
Abstract
Triple-negative breast cancer (TNBC) is one of the most aggressive types of cancer. Despite decades of intense investigation, treatment options remain limited, and rapid recurrence with distant metastases remains a significant challenge. Cancer cell-intrinsic production of cytokines such as type I interferons (IFN-I) is a known potent modulator of response to therapy in many cancers, including TNBC, and can influence therapeutic outcome. Here, we report that, in TNBC systems, the aryl hydrocarbon receptor (AhR) suppresses IFN-I expression via inhibition of STImulator of Interferon Genes (STING), a key mediator of interferon production. Intratumoral STING activity is essential in mediating the efficacy of PARP inhibitors (PARPi) which are used in the treatment of cancers harboring BRCA1 deficiency. We find that, in TNBC cells, PARPi treatment activates AhR in a BRCA1 deficiency-dependent manner, thus suggesting the presence of a negative feedback loop aimed at modulating PARPi efficacy. Importantly, our results indicate that the combined inhibition of PARP and AhR is superior in elevating IFN-I expression as compared to PARPi-alone. Thus, AhR inhibition may allow for enhanced IFN-I production upon PARPi in BRCA1-deficient breast cancers, most of which are of TNBC origin, and may represent a therapeutically viable strategy to enhance PARPi efficacy.
Similar content being viewed by others


Introduction
Triple-negative breast cancer (TNBC) is a highly aggressive form of breast cancer that lacks the expression of targetable molecular features, mainly ER, PR and HER2 receptors1. These tumors account for roughly 15–20% of all breast cancer cases and due to the relative lack of consistent genetic or molecular features, treatment options remain limited and mainly consists of surgery and highly toxic chemotherapy2. Furthermore, TNBC displays faster rates of recurrence with distant metastasis following initial treatment of any breast cancer subtype and is associated with the worst prognosis2,3. Throughout the course of the two most recent decades, there has been a concerted effort aimed at the identification of novel therapeutic options for this disease. Importantly, the use of immunotherapy (atezolizumab, an anti-PD-L1 monoclonal antibody) in combination with chemotherapy was recently approved for use as first-line therapy in metastatic TNBC tumors that overexpress PD-L1, owing to the potential for immunomodulation as a therapeutic opportunity for this indication4. However, only a fraction of patients responds to immune checkpoint blockade and most of these patients will relapse with distal recurrent disease, highlighting the importance to improve therapeutic responses5.
Tumors that harbor defects in homologous recombination repair (HRR), mainly homozygous BRCA1/2 mutations, have benefited from the introduction of inhibitors of the Poly-ADP Ribose Polymerase-1 (PARP1), an enzyme involved in DNA break repair6. The majority of hereditary BRCA-associated breast cancer tumors have the molecular features of TNBC7 and typically receive a regimen of chemotherapy centered around the use of PARP1 inhibitors (PARPi)8. While the use of PARPi has significantly improved the progression-free survival of patients with advanced HRR-deficient ovarian cancer9, the long-term response rates among breast cancer patients remain suboptimal10. Understanding the molecular mechanisms behind the lack of PARPi efficacy in breast cancer could lead to the identification of novel therapeutic combinations.
Several recent studies have suggested a mechanism in which, upon PARPi treatment, the cancer cells induce alterations to the tumor microenvironment (TME), particularly the tumor-infiltrating lymphocytes, and these changes appear to be essential in mediating PARPi anti-tumor response in BRCA1-mutated breast cancer tumors and the overall efficacy of PARPi11,12,13. Specifically, PARPi-induced genetic instability has been shown to activate the cytosolic pattern recognition receptor cGAS and its associated pathway STImulator of Interferon Genes (cGAS-STING14) within the BRCA1-mutant cancer cells, resulting in the initiation of type I IFN production, cytokine-mediated T cell infiltration, and subsequent anti-tumor immune response11. Interestingly, STING activity is suppressed in several cancer types, likely attributed to its role as a tumor suppressor15. Therefore, improving the PARPi-mediated STING activation and subsequent type I interferon (IFN-I) expression may lead to enhanced immunogenicity of PARP inhibitors in breast cancer, improving overall efficacy11,12.
