
Abstract
Smoking exposure during adulthood can disrupt oocyte development in women, contributing to infertility and possibly adverse birth outcomes. Some of these effects may be reflected in epigenome profiles in granulosa cells (GCs) in human follicular fluid. We compared the epigenetic modifications throughout the genome in GCs from women who were former (N = 15) versus never smokers (N = 44) undergoing assisted reproductive technologies (ART). This study included 59 women undergoing ART. Smoking history including time since quitting was determined by questionnaire. GCs were collected during oocyte retrieval and DNA methylation (DNAm) levels were profiled using the Infinium MethylationEPIC BeadChip. We performed an epigenome-wide association study with robust linear models, regressing DNAm level at individual loci on smoking status, adjusting for age, ovarian stimulation protocol, and three surrogate variables. We performed differentially methylated regions (DMRs) analysis and over-representation analysis of the identified CpGs and corresponding gene set. 81 CpGs were differentially methylated among former smokers compared to never smokers (FDR < 0.05). We identified 2 significant DMRs (KCNQ1 and RHBDD2). The former smoking-associated genes were enriched in oxytocin signaling, adrenergic signaling in cardiomyocytes, platelet activation, axon guidance, and chemokine signaling pathway. These epigenetic variations have been associated with inflammatory responses, reproductive outcomes, cancer development, neurodevelopmental disorder, and cardiometabolic health. Secondarily, we examined the relationships between time since quitting and DNAm at significant CpGs. We observed three CpGs in negative associations with the length of quitting smoking (p < 0.05), which were cg04254052 (KCNIP1), cg22875371 (OGDHL), and cg27289628 (LOC148145), while one in positive association, which was cg13487862 (PLXNB1). As a pilot study, we demonstrated epigenetic modifications associated with former smoking in GCs. The study is informative to potential biological pathways underlying the documented association between smoking and female infertility and biomarker discovery for smoking-associated reproductive outcomes.
Similar content being viewed by others


Introduction
Cigarette smoking has been well associated with female infertility1,2. Both women who are current smokers and those who have quit smoking before conception were found to have lower natural fertility3. Previous studies have well-associated smoking with ovarian dysfunction as reflected clinically by lower ovarian reserve, poorer response to ovarian stimulation, and earlier age at menopause4. However, the underlying biological mechanisms remained to be further investigated.
Epigenetics is a useful tool for understanding the biological responses of environmental exposure to adverse health impacts and providing insights into biomarker development. Mounting studies associated smoking with changes in DNA methylome in a variety of biosamples, including the placenta, fetal cord blood, peripheral blood, and lung tissue, some of which were found to be persistent into childhood and adolescence5,6,7,8,9,10,11. To our best knowledge, none has investigated the smoking-associated epigenetic modifications in human granulosa cells (GCs) obtained from follicular fluid (FF).
Human FF is responsible for the constant and sufficient supply of nutrients to the oocyte, contributing to its maturation and good quality, which is a key element for quality of embryo and subsequent success of in vitro fertilization (IVF) treatment12,13. IVF is the most common assisted reproductive treatment (ART) procedure performed. The sources of FF mainly include the secretory activity of GCs and thecal cells, and blood plasma composition that crosses the thecal capillaries14. A recent study within women undergoing IVF reported that cigarette smoking was associated with de-regulation of 26 oxidative stress-related genes in cumulus cells originating from relatively undifferentiated GCs15. They also observed significantly lower methylation at promoter of interleukin-6 (IL-6) which is an important inflammation factor. It is plausible that DNAm altered by smoking exposure in these cells may affect the FF microenvironment, and as a consequence having influences on oocyte dysfunction. Therefore, interrogation on the association of smoking with DNAm in GCs may be informative about biological mechanisms underlying smoking-associated female infertility demonstrated by oocyte quality and dysfunction. In addition, the aspiration of oocytes from follicle is accompanied by FF during IVF treatment. Thus, GCs in FF is an ideal and reasonable biosample for screening of oocyte quality and dysfunction, particularly among those undergoing IVF. There are growing number of studies focused on discovery of protein and microRNA biomarkers for IVF in FF but less on DNAm biomarkers13,16,17.
We performed a pilot study consisting of 59 women undergoing ART who were non-active smokers at recruitment. This provides a unique opportunity to study the impacts of smoking, after cessation, on DNAm in GCs. We aimed to investigate the associations of former smokers versus never smokers with DNAm differences, which may have implications for better understanding the biological pathways through which prior smoking leads to ovarian dysfunction and providing insights into biomarker discovery for smoking-associated reproductive outcomes.
