. 12(1), 64. https://doi.org/10.1186/s13287-020-02117-4 (2021).
Yu, D. S. et al. Salvianolic acid A ameliorates the integrity of blood–spinal cord barrier via miR-101/Cul3/Nrf2/HO-1 signaling pathway. Brain Res. 1657, 279–287. https://doi.org/10.1016/j.brainres.2016.12.007 (2017).
Grieco, F. A. et al. The miRNAs miR-211-5p and miR-204-5p modulate ER stress in human beta cells. J. Mol. Endocrinol. 63(2), 139–149. https://doi.org/10.1530/JME-19-0066 (2019).
Kassan, M. et al. MicroRNA-204 promotes vascular endoplasmic reticulum stress and endothelial dysfunction by targeting Sirtuin1. Sci. Rep. 7(1), 9308. https://doi.org/10.1038/s41598-017-06721-y (2017).
Zhu, X. et al. LncRNA LYPLAL1-DT screening from type 2 diabetes with macrovascular complication contributes protective effects on human umbilical vein endothelial cells via regulating the miR-204-5p/SIRT1 axis. Cell Death Discov. 8(1), 245. https://doi.org/10.1038/s41420-022-01019-z (2022).
Guo, Y. et al. 2,3,5,4’-Tetrahydroxystilbene-2-O-β-D-glucoside modulated human umbilical vein endothelial cells injury under oxidative stress. Korean J. Physiol. Pharmacol. 24(6), 473–479. https://doi.org/10.4196/kjpp.2020.24.6.473 (2020).
Wu, Z., Wang, H., Fang, S. & Xu, C. Roles of endoplasmic reticulum stress and autophagy on H2O2-induced oxidative stress injury in HepG2 cells. Mol. Med. Rep. 18(5), 4163–4174. https://doi.org/10.3892/mmr.2018.9443 (2018).
Chen, Z. et al. INF2 regulates oxidative stress-induced apoptosis in epidermal HaCaT cells by modulating the HIF1 signaling pathway. Biomed. Pharmacother. 111, 151–161. https://doi.org/10.1016/j.biopha.2018.12.046 (2019).
Guo, G. & Dong, J. Diosmetin attenuates oxidative stress-induced damage to lens epithelial cells via the mitogen-activated protein kinase (MAPK) pathway. Bioengineered 13(4), 11072–11081. https://doi.org/10.1080/21655979.2022.2068755 (2022).
Cao, Y. et al. Punicalagin prevents inflammation in LPS-induced RAW264.7 macrophages by inhibiting FoxO3a/autophagy signaling pathway. Nutrients 11(11), 2794. https://doi.org/10.3390/nu11112794 (2019).
Tai, G. J. et al. NLRP3 inflammasome links vascular senescence to diabetic vascular lesions. Pharmacol. Res. 178, 106143. https://doi.org/10.1016/j.phrs.2022.106143 (2022).
Lin, Y. C. et al. Tumor suppressor miRNA-204-5p promotes apoptosis by targeting BCL2 in prostate cancer cells. Asian J. Surg. 40(5), 396–406. https://doi.org/10.1016/j.asjsur.2016.07.001 (2017).
Deng, W. et al. Inhibition of PLK3 attenuates tubular epithelial cell apoptosis after renal ischemia-reperfusion injury by blocking the ATM/P53-mediated DNA damage response. Oxid. Med. Cell. Longev. 2022, 4201287. https://doi.org/10.1155/2022/4201287 (2022).
Bu, L. L. et al. New dawn for atherosclerosis: Vascular endothelial cell senescence and death. Int. J. Mol. Sci. 24(20), 15160. https://doi.org/10.3390/ijms242015160 (2023).
Xu, S. et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: From mechanism to pharmacotherapies. Pharmacol. Rev. 73(3), 924–967. https://doi.org/10.1124/pharmrev.120.000096 (2021).
Aboutaleb, N., Jamali, H., Abolhasani, M. & Pazoki Toroudi, H. Lavender oil (Lavandula angustifolia) attenuates renal ischemia/reperfusion injury in rats through suppression of inflammation, oxidative stress and apoptosis. Biomed. Pharmacother. 110, 9–19. https://doi.org/10.1016/j.biopha.2018.11.045 (2019).
Zhang, H. F. et al. Salvianolic acid A protects the kidney against oxidative stress by activating the Akt/GSK-3β/Nrf2 signaling pathway and inhibiting the NF-κB signaling pathway in 5/6 nephrectomized rats. Oxid. Med. Cell. Longev. 2019, 2853534. https://doi.org/10.1155/2019/2853534 (2019).