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor and a member of the bHLH superfamily of transcription factors. Initially discovered as the main mediator of xenobiotics metabolism and clearance, it has subsequently become apparent that AhR can respond to a variety of endogenous cellular ligands that mediate its functions16,17,18. Active AhR harbors a potent ability to influence gene expression dynamics at both the genetic and epigenetic levels19,20,21,22. In the inactive form, AhR is kept sequestered in the cytoplasm in complex with chaperone proteins. Ligand binding triggers its nuclear localization where it dissociates from its retention complex and dimerizes with its transcriptional coactivator ARNT. AhR-ARNT then facilitates the recruitment of transcription complexes to several loci throughout the genome18. Among the most prominent AhR target genes are the Cytochrome P450, family 1, subfamily A/B, polypeptide 1 (CYP1A1 and CYP1B1), which are crucial players in the metabolism of xenobiotic agents such as dioxin23. Importantly, AhR is known to be a potent modulator of immunity and has consequential roles in several disease settings including autoimmunity and cancer24,25,26,27,28,29,30,31.
In several tumor types, the production of endogenous AhR ligands accelerates during disease progression, resulting in elevated intratumoral AhR activity in advanced disease. The consequences of elevated AhR activity are context dependent as it has been shown to harbor both pro- and anti-tumorigenic functions. For example, AhR has been shown to have tumor suppressor activity in various cancer types including intestinal, prostate, lung, and liver (reviewed in32). In breast cancer, AhR suppresses the transcriptional activity of ER, thereby inhibiting hormone-mediated proliferation and hampering the disease progression of ER + breast tumors33. Furthermore, AhR ligands have been shown to augment chemotherapy efficacy, enhancing the anti-tumor effects of treatment34,35,36. On the other hand, AhR activity is known to enhance pro-tumor functions such as invasion, migration20,37, resistance to apoptosis38, and cancer stem cell development19 in various tumor types. AhR activity can also influence the immune microenvironment, where it promotes an immunosuppressive TME, favoring disease progression16. Importantly, several recent studies have revealed a role for AhR in the inhibition of cGAS-STING pathway activity39,40,41,42. Direct investigations aimed at elucidating the precise role of AhR in specific disease settings are needed to fully understand its role in cancer. In line with this notion, the consequences of interfering with AhR signaling with respect to STING activity and immunomodulation in TNBC has not yet been investigated to date.
Here, we provide evidence that AhR activity suppresses STING-mediated IFN-I expression in a panel of TNBC cell lines. Additionally, we identify a novel AhR-mediated negative feedback loop that modulates PARPi-induced IFN-I expression in BRCA1-deficient TNBC cells. We found that PARPi treatment activates AhR, a phenomenon further accentuated in the context of BRCA1-deficiency, resulting in the induction of key AhR target genes among which is TIPARP, a known antagonist of STING-induced IFN response39. Consistently, we found that addition of an AhR antagonist augmented the IFN-I expression upon PARPi treatment in a cell line model of BRCA1-deficient TNBC. Thus, our study identifies AhR as a promising target to elevate PARPi-induced IFN-I expression, potentially enhancing the immunogenicity and overall efficacy of PARPi in HRR-deficient TNBC.
Results
AHR levels associate with poor survival in TNBC tumors
AhR has been proposed to act as both a tumor suppressor32,43 and tumor promoter19,20,37,44 in breast cancer. To better understand the role of AhR in breast cancer, we queried the University of Alabama at Birmingham Cancer (UALCAN) data analysis portal45, which hosts a collection of publicly available cancer omics, including protein data from the Clinical Proteomic Tumor Analysis Consortium (CPTAC)46,47,48. The analysis revealed that AhR protein levels are slightly increased in the primary breast tumor cohort as compared to normal tissue (Fig. 1A). This differential protein abundance is mainly driven by increased levels in the triple negative (TNBC) subtype (Fig. 1B) suggesting that AhR may have a specific role in this subtype, corroborating previous reports. Consistently, Kaplan–Meier estimation, via datasets available at KMplot.com49, revealed that AHR levels displayed an inverse association with overall survival (OS) only in TNBC patients (Fig. 1C and Supplemental Fig. S1A–C). To further delve into the role of AhR in TNBC cells, human MDA-MB-231 cells were depleted of AHR via two independent, previously validated shRNAs41, and RNAseq was performed. Gene Set Enrichment Analysis (GSEA) of the differentially expressed genes revealed that AHR depletion led to an increase in inflammatory response and various immune-related pathways (Fig. 1D and Supplemental Fig. S2).