Methods
Study population
The study population in our analysis was a subset of participants in the Environment and Reproductive Health (EARTH) Study, a prospective cohort that was established for evaluating factors that influence human fertility and pregnancy outcomes18. Specifically, the participants consisted of women between 18 and 45 years of age who sought fertility evaluation and treatment at the Massachusetts General Hospital (MGH) Fertility Center (2004–2019). Infertility insurance coverage in Massachusetts requires smoking cessation prior to treatment. Therefore, current smokers could enroll in the EARTH Study (prior to starting infertility treatment) but were excluded in this analysis since it was unclear how recently they had quit smoking between joining EARTH and prior to starting IVF. At enrollment, participants completed questionnaires for demographics, lifestyle, and medical and reproductive history. Anthropometric measurements were collected by trained research staff and used to determine body mass index (BMI). For this pilot study, eligible participants were women who underwent an ART cycle between 2006 and 2016 and provided a FF sample during oocyte retrieval process. The details of participant inclusion criteria for this study are provided elsewhere19.
The institutional review boards of the Massachusetts General Hospital and Harvard T.H. Chan School of Public Health approved this study and all subjects met with trained study staff before providing written informed consent. All methods in the study were conducted in adherence to relevant guidelines and regulations.
Follicular fluid collection
Oocyte retrieval occurred after 9–14 days of controlled ovarian stimulation. FF was retrieved from the first three follicles using a 16 G needle, with separate tubes for each sample containing 1 ml of flushing media. FF was then centrifuged to separate the supernatant and cellular pellet, and stored at − 80 °C. The stored pellet from the first aspirated follicle was shipped to the Emory Integrated Genomics Core on dry ice.
Quality control and processing of DNA methylation data
DNA was extracted using the QIAamp UCP DNA Micro Kit (Qiagen, Hilden, Germany), and quantification was performed with Quant-iT dsDNA broad range assay kit (ThermoFisher, Waltham, MA), and assessed for quality on a 2% agarose gel. DNA preparation followed the Illumina Infinium HD Assay Methylation Protocol Guide. To limit potential batch effects, samples were randomly distributed across the plate. The Emory Integrated Genomics Core performed bisulfite modification with the EZ DNA Methylation Kit (Zymo Research, Irvine, CA), DNAm was quantified across the genome with the Illumina MethylationEPIC Beadarray (Illumina, San Diego, CA) following the manufacturer’s protocol. We utilized ‘minfi’ package for quality control and pre-processing, and applied their method for detection p-values20. To reduce technical artefacts and probe type bias, we performed functional normalization and beta-mixture quantile (BMIQ) normalization21,22. We excluded probes on the X and Y chromosomes and those exhibiting low variability (with a standard deviation of beta-values < 0.02)23.
Accounting for unmeasured confounding
We also aimed to control for technical confounders and cellular composition in our data, but given the small sample size we only had one batch and there is no reference methylome for GCs at this time. Thus, we utilized surrogate variable analysis (sva package in R) to estimate major sources of variation in our data, some of which are likely reflective of differences in cellular composition between samples, technical artefacts, or other unmeasured confounders. The first three surrogate variables were included in our models.
Statistical analyses
Epigenome-wide association study (EWAS): identification of former smoking-associated CpGs
We performed an epigenome-wide association study (EWAS) using robust linear regressions for each CpG sites, with the methylation beta-values (continuous) as dependent variables and the smoking history (2-level factor: former vs. never smokers) as the independent variable, while adjusting for age, ovarian stimulation protocol (3-level factor: antagonist, flare, and luteal phase agonist) and three surrogate variables. A Manhattan plot was generated to summarize the EWAS results. Multiple tests were corrected by Benjamini-Horchberg (BH) procedure. Associations within false discovery rate (FDR) of 5% were considered statistically significant.
To test the robustness of the EWAS results, we performed sensitivity analyses that included additional adjustments for education levels (3-level factor: did not graduate from college, college degree, and graduate degree) and BMI (continuous, kg/m2) and compared the effect coefficients before and after the further adjustments. We limited this sensitivity analysis to the CpG sites that met the FDR of 5% from the main analysis. All analyses were performed in R version 4.1.0.
Identification of differentially methylated regions (DMRs)
Differentially methylated regions (DMRs) comprised multiple consecutive methylated CpG sites deemed under similar regulatory control, which has been suggested to be more informative for diseases etiologies. We conducted DMR analysis with the CpG sites associated with smoking history at raw p-value < 0.001 with the R package ‘dmrff’24. We specified the maximum distance to 1500 bp for features considered to be proximal consecutive CpG sites for candidate DMRs. To identify the regional differential methylation among candidate regions, dmrff uses an extension of inverse-variance weighted meta-analysis to produce a weighted average difference in DNAm for each candidate DMR. Regional test-statistics were performed and those DMRs with FDR < 0.05 were considered to be statistically significant.
Identification of over-representation of genes in biological pathways and gene ontology (GO) terms
To gain more context of the biological activity of former smoking-associated CpG sites, we performed over-representation analyses to identify enrichment pathways and GO terms among the genes annotated to CpG sites with suggestive association at raw p-value < 10–4 from EWAS. R package ‘org.Hs.eg.db’ was used to convert genes to Entrez IDs. We used R packages ‘clusterProfiler’, ‘ReactomePA’, and ‘DOSE’ for KEGG pathway25 and GO term analysis, REACTOME pathway analysis, and disease ontology semantic and enrichment analysis, respectively. Results within a 10% BH-corrected FDR were considered significant over-representation.