Li, X. L., Fan, J. P., Liu, J. X. & Liang, L. N. Salvianolic acid A protects neonatal cardiomyocytes against hypoxia/reoxygenation-induced injury by preserving mitochondrial function and activating Akt/GSK-3β signals. Chin. J. Integr. Med. 25(1), 23–30. https://doi.org/10.1007/s11655-016-2747-z (2019).
Omidifar, N. et al. Trends in natural nutrients for oxidative stress and cell senescence. Oxid. Med. Cell Longev. 2021, 7501424. https://doi.org/10.1155/2021/7501424 (2021).
Gholami, A., Mohammadi, F., Ghasemi, Y., Omidifar, N. & Ebrahiminezhad, A. Antibacterial activity of SPIONs versus ferrous and ferric ions under aerobic and anaerobic conditions: A preliminary mechanism study. IET Nanobiotechnol. 14(2), 155–160. https://doi.org/10.1049/iet-nbt.2019.0266 (2020).
Chen, J. et al. Chloroquine regulates the proliferation and apoptosis of palate development on mice embryo by activating P53 through blocking autophagy in vitro. In Vitro Cell Dev. Biol. Anim. 58(7), 558–570. https://doi.org/10.1007/s11626-022-00704-8 (2022).
Yang, F., Bian, Z., Xu, P., Sun, S. & Huang, Z. MicroRNA-204-5p: A pivotal tumor suppressor. Cancer Med. 12(3), 3185–3200. https://doi.org/10.1002/cam4.5077 (2023).
Kang, Y. et al. Long noncoding RNA KCNQ1OT1 promotes the progression of non-small cell lung cancer via regulating miR-204-5p/ATG3 axis. Onco Targets Ther. 12, 10787–10797. https://doi.org/10.2147/OTT.S226044 (2019).
Song, S., Fajol, A., Tu, X., Ren, B. & Shi, S. MiR-204 suppresses the development and progression of human glioblastoma by targeting ATF2. Oncotarget 7(43), 70058–70065. https://doi.org/10.18632/oncotarget.11732 (2016).
Houzelle, A. et al. MicroRNA-204-5p modulates mitochondrial biogenesis in C2C12 myotubes and associates with oxidative capacity in humans. J. Cell. Physiol. 235(12), 9851–9863. https://doi.org/10.1002/jcp.29797 (2020).
Yang, S. et al. miR-204-5p promotes apoptosis and inhibits migration of gastric cancer cells by targeting HER 2. Mol. Med. Rep. 22(4), 2645–2654. https://doi.org/10.3892/mmr.2020.11367 (2020).
Wang, H., Guo, M., Wei, H. & Chen, Y. Targeting p53 pathways: mechanisms, structures, and advances in therapy. Signal Transduct. Target Ther. 8(1), 92. https://doi.org/10.1038/s41392-023-01347-1 (2023).
Yu, G. et al. Loss of p53 sensitizes cells to palmitic acid-induced apoptosis by reactive oxygen species accumulation. Int. J. Mol. Sci. 20(24), 6268. https://doi.org/10.3390/ijms20246268 (2019).
White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 6(4), a026120. https://doi.org/10.1101/cshperspect.a026120 (2016).
Dong, Y. et al. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J. Mol. Cell Cardiol. 136, 27–41. https://doi.org/10.1016/j.yjmcc.2019.09.001 (2019).
Zając, A. et al. The role of Bcl-2 and Beclin-1 complex in “switching” between apoptosis and autophagy in human glioma cells upon LY294002 and Sorafenib treatment. Cells 12(23), 2670. https://doi.org/10.3390/cells12232670 (2023).
Acknowledgements
We are very grateful to the Public Platform of Medical Research Center, Academy of Chinese Medical Science, Zhejiang Chinese Medical University, and reviewers for their valuable help in completing this article.
Funding
This work was supported by the Natural Science Foundation of China (Nos. 82374071 and 82304763); the Natural Science Foundation of Zhejiang Province, China (LY23H280006) and the Scientific Research Foundation of Zhejiang Chinese Medical University (2022GJYY024).
Author information
Authors and Affiliations
Contributions
B.J. conceived and designed the study. X.L.Q., S.Y.C., S.Y.Ch. and Y.G. performed the experiments, acquired and analyzed the data. X.L.Q. wrote the manuscript. N.P.C. and Y.Z. contributed to the critical revision of the manuscript. All authors have read and approved the final manuscript. All authors agree to be accountable for all aspects of work ensuring integrity and accuracy.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qiao, X., Cao, S., Chen, S. et al. Salvianolic acid A alleviates H2O2-induced endothelial oxidative injury via miR-204-5p. Sci Rep 14, 11931 (2024). https://doi.org/10.1038/s41598-024-62556-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-62556-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