AHR is a negative prognostic indicator in triple-negative breast cancer. (A) Expression levels of AhR protein in Normal and Primary Breast Cancer Tumor Samples. Analysis performed at https://ualcan.path.uab.edu using CPTAC samples. (B) Analysis as in A but with separation of the breast cancer cases into the four major subtypes. Statistics by Welch’s T-test. (C) Kaplan Meier analysis for overall survival of TNBC patients, stratified on median level of AHR expression performed at https://kmplot.com. Statistics by LogRank Test. (D) GSEA pathway enrichment analysis of DEGs from MDA-MB-231 shAHR RNA-seq for several Immune-related pathways.

AHR suppresses Type I Interferon expression in TNBC cells. qRT-PCR analysis of indicated genes in MDA-MB-436 (A) and MDA-MB-231 (B) cells with BAY-2416964 (BAY, 20 µM, 48 h). Data (n ≥ 3 biological replicates) is average −/+ std dev. Statistics by unpaired t test. (C) Immunoblot for AhR in MDA-MB-436 cells transduced with two independent shRNA toward AHR or a non-targeting control vector (pLKO.GFP). GAPDH is used as a loading control. Quantification performed with ImageJ. Membranes were cut prior to hybridization with antibodies. (D) qRT-PCR analysis of indicated genes in cells as in (C). pLKO-GFP = black bars, shAHR #1 = red bars, shAHR #2 = blue bars. Data (n = 3 biological replicates) is average −/ + std dev. Statistics by 2-way ANOVA. (E) qRT-PCR analysis of indicated genes in MDA-MB-436 treated with TCDD (50 nM, 96 h) or vehicle control (DMSO). Data (n = 3 biological replicates) is average −/+ std dev. Statistics by unpaired t test. *p < 0.05; **p < 0.01; ***p < 0.005; ****p < 0.0005.
Altogether, these data support previous claims that AhR is detrimental for TNBC19,20,37,44 and suggest a potential role in suppressing immune pathways within the cancer cells.
AhR suppresses IFN-I expression in TNBC cell lines
Recent work suggests that AhR activity antagonizes STING-mediated IFN-I expression in multiple cancer types including lung and muscle-invasive bladder cancer39,40. Given the increase in immune pathway-related signatures upon AHR depletion in MDA-MB-231 TNBC cell line (Fig. 1D), we asked whether these findings represented a common response in TNBC. To this end, a panel of human TNBC cell lines (MDA-MB-231, MDA-MB-436, and HCC1937) was treated with the AhR antagonists BAY-2416964 (herein referred to as BAY), which is currently being tested in clinical trials for treatment of advanced solid tumors (NCT04069026 and NCT04999202). Due to the presence of intrinsic AhR activity within these cells, potentially due to the endogenous production of AHR ligands41,50, the treatment resulted in reduction of AhR established targets CYP1A123 and TIPARP51. Importantly, AHR antagonism also resulted in a significant induction of the expression of key type I IFNs, IFNB1, CXCL10, and CCL5, in all TNBC cell lines tested (Fig. 2A, B and Supplemental Figure S3A). Importantly, this was not observed in either the immortalized breast epithelial non-tumor MCF10A cells (Supplemental Figure S3B) or the immortalized human fibroblast cell line WI-38 (Supplemental Figure S3C), suggesting that this phenomenon could be restricted to tumor cells.
Consistent results were obtained with AhR genetic, rather than pharmacologic, suppression. MDA-MB-436 cells were depleted of AHR via shRNA as above (Fig. 2C) and a reduction in AhR target genes CYP1A1 and TIPARP was observed (Fig. 2D) concomitantly with a significant increase in the expression of IFNB1, CXCL10, and CCL5 (Fig. 2D). On the converse side, activation of AhR with its classical agonist TCDD17 resulted in an induction of the expression of target genes CYP1A1 and TIPARP (Fig. 2E and Supplemental Figure S4A,B), as well as in a mild but very consistent reduction in the expression of IFNB1, CXCL10, and CCL5 in MDA-MB-436 cells (Fig. 2E).