Examination of correlation between the time since quitting smoking and DNAm at the significant CpG sites among former smokers
We also examined the association between DNAm at individual genome-wide significant CpG sites and time since smoking cessation among the former smokers. We regressed methylation levels at each significant CpG on time since quitting smoking (years) and three surrogate variables. We then used “visreg” package to visualize the partial correlation between each CpG and time since quitting smoking, conditioning on three surrogate variables.
Ethics approval and consent to participate
The institutional review boards of the Massachusetts General Hospital and Harvard T.H. Chan School of Public Health approved this study. All subjects met with trained study staff before providing written informed consent.
Results
Characteristics of the study population
A total of 132 women participating in the EARTH study had a preserved FF sample available for analysis. One sample was damaged during transit. DNA extraction yielded from the remaining 131 samples exhibited a range from 0 to 2896 ng. Only 60 women had DNA extractions of ≥ 200 ng, while 16 had extractions ranging from 100 to 199 ng. We included 64 samples on the DNAm array, including four with DNA extractions yielded between 100 and 199 ng. We excluded two samples that had more than 5% of probes yielding detection p-values > 0.001 and another sample that was an outlier in principal components analyses. Furthermore, we excluded one participant who was a current smoker at enrollment into EARTH and one participant with missing covariate data. Consequently, the analytic dataset comprised 474,545 probes from 59 samples.
The 59 women in our study had a mean (range) age of 35 (28–44) years at enrollment, of which 86% were white. The mean BMI was 25 kg/m2. 75% were never smokers and 25% were former smokers. The time since smoking cessation among the 15 former smokers ranged from 1 to 17 years. 73%, 17%, and 10% of participants received luteal phase agonist, flare, and antagonist protocols, respectively. The overall education level was high, with 37% of women having completed a college degree and 56% having completed a graduate degree (Table 1).
Identification of former smoking-associated CpGs
We identified 81 CpGs that were differentially methylated with former smoking at a 5% FDR (Excel Table S1), which were spread throughout the genome (Fig. 1). Comparing former smokers to never smokers, 63 CpGs had higher methylation levels with the effect sizes ranging between 1.0 and 8.0%, while 18 had lower methylation with the effect sizes ranging from − 2.1 to − 7.1% in former smokers. Except for 12 CpGs that were intergenic, the remaining 69 CpGs were within different genes. We also explored whether adjustment for additional potential confounders may explain our findings. Further adjustment for education levels and BMI did not have large impacts on effect size of the 81 significant probes (Fig. S1). Among the top 10 most statistically significant associations from EWAS, two CpGs were intergenic while the other eight CpGs were within PLA2G4A, ACAP3, SLC22A18, CHST8, ADGRG1, ZAR1L, KCNK9, and AK7 (Table 2). The full EWAS results are provided (Excel Table S2).

A Manhattan plot of the epigenome-wide association study (EWAS) results. All models were adjusted for age at enrollment, ovarian stimulation protocol, and three surrogate variables. The blue line represents the false discovery rate (FDR) of 5%. The annotated dots include the top 10 most statistically significant former smoking-associated CpGs (gene).
Identification of DMRs
We identified 16 candidate DMR regions with at least two differentially methylated CpGs, two of which were significant DMRs with FDR < 0.05 and additional five of which were with raw p-value < 10–5. The most significant DMR (FDR = 4.01 × 10–11) was within KCNQ1 gene. The other significant DMR (FDR = 1.01 × 10–6) was intergenic, but was 4 kb upstream of RHBDD2. The additional five DMRs with raw p-value < 10–5 were within genes DEGS2, ELMO3, C10orf41, MAS1L, and LMNTD1 (Table 3). Significant smoking-associated CpGs within these identified DMRs were all hypermethylated among former versus never smokers (Fig. 2, Excel Table S3).

A volcano plot of the beta coefficients and -log10 (p-values) from the epigenome-wide association study (EWAS). All models were adjusted for age at enrollment, ovarian stimulation protocol, and three surrogate variables. Grey dots denote CpGs that were associated with former smoking at raw p-value < 0.001 from EWAS. Different colors highlight significant CpGs within two significant differentially methylated regions (DMRs) with FDR < 0.05 and five additional DMRs with raw p-value < 10–5. Different shapes represent the regulatory annotations for the CpGs that are highlighted from the DMR analysis and * denotes the DMRs with FDR < 0.05.
Identification of biological pathways and GO terms over-represented in gene set
For the pathway enrichment analyses, we used a list of genes annotated to the CpGs that yielded p-value < 0.0001, among which 260 matched to Entrez gene IDs. Five KEGG pathways and 24 GO terms were significantly over-represented (FDR < 0.1). The five KEGG pathways are oxytocin signaling pathway (hsa: 04921), adrenergic signaling in cardiomyocytes (hsa: 04261), platelet activation (hsa: 04611), axon guidance (hsa: 04360), and chemokine signaling pathway (hsa: 04062), which contained 9, 8, 7, 8, and 8 genes from our gene set, respectively (Table 4). 24 GO terms were related to general biological processes and molecular functions, which were mainly channel activity, membrane transporter and antiporter activity, oxidoreductase activity, and G protein activity (Fig. S2, Excel Table S4).