Thus, AhR has the capability of modulating the expression of IFN-I -related genes in TNBC cells.
AhR suppresses STING-dependent IFN-I expression
TNBC tumors are known to exhibit high genomic instability, and therefore high likelihood of enhanced basal cGAS-STING activity15. AhR has been proposed to have antagonistic functions toward STING in other cancer types39,40. Thus, to assess whether the changes in IFN-I genes we observed upon AhR manipulation in TNBC are dependent on STING activity, we took a two-fold approach as above. STING was genetically depleted in the MDA-MB-436 cells using shRNAs (Fig. 3A); empty vector control (pLKO) cells and STING-knockdown cells were treated with BAY for 72 h and the expression of IFN-I genes was assessed. As before, a significant increase in IFNB1, CXCL10 and CCL5 expression, as well as the interferon-stimulated gene ISG15, was observed upon BAY treatment in control cells (Fig. 3B and Supplemental Figure S5A). However, this increase was blunted by STING depletion (Fig. 3B and Supplemental Fig. S5A), suggesting that BAY-mediated IFN-I expression is STING-dependent. These results were then confirmed using the STING-specific small molecule inhibitor H-15152. Similarly to the genetic depletion, inhibition of STING with H-151 suppressed the interferon gene induction promoted by BAY (Fig. 3C and Supplemental Figure S5B). Furthermore, immunoblot analysis of cells treated as above revealed that AhR inhibition with BAY induced TBK1 phosphorylation at S172, an event known to be STING-dependent53 and necessary to mediate IFN gene production in response to STING activation15. Concomitant treatment with H-151 attenuated this effect (Fig. 3D and Supplementary Figure S5C).

AHR antagonist-mediated IFN-I expression is STING-dependent. (A) Immunoblot for STING in MDA-MB-436 cells transduced with shRNA toward STING or a non-targeting control vector (pLKO.GFP). ß-Actin is used as a loading control. Quantification performed with ImageJ. Membranes were cut prior to hybridization with antibodies. (B) qRT-PCR analysis of indicated genes in MDA-MB-436 cells (pLKO = white bars, shSTING = grey bars – 20 µM BAY, 72 h). Data (n = 3 biological replicates) is average −/ + std dev. Statistics by 2-way ANOVA. C. qRT-PCR analysis of indicated genes in MDA-MB-436 cells (10 µM H-151, 20 µM BAY2416964, or the combination, 72 h). Data (n = 2 biological replicates) is average −/ + std dev. Statistics by 2-way ANOVA *p < 0.05; **p < 0.01, ***p < 0.005. (D) Representative immunoblot for pTBK1 (S172), total TBK1, and ß-Actin in MDA-MB-436 cells treated as in panel B. Quantification performed with ImageJ (figure shows representative image of 3 biological replicates). Membranes were cut prior to hybridization with antibodies. (E) Relative expression levels of IFNB1 and ISG15 in TCGA (top) and METABRIC (bottom) databases in patients split on TIPARP median levels. Statistics by Wilcoxon signed-rank test.
To gain more insight into the relationship between AhR and STING activity in TNBC tumors, we analyzed the TCGA and METABRIC datasets by parsing expression of known AHR target genes and known STING pathway-associated genes. The analysis revealed inverse expression levels between TIPARP (a key target of AhR) and IFNB1 or ISG15 (known to be influenced by STING activity14 (Fig. 3E), suggesting that tumors with high AhR activity (represented by high TIPARP) may have reduced interferon-based response.
Taken together, these results support previous findings that AhR antagonism increases IFN-I expression, and expand on this concept, by proposing a STING-dependent mechanism.
AhR suppresses PARPi-induced IFN-I expression
STING-mediated production of IFN-I in a cancer cell-intrinsic fashion has been recently shown to be essential for the anti-tumor effects of PARPi treatment in BRCA1-deficient TNBC11. To assess whether any relationship exists between PARPi and AhR, the BRCA1-mutant MDA-MB-436 cells54 were transduced with control vector or shAHR and treated with the PARP inhibitor Talazoparib (an FDA approved PARP inhibitor currently being used in the clinic for BRCA1/2-mutant TNBC and ovarian cancer55; herein referred to as TALA). Surprisingly, we found that TALA treatment in these cells resulted in AhR induction and an AhR-dependent increase of its target genes CYP1A1 and TIPARP, thus suggesting that PARPi induces AhR activation (Fig. 4A). To assess whether this phenomenon was dependent on BRCA1 activity, we expanded to a second cell line model by performing BRCA1-knockdown via shRNA in MDA-MB-231 cells, a well-established BRCA1WT TNBC54 cell line (Fig. 4B) and treating cells with TALA. As expected, shBRCA1 MDA-MB-231 cells displayed increased sensitivity to PARP inhibition (Supplemental Figure S6)6. Importantly, TALA treatment caused a robust induction in expression of AHR, CYP1A1, and TIPARP in the BRCA1 knockdown cells (Fig. 4C), while these effects were markedly reduced in control cells.

AHR suppresses PARPi-induced IFN-I expression in BRCA1-deficient TNBC. (A) qRT-PCR analysis of indicated genes in MDA-MB-436 cells (pLKO = light grey bars, shAHR #2 = dark grey bars—1 µMTalazoparib, 96 h). Data (n = 4 biological replicates) is average −/ + std dev. Statistics by 2-way ANOVA. (B) Immunoblot for BRCA1 in MDA-MB-231 cells transduced with shRNA toward BRCA1 or a non-targeting control vector (pLKO.GFP). Vinculin is used as a loading control. Membranes were cut prior to hybridization with antibodies. (C) qRT-PCR analysis of indicated genes in MDA-MB-231 cells (pLKO = light grey bars, shBRCA1 = dark grey bars—1 µMTalazoparib, 96 h). Data (n = 3 biological replicates) is average −/ + std dev. Statistics by 2-way ANOVA. (D) qRT-PCR analysis of indicated genes in MDA-MB-436 cells (1 µM Talazoparib, 20 µM BAY2416469, or the combination, 96 h). Data (n ≥ 3 biological replicates) is average −/ + std dev. Statistics by 2-way ANOVA. (E) qRT-PCR analysis of indicated genes in MDA-MB-436 cells (1 µM Talazoparib, 50 nM TCDD, or the combination, 96 h). Data (n = 2 biological replicates) is average −/ + std dev. Statistics by 2-way ANOVA. *p < 0.05; **p < 0.01, ***p < 0.005, ****p < 0.0005.
Thus, our data suggest that in conditions of BRCA deficiency, PARP inhibition results in AhR activation. This, combined with our previous data, leads to a model in which AhR activation by PARPi may create a self-regulatory loop that suppresses PARPi induction of IFN-I genes, thereby alleviating the effects of PARP inhibition.
To test this, MDA-MB-436 cells were treated with either BAY (AhR antagonist), TALA (PARP inhibitor), or a combination of the two and the expression of AhR targets and IFN-I genes was assessed. As before, TALA by itself induced CYP1A1 and TIPARP, which could be suppressed by concomitant BAY treatment (Fig. 4D, first two panels) and both BAY and TALA induced increases in the levels of IFN-I genes IFNB1, CXCL10, and CCL5 (Fig. 4D, last three panels). However, the combination of TALA + BAY resulted in a greater induction of these IFN-I genes as compared to either drug alone, thus suggesting that AhR antagonism may potentiate the effects and durability of PARPi. Conversely, AhR activation with its prototypical ligand TCDD, resulted in the opposite effect, blunting the induction of IFN-I genes by TALA (Fig. 4E).
Altogether, our work reveals a novel function for AhR in the suppression of tumor-intrinsic, STING-mediated IFN-I production and helps to shed light on the lack of efficacy of PARPi in TNBC.