Correlation between the time since quitting smoking and DNAm at the significant CpG sites among former smokers
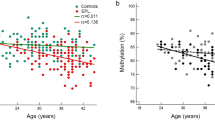
Last, we explored whether the DNAm levels for each of 81 significant CpGs was associated with time since smoking cessation, among the subset of former smokers (n = 15). We regressed the methylation levels at each significant CpG on the time since quitting (years), while controlling for three surrogate variables using linear regression. As a result, four CpGs were significantly associated with the time since smoking cessation while controlling for three surrogate variables (p < 0.05), which were cg04254052 (KCNIP1) (\(\beta\)= − 0.006, p = 0.004), cg13487862 (PLXNB1) (\(\beta\) = 0.003, p = 0.007), cg22875371 (OGDHL) (\(\beta\) = − 0.004, p = 0.022), cg27289628 (LOC148145) (\(\beta\) = − 0.004, p = 0.025) (Excel Table S5). In other words, a one-year increase in the time since smoking cessation was associated with 0.6%, 0.4%, and 0.4% decrease in the methylation levels at cg04254052 (KCNIP1), cg22875371 (OGDHL), and cg27289628 (LOC148145) respectively, and a 0.3% increase in methylation levels at cg13487862 (PLXNB1). For all four of the CpGs where we observed associations with time since quitting (Fig. 3), former smokers had higher DNA methylation levels than never smokers. For three of these four CpGs (annotated to KCNIP1, OGDHL, and LOC148145), participants that quit smoking more recently also had higher DNA methylation levels than those with increased time since cessation.

Partial correlations between each of four former smoking-associated CpGs and the time since smoking cessation among 15 former smokers. The model was conditioned on three surrogate variables.
Discussion
We performed an epigenome-wide association study to evaluate the former smoking-associated variations in DNA methylation that occur in GCs among 59 women undergoing ART. We identified 81 CpGs that were differentially methylated among former smokers compared to never smokers. Additionally, we observed evidence of regional differences in DNAm at the KCNQ1 gene and in one intergenic region upstream of RHBD2. Five KEGG pathways were over-represented among the gene set, including oxytocin signaling pathway, adrenergic signaling in cardiomyocytes, platelet activation, axon guidance, and chemokine signaling pathway. Overall, these epigenetic changes have been implicated in the perturbation of biological processes, including those associated with inflammatory responses, reproductive outcomes, cancer development, neurodevelopmental disorders, and cardiometabolic health. To our knowledge, this is the first epigenome-wide association study on prior smoking exposure in human GCs.
Notably, multiple genes annotated to the former smoking associated CpGs that we identified have been reported in association with former and/ or current smoking in a meta-analysis on epigenetic modification of cigarette smoking with 15,907 blood derived samples from participants in 16 cohorts11. These genes include ACAP3, KCNK9, CHST8, PARD3, TNRC6B, CD37, and several others. We observed a nominal association between former smoking and DNAm changes on cg05575921 within AHRR, a well-documented smoking-associated gene11,26,27, with a p-value of 0.037 (Excel Table S2), but this did not survive the false discovery rate correction threshold (FDR < 0.05). Additionally, we observed the association between time of quitting smoking and methylation differences at five other CpGs within the AHRR gene (p-value < 0.05) (results not shown), but none of them were significantly associated with former smoking in EWAS (FDR < 0.05). This may implicate a power issue. The other possibility could be the uniqueness of our tissue sample, as many DNAm associations are tissue-specific28. Results should be interpreted with caution. Future large-scale investigations on the influences of smoking on DNAm in GC cells should be performed to validate our findings.
The genes annotated to the top 10 statistically significant CpGs (two were intergenic), were PLA2G4A, ACAP3, SLC22A18, CHST8, KCNK9, ZAR1L, ADGRG1, and AK7, which may be indicative to potential disruption of several biological processes. PLA2G4A is crucial for the composition of eicosanoids including prostaglandins and leukotrienes that play important role in inflammatory responses and hemodynamics regulation29,30. ACAP3 has been observed to be involved in regulating neuronal migration in mice, which suggests its important role in brain development31. SLC22A18 is located in 11p15.5, an important tumor-suppressor gene region, together with other 21 genes including KCNQ1 that was found within one identified significant DMR. SLC22A18 encodes a membrane protein, which has been suggested as a tumor suppressor gene in multiple cancers32,33,34, but its function has not been well established. CHST8 (GalNAc-4-ST1) is primarily expressed in the pituitary gland, which is required for the biosynthesis of glycoprotein hormone such as lutropin (LH) and thyrotropin (TSH) by catalyzing sulfate addition of the carbohydrate structures35. KCNK9 encodes an oncogenic potassium (K+) channel, which was found overexpressed in both breast, lung, and colorectal cancers36,37. It has been suggested to promote tumor growth and metastasis in cancer cell lines38. ZAR1L (ZAR2) was recognized as a maternal factor with function of maintaining the stability of maternal transcriptome39, thereby is important for oogenesis and embryogenesis40. Besides, ZAR1L has been suggested potential associated with tumor suppressors, which can repress the transcription of BACA2 to repress breast cancer cells41. ADGRG1 (GPR56) is a representative tumor-related G protein-coupled receptors (GPCRs) member, which plays an important role in tumor cell growth, migration, angiogenesis, and metastasis42,43. AK7 is known to maintain ciliary structure and function whose dysfunction may lead to primary ciliary dyskinesia including bronchiectasis and reduced fertility44,45,46. In summary, the smoking-associated epigenetic modifications have been associated with tumor development, inflammation, reproductive health, and neurodevelopmental disorders.