Discussion
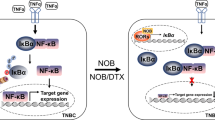
The cGAS-STING pathway has received much attention as a promising immunomodulatory therapeutic target because it represents a mechanistic link between cellular stress, a hallmark of cancerous cells and chemotherapeutic mechanisms of action, and the activation of the innate immune system14,15,53,56,57. Recent findings highlight a key role for this pathway in mediating anti-tumor immunity and immune checkpoint inhibitor efficacy58,59. Importantly, STING activity has also been shown to be essential for the anti-tumor effects of various chemotherapeutics60, including PARPi in BRCA1-deficient TNBC12,13. Therefore, it has been hypothesized that overall tumor immunogenicity and therapeutic efficacy, including that of PARPi, can be improved by enhancing cancer cell-intrinsic STING activation and subsequent IFN-I production. Here, we provide evidence that suggests that AhR activity suppresses STING-mediated IFN-I in TNBC, especially in the context of PARPi treatment of BRCA1-deficient cells, and that its inhibition represents a therapeutically viable method to enhance cancer cell-intrinsic STING activity, both alone and in combination with PARPi (Fig. 5).

Model of AhR suppression of STING-mediated Type I IFN expression upon PARPi. (TOP) In TNBC cells under basal conditions, AHR activity suppresses STING-mediated IFN-I production. (BOTTOM) In BRCA1-deficient TNBC cells, PARPi stimulates STING activity but at the same times it induces AhR activation, which in turns acts as a feedback mechanism to reduced STING activity and its induction of Type I interferon genes.
TNBC cells are characteristically more proliferative and harbor higher levels of genomic instability than other subtypes of breast cancer 61. As a result, these tumors tend to have higher mutational burdens, elevated expression of neoantigens, and greater immunogenicity than other subtypes62,63,64. Furthermore, high genomic instability is known to activate cGAS-STING and lead to IFN-I production and immune cell infiltration65, highlighting another intrinsically immunostimulatory characteristic of TNBC tumors. Therefore, in order to remain viable, it is likely that TNBC tumors must adopt mechanisms that suppress the immune system and overcome genomic instability-associated immunity. Interestingly, AhR is known to promote immunosuppression in several tumor types through various mechanisms, including the inhibition of STING activity28,30,31,40,66. This may partly account for why TNBC tumors have higher levels of AhR and why high AHR expression is a negative prognostic indicator, only in TNBC (Fig. 1). It is likely that without sufficient AhR activity, anti-tumor immunity would act as a barrier to tumor formation. In line with this, RNAseq analysis upon AHR depletion in the TNBC cell line MDA-MB-231, revealed significant enrichment of immune-related signatures suggesting an AhR-mediated suppression of immunity in this context. Furthermore, gene specific qPCR showed that AhR inhibition alone was sufficient to upregulate key IFN-I genes including IFNB1, CXCL10, and CCL5 while AhR activation suppressed them, in multiple cell line systems of TNBC, but not non-tumorigenic cell lines (Fig. 2 and Supplementary Figure S3).
The transcriptional activation of IFN-I genes involves a complex cellular regulatory network that is not limited to the cGAS-STING pathway67. However, recent findings have indicated a role for AhR in the suppression of STING39,40,41,42. For example, the AhR-target PARP7 (product of the gene TIPARP), can directly suppress STING activity by facilitating the degradation of TBK1, a key downstream effector kinase of STING, thereby suppressing IFN-I expression and subsequent anti-tumor immunity39. AhR has also been shown to regulate the synthesis of the polyamines spermine and spermidine41 which have recently been implicated in the attenuation of cGAS activity via alterations to the DNA ligands in the cytoplasm42. Additionally, a recent study showed that AhR itself may suppress IFN-I expression in muscle invasive bladder cancer by directly interacting with and mediating the degradation of STING40. Therefore, the upregulation of IFN-I genes upon AHR depletion or inhibition may be due to an alleviation of STING suppression. Consistent with this hypothesis, genetic and pharmacologic depletion of STING revealed that IFN-I upregulation upon AhR inhibition is STING-dependent (Fig. 3A–D). Importantly, analysis of gene expression data from patient tumors revealed a negative correlation between the key AhR target gene TIPARP and STING-associated IFN genes, IFNB1 and ISG15 indicating that AhR inhibition of STING is likely occurring in patient tumors. Further studies must be performed to identify the mechanistic basis of this regulation.