We further examined whether the amount of time since smoking cessation correlated with DNAm at the identified CpG sites. Four CpGs showed significant correlations with time since cessation, which were cg04254052 (KCNIP1), cg13487862 (PLXNB1), cg22875371 (OGDHL), and cg27289628 (LOC148145). KCNIP1 is an important component of potassium channel complexes, which regulates neuronal membrane excitability47,48. PLXNB1 is involved in extensive biological processes including cell adhesion, cell migration, and cell shape regulation49,50,51,52. OGDHL is a component of the oxoglutarate dehydrogenase complex that is involved in degradation of glucose and glutamate, and has been associated with tumor development and progression53,54. The functions of LOC148145 are not well characterized at this time. For three of these CpGs (annotated to KCNIP1, OGDHL, and LOC148145), the direction of this association implies that methylation levels may revert back to levels that are consistent with never smokers with increasing time since smoking cessation, which aligns with the findings from studies of other tissue samples. Consistently, previous studies have also reported a gradual reversal of methylation levels at certain smoking-relate CpG sites after quitting smoking, while other CpGs exhibit a more persistent smoking-associated modification55,56,57. Several other CpGs showed potential associations (p < 0.1) in our analysis, but these were not statistically significant. We expected more CpGs with reversible methylation after quitting smoking could be identified in a larger sample of former smokers.
We identified two significant smoking-associated DMRs. One DMR (chr11: 2,746,420–2,747,204) contained KCNQ1 gene. KCNQ1 contributes significantly to repolarization process, which has been closely linked to cardiometabolic outcomes including cardiac arrhythmias and type 2 diabetes58,59. The other DMR (chr7: 75,522,467–75,523,398) is at an intergenic region, upstream of RHBDD2, which was found to be overexpressed in breast cancers60. In addition, over-representation analysis identified five enriched pathways, including oxytocin signaling pathway, adrenergic signaling in cardiomyocytes, platelet activation, axon guidance, and chemokine signaling pathway. The oxytocin signaling pathway plays a critical role in various aspects of reproductive health61,62,63,64. Specifically, an increase in synthesis and release oxytocin contributes to the maternal behavior and lactation during pregnancy, and expedites fetal expulsion and reduces postpartum hemorrhage in parturition. Additionally, intermittent oxytocin secretion regulates milk-ejection reflex and initiates maternal behavior in lactation, while disorders in oxytocin secretion have been associated with maternal depression61,62. Platelet activation and its controlled activation are crucial for processes including folliculogenesis, ovulation, placental development, implantation, and embryo devlopment65. The chemokine signaling pathway is a key process in inflammation. Previous studies have associated several chemokines in FF with ovarian function and successful pregnancy following IVF treatment66,67. The smoking-associated CpGs and their corresponding genes identified within these pathways warrants further investigation, which has potential to be developed as screening biomarkers and therapeutic targets for smoking-induced reproductive outcomes. In addition, adrenergic signaling in cardiomyocytes and platelet activation have been associated with adverse cardiovascular outcomes68,69,70, while axon guidance is associated with neuroinflammation71. In summary, these pathways were informative to inflammatory responses, reproductive health, neurodevelopmental, and cardiovascular outcomes.
Overall, our analysis identified former smoking-associated CpGs, DMRs, and gene sets that are involved in several biological processes, including inflammation, reproduction, neurodevelopment, cancer, and cardiovascular health. Whether, or how, such epigenetic modifications in GCs may relate to the health outcomes is not yet clear. But our findings align with other published literature regarding smoking-associated DNAm changes in other biosamples including peripheral blood and placenta6,11, demonstrating that tobacco smoke has a substantial impact on the epigenome, some of which persists even after smoking cessation, and our study adds that such a signature is detectable in GCs which may have important implications for reproductive health.