IFN-I cytokines harbor potent immunomodulatory properties and function to influence the recruitment and activity of immune cells67 and are known to have both pro- and anti-tumor functions68. Importantly, a foundational study published in 2015 analyzed hundreds of TNBC patient tumors and found that tumors with a high “immunostimulatory” gene expression signature were associated with the best prognosis and longest overall survival of any TNBC subtype69. One of the key characteristics of the “immunostimulatory” subtype was the enrichment for genes sets including: “JAK-STAT-associated cytokines’, “NF-kB activation by viruses”, and “Interferon Signaling”, which are all related to the cGAS-STING pathway and IFN-I gene signatures14,15. Furthermore, CCL5 expression levels in TNBC have been associated with a better prognosis and increased recruitment of CD8 T cells, CD4 activated T cells, NK cells and anti-tumor macrophages70. These data heavily imply that IFN-I cytokines have potent anti-tumor activity in TNBC and justify the pursuit of therapeutic strategies to increase their intratumoral abundance. AhR antagonism as a method to achieve this end has not been directly assessed, and future studies are warranted.
cGAS-STING activity is vital for the anti-tumor effects of various chemotherapeutics including PARPi11,12,60. Specifically, PARPi-induced cGAS-STING activation within BRCA1-deficient cancer cells results in the intratumoral production of IFN-I, subsequent immune cell infiltration, and T cell-dependent anti-tumor immunity. STING-depleted cancer cells fail to elicit this response, which significantly diminishes the therapeutic effect of PARPi11. This implies that STING activity within the cancer cells themselves may potentially be important for the therapeutic effects of PARPi and that PARPi efficacy may be enhanced by increasing intracellular STING activity. Interestingly, we found that treatment with Talazoparib (an FDA approved PARP inhibitor currently used in the clinic55) activated AhR in TNBC cells, a phenomenon that was accentuated in the BRCA1-deficient setting (Fig. 4A–C). Being that AhR is a ligand-activated transcription factor with affinity to aromatic hydrocarbons18, it is possible that the chemical structure of Talazoparib itself could activate AhR. However, the fact that BRCA1 knockdown accentuated the activity of AhR implies some intracellular response to PARPi treatment that is augmented by BRCA loss, potentially apoptosis, is required for AhR activation. Regardless of the mechanistic basis, the fact that AhR activity is stimulated by PARP inhibition hints at the existence of a self-regulatory mechanism in which the production of IFN-I upon PARPi treatment is diminished by AhR; a mechanism that can be overcome with the addition of an AhR antagonist. Indeed, we found that the addition of the AhR antagonist BAY-2416964 enhanced the PARPi-mediated IFN-I production in a BRCA1-mutant TNBC cell line, suggesting that AhR antagonism may be a means to enhance the immunogenicity and overall efficacy of PARPi in the clinic.
Additionally, these findings have significant implications with regards to immune checkpoint inhibitor therapy, as STING-mediated IFN-I production is known to enhance ICI efficacy. Treatment with STING agonists has been tested as a way to enhance ICI therapy14,56,57,71; however, systemic delivery of STING agonists has failed due to poor stability of the specific agents in circulation or the emergence of severe side effects which limits the clinical viability of this approach56,72. Our results suggest that AhR antagonism may indirectly result in the activation of STING specifically within the TNBC cells. The use of AhR antagonists to enhance ICI therapy has not been tested to date, though AhR antagonists are currently being investigated for their immunomodulatory potential in several cancer types (NCT04069026 and NCT04999202).