The above findings should be interpreted in the contexts of the strengths and limitations of this study. First, GCs are a promising biosample for investigating the associations between environmental exposures and female fertility, because these cells are critical mediators of oocyte health and development. Second, current insurance mandates in Massachusetts, which require women to stop smoking prior to initiating ART, meant that we did not have any current smokers in our study. Nevertheless, our pilot study provided a unique opportunity to study the short- and long-term impacts of smoking cessation on DNAm patterns in GCs. This also allowed us to establish temporality between smoking and the observed variations in DNAm. However, there are some limitations of this analysis. First, we were not able to perform cell sorting or to adjust for cellular composition given that there is no epigenome reference for FF. We also did not adjust for some potential confounders such as alcohol or other substance usage. Rates of alcohol and substance use were too low in our sample to have impacted our findings, but are likely considerations for future studies with larger sample sizes. However, we performed surrogate variable analysis, to capture and adjust for the potential impacts of cellular composition and other unmeasured confounders, which is a common approach for EWAS performed on tissues where reference methylomes are not available. Second, this pilot study has a small sample size which limited statistical power, and this is particularly apparent for our exploratory correlation analyses of time since cessation. Therefore, the interpretation and extrapolation of the results should be cautious. Further investigation on reversibility or persistence of smoking-induced epigenetic changes is necessary. Additionally, due to the uniqueness of our samples, we could not identify an appropriate data source for external validation, but we did use bootstrapping to examine the internal validity of our most significant findings, and found that our results were robust and consistent within our small sample. Future studies could incorporate additional measures of biological activity, such as gene or protein expression, to better delineate the affected underlying biological process, which currently remain unclear. Findings from our analysis should also be replicated or expanded upon in studies with larger sample sizes. It will also be of interest to include current smokers in future studies.
Conclusions
In conclusion, our study provides evidence regarding the associations between former smoking with DNAm in GCs. The observed smoking-associated epigenetic modifications in GCs have been associated with similar traits as those reported in other studies with different biosamples, including inflammatory responses, reproductive outcomes, cancer development, neurodevelopmental disorders, and cardiometabolic health. Findings are informative to the potential biological pathways that may underlie the documented association between smoking and female infertility. It is also possible our results have implications for biomarker discovery for smoking-associated reproductive health. Though it is unclear whether the DNAm differences that we detected in GCs have direct influences on pregnancy outcomes or offspring health, future investigations are encouraged.
Data availability
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
Penzias, A. et al. Smoking and infertility: A committee opinion. Fert. Sterility. 110(4), 611–618 (2018).
Augood, C., Duckitt, K. & Templeton, A. Smoking and female infertility: A systematic review and meta-analysis. Hum. Reprod. (Oxford, England). 13(6), 1532–1539 (1998).
de Angelis, C. et al. Smoke, alcohol and drug addiction and female fertility. Reprod. Biol. Endocrinol. 18(1), 21 (2020).
Reproductive Medicine. Electronic address: asrm@asrm.org, Practice Committee of the American Society for Reproductive Medicine. Smoking and infertility: a committee opinion. Fertil. Steril. 110(4), 611–618 (2018).
Fragou, D., Pakkidi, E., Aschner, M., Samanidou, V. & Kovatsi, L. Smoking and DNA methylation: Correlation of methylation with smoking behavior and association with diseases and fetus development following prenatal exposure. Food Chem. Toxicol. 129, 312–327 (2019).
Everson, T. M. et al. Placental DNA methylation signatures of maternal smoking during pregnancy and potential impacts on fetal growth. Nat. Commun. 12(1), 5095 (2021).
Joubert Bonnie, R. et al. DNA methylation in newborns and maternal smoking in pregnancy: Genome-wide consortium meta-analysis. Am. J. Hum. Genet. 98(4), 680–696 (2016).
Chhabra, D. et al. Fetal lung and placental methylation is associated with in utero nicotine exposure. Epigenetics. 9(11), 1473–1484 (2014).
Lee, K. W. K. et al. Prenatal exposure to maternal cigarette smoking and DNA methylation: Epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age. Environ. Health Perspect. 123(2), 193–199 (2015).
Ladd-Acosta, C. et al. Presence of an epigenetic signature of prenatal cigarette smoke exposure in childhood. Environ. Res. 144, 139–148 (2016).
Joehanes, R. et al. Epigenetic signatures of cigarette smoking. Circ. Cardiovasc. Genet. 9(5), 436–447 (2016).
Basuino, L. & Silveira, C. F. Jr. Human follicular fluid and effects on reproduction. JBRA Assist. Reprod. 20(1), 38–40 (2016).
Shen, X. et al. Proteomic analysis of human follicular fluid associated with successful in vitro fertilization. Reprod. Biol. Endocrinol. 15(1), 58 (2017).
Fortune, J. E. Ovarian follicular growth and development in mammals1. Biol. Reprod. 50(2), 225–232 (1994).
Konstantinidou, F. et al. Impact of cigarette smoking on the expression of oxidative stress-related genes in cumulus cells retrieved from healthy women undergoing IVF. Int. J. Mol. Sci. 22(23), 1 (2021).
Chen, F., Spiessens, C., D’Hooghe, T., Peeraer, K. & Carpentier, S. Follicular fluid biomarkers for human in vitro fertilization outcome: Proof of principle. Proteome Sci. 14, 17 (2016).
Qasemi, M. & Amidi, F. Extracellular microRNA profiling in human follicular fluid: New biomarkers in female reproductive potential. J. Assist. Reprod. Genet. 37(8), 1769–1780 (2020).
Messerlian, C., Williams, P. L., & Ford, J. B., et al. The Environment and Reproductive Health (EARTH) Study: A prospective preconception cohort. Hum. Reprod. Open. 2018(2), hoy001 (2018).
Gaskins, A. J. et al. Traffic-related air pollution and supplemental folic acid intake in relation to DNA methylation in granulosa cells. Clin. Epigenet. 15(1), 84 (2023).
Fortin, J.-P., Triche, T. J. Jr. & Hansen, K. D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 33(4), 558–560 (2016).
Teschendorff, A. E. et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics (Oxford, England). 29(2), 189–196 (2013).
Pidsley, R. et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 17(1), 208 (2016).
Logue, M. W. et al. The correlation of methylation levels measured using Illumina 450K and EPIC BeadChips in blood samples. Epigenomics. 9(11), 1363–1371 (2017).
Suderman, M., Staley, J. R., French, R., Arathimos, R., Simpkin, A., & Tilling, K. dmrff: Identifying differentially methylated regions efficiently with power and control. bioRxiv. 2018:508556.
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51(D1), D587–D592 (2022).
Opitz, C. A. et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 478(7368), 197–203 (2011).
Heikkinen, A., Bollepalli, S. & Ollikainen, M. The potential of DNA methylation as a biomarker for obesity and smoking. J. Intern Med. 292(3), 390–408 (2022).
Gutierrez-Arcelus, M. et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 11(1), e1004958 (2015).
Burke, J. E. & Dennis, E. A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 50(Suppl 1), S237–S242 (2009).
Leslie, C. C. Cytosolic phospholipase A2: Physiological function and role in disease. J. Lipid Res. 56(8), 1386–1402 (2015).
Miura, Y. & Kanaho, Y. ACAP3, the GTPase-activating protein specific to the small GTPase Arf6, regulates neuronal migration in the developing cerebral cortex. Biochem. Biophys. Res. Commun. 493(2), 1089–1094 (2017).
Chu, S. H. et al. Correlation of low SLC22A18 expression with poor prognosis in patients with glioma. J. Clin. Neurosci. 19(1), 95–98 (2012).
Schwienbacher, C. et al. Gain of imprinting at chromosome 11p15: A pathogenetic mechanism identified in human hepatocarcinomas. Proc. Natl. Acad. Sci. 97(10), 5445–5449 (2000).
Jung, Y. et al. Characterization of SLC22A18 as a tumor suppressor and novel biomarker in colorectal cancer. Oncotarget 6(28), 1 (2015).
Baenziger, J. U. Glycoprotein hormone GalNAc-4-sulphotransferase. Biochem. Soc. Trans. 31(2), 326–330 (2003).
Mu, D. et al. Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell. 3(3), 297–302 (2003).
Kim, C. J. et al. Altered expression of KCNK9 in colorectal cancers. Apmis. 112(9), 588–594 (2004).
Sun, H. et al. A monoclonal antibody against KCNK9 K(+) channel extracellular domain inhibits tumour growth and metastasis. Nat. Commun. 7, 10339 (2016).
Rong, Y. et al. ZAR1 and ZAR2 are required for oocyte meiotic maturation by regulating the maternal transcriptome and mRNA translational activation. Nucleic Acids Res. 47(21), 11387–11402 (2019).
Wu, Y.-K. & Fan, H.-Y. Revisiting ZAR proteins: The understudied regulator of female fertility and beyond. Cell. Mol. Life Sci. 79(2), 92 (2022).
Misra, S. et al. Cell cycle-dependent regulation of the bi-directional overlapping promoter of human BRCA2/ZAR2 genes in breast cancer cells. Mol. Cancer. 9, 50 (2010).
Ng, K. F., Chen, T. C., Stacey, M. & Lin, H. H. Role of ADGRG1/GPR56 in tumor progression. Cells. 10(12), 1 (2021).
Singh, A. K. & Lin, H.-H. The role of GPR56/ADGRG1 in health and disease. Biomed. J. 44(5), 534–547 (2021).
Panayiotou, C., Solaroli, N. & Karlsson, A. The many isoforms of human adenylate kinases. Int. J. Biochem. Cell Biol. 49, 75–83 (2014).
Fernandez-Gonzalez, A., Kourembanas, S., Wyatt, T. A. & Mitsialis, S. A. Mutation of murine adenylate kinase 7 underlies a primary ciliary dyskinesia phenotype. Am. J. Respir. Cell Mol. Biol. 40(3), 305–313 (2009).
Badano, J. L., Mitsuma, N., Beales, P. L. & Katsanis, N. The ciliopathies: An emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 7, 125–148 (2006).
An, W. F. et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 403(6769), 553–556 (2000).
Hoffman, D. A., Magee, J. C., Colbert, C. M. & Johnston, D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 387(6636), 869–875 (1997).
Kumanogoh, A. & Kikutani, H. Semaphorins and their receptors: novel features of neural guidance molecules. Proc. Japan Acad. Ser. B. 86(6), 611–620 (2010).
Tamagnone, L. et al. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell. 99(1), 71–80 (1999).
Malik, M. F. A. et al. Role of plexin B1 in a breast cancer cohort of pakistani patients and its contribution towards cancer metastasis as indicated by an in vitro model. Anticancer Res. 37(8), 4483–4488 (2017).
Tong, Y. et al. Structure and function of the intracellular region of the plexin-b1 transmembrane receptor. J. Biol. Chem. 284(51), 35962–35972 (2009).
Khalaj-Kondori, M., Hosseinnejad, M., Hosseinzadeh, A., Sharif, S. B. & Hashemzadeh, S. Aberrant hypermethylation of OGDHL gene promoter in sporadic colorectal cancer. Curr. Probl. Cancer. 44(1), 100471 (2020).
Liu, Y. et al. A novel oxoglutarate dehydrogenase-like mediated miR-214/TWIST1 negative feedback loop inhibits pancreatic cancer growth and metastasis. Clin. Cancer Res. 25(17), 5407–5421 (2019).
Zhang, Y., Yang, R., Burwinkel, B., Breitling, L. P. & Brenner, H. F2RL3 methylation as a biomarker of current and lifetime smoking exposures. Environ. Health Perspect. 122(2), 131–137 (2014).
Wan, E. S. et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum. Mol. Genet. 21(13), 3073–3082 (2012).
Guida, F. et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum. Mol. Genet. 24(8), 2349–2359 (2015).
Dixit, G., Dabney-Smith, C. & Lorigan, G. A. The membrane protein KCNQ1 potassium ion channel: Functional diversity and current structural insights. Biochim. Biophys. Acta Biomembr. 1862(5), 183148 (2020).
Yasuda, K. et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat. Genet. 40(9), 1092–1097 (2008).
Abba, M. C. et al. Rhomboid domain containing 2 (RHBDD2): A novel cancer-related gene over-expressed in breast cancer. Biochim. Biophys. Acta. 1792(10), 988–997 (2009).
Liu, N., Yang, H., Han, L. & Ma, M. Oxytocin in women’s health and disease. Front. Endocrinol. (Lausanne). 13, 786271 (2022).
Chatterjee, O. et al. An overview of the oxytocin-oxytocin receptor signaling network. J. Cell Commun. Signal. 10(4), 355–360 (2016).
Arrowsmith, S. & Wray, S. Oxytocin: Its mechanism of action and receptor signalling in the myometrium. J. Neuroendocrinol. 26(6), 356–369 (2014).
Monks, D. T. & Palanisamy, A. Oxytocin: At birth and beyond: A systematic review of the long-term effects of peripartum oxytocin. Anaesthesia. 76(11), 1526–1537 (2021).
Nagy, B., Kovács, K., Sulyok, E., Várnagy, Á. & Bódis, J. Thrombocytes and platelet-rich plasma as modulators of reproduction and fertility. Int. J. Mol. Sci. 24(24), 17336 (2023).
Sarapik, A. et al. Follicular proinflammatory cytokines and chemokines as markers of IVF success. Clin. Dev. Immunol. 2012, 606459 (2012).
Koks, S. et al. The differential transcriptome and ontology profiles of floating and cumulus granulosa cells in stimulated human antral follicles. Mol. Hum. Reprod. 16(4), 229–240 (2010).
Santulli, G. & Iaccarino, G. Adrenergic signaling in heart failure and cardiovascular aging. Maturitas. 93, 65–72 (2016).
Khodadi, E. Platelet function in cardiovascular disease: activation of molecules and activation by molecules. Cardiovasc. Toxicol. 20(1), 1–10 (2020).
Rubenstein, D. A. & Yin, W. Platelet-activation mechanisms and vascular remodeling. Compr. Physiol. 8(3), 1117–1156 (2018).
Lee, W. S., Lee, W. H., Bae, Y. C. & Suk, K. Axon guidance molecules guiding neuroinflammation. Exp. Neurobiol. 28(3), 311–319 (2019).
Funding
This work was supported by several NIH Grants including R01 ES009718, R01 ES022955, K99/R00 ES026648, T32 ES012870, R01 DA056787, and a HERCULES Pilot Award (P30 ES019776).
Author information
Authors and Affiliations
Contributions
A.J.G., J.B.F., R.H., and A.K.S. managed the EARTH study and acquired the data. Z.T. performed the analysis. Z.T. and T.M.E. interpreted the data. The first draft was completed by Z.T. and T.M.E. All authors were involved in revising the manuscript. All authors read and approve the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tang, Z., Gaskins, A.J., Hood, R.B. et al. Former smoking associated with epigenetic modifications in human granulosa cells among women undergoing assisted reproduction. Sci Rep 14, 5009 (2024). https://doi.org/10.1038/s41598-024-54957-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54957-2
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