Conversely, treatment with the AhR agonist TCDD suppressed the expression of STING-associated IFN-I genes (Fig. 2E). While the TCDD-induced IFN-I suppression was mild (~ 15–20% reduction) the data were very consistent. This is particularly relevant because several studies have reported that microbial metabolism contributes to the accumulation of AhR ligands such as indoles from tryptophan metabolism within the tumor microenvironment of multiple cancer types (i.e. PDAC31 and melanoma25), which mediate AhR-dependent immunosuppressive processes within the immune cells. Our data suggest that these ligands may also negatively influence intratumoral STING activity in an AhR-dependent manner. Given that STING activation is essential for proper PARPi-mediated anti-tumor efficacy, it would be interesting to investigate if microbiome-dependent AhR activity contributes to the disparities among patient responses to PARPi treatment. For example, dietary habits that alter microbiome composition, such as diets high in tryptophan 25, may result in an AhR-mediated STING-suppressed pro-tumorigenic microenvironment that hinders the immunogenicity of PARPi treatment affecting overall efficacy. This raises the possibility of investigating the potential to augment PARPi + ICI efficacy with dietary alterations or with the use of probiotic adjuvants to influence the composition of the microbiome towards a low AhR ligand-producing metabolic state.
Although limited by the fact that our studies have been conducted only in cell lines, our data strongly suggest that AhR activity suppresses STING-mediated IFN-I expression in TNBC, especially in the context of PARPi treatment of BRCA1-mutant cells (Fig. 5). Further implications, that will need to be address in vivo with the use of xenografts and mouse models, are that AhR antagonism may be a viable therapeutic strategy to enhance the overall immunogenicity of TNBC tumors and improve PARPi efficacy.
Materials and methods
Cell lines
MDA-MB-436 and HCC1937 cells were a kind gift from Dr. Eric Knudsen (Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA). HEK-293T cells were a kind gift from Dr. Irwin Gelman (Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA). MDA-MB-436 and HCC1937 cells were cultured in RPMI media (Invitrogen, Carlsbad, CA, USA). WI-38, MDA-MB-231 and HEK293T were cultured in DMEM media (Invitrogen). Both media were supplemented with 10% fetal bovine serum (Invitrogen) and 1% antibiotic–antimycotic (Invitrogen). MCF10A cells were incubated in a 50/50 DMEM/F-12 formulation (Invitrogen #11330-032) with the following supplements: Horse serum (Invitrogen #16050-122; 5% final), EGF (20 ng/mL final), Hydrocortisone (0.5 mg/mL final), Cholera Toxin (100 ng/mL final), antibiotic–antimycotic (1% final). All cell lines were authenticated via short tandem repeat (STR) sequencing at the Roswell Park Genomics Shared Resource and routinely tested for mycoplasma contamination.
Antibodies and other reagents
Primary antibody information
Rabbit monoclonal antibodies to AHR ((D5S6H) Cat. No. 83200S), GAPDH ((D4C6R) Cat. No. 97166T), pTBK1/NAK (S172) ((D52C2) XP (R) Cat. No. 5483T), and TBK1/NAK (Cat. No. 3013S) were purchased from Cell Signaling (Cell Signaling Technology, Danvers, MA, USA). Mouse monoclonal antibodies to BRCA1 ((D-9) Cat. No. sc-6954), and Vinculin ((7F9) Cat. No. sc-73614) were purchased from Santa Cruz (Santa Cruz, CA, USA). Mouse monoclonal antibody to ß-Actin (Cat No. 66009-1-1g) was purchased from Proteintech (Rosemont, IL, USA). αMouse-HRP secondary antibody (Goat Anti-Mouse IgG (H + L)-HRP Conjugate cat. No. 172-1011) and αRabbit-HRP rabbit secondary antibody (Goat Anti-Rabbit IgG (H + L)-HRP Conjugate cat. No. 170-6515) were purchased from Bio-Rad (Bio-Rad, Hercules, CA, USA). AhR antagonist BAY-2416964 (Cat. No.: HY-135829), PARP inhibitor Talazoparib (No. S7048), and STING inhibitor H-151 (cat. No. S6652) were purchased from Selleckchem (Houston, Texas, USA).
Cell viability assay
Cells were seeded (3000–5000 cells per well in a 6-well plate) and allowed to incubate overnight at 37 °C and treated with increasing concentrations of Talazoparib for 72 h. At the end of treatment, 10µL of Cell counting kit-8 (WST-8/CCK8; abcam cat. No. ab228554) reagent was added to each well and cells were incubated for 1–3 h. Total absorbance was measured at 450 nm on a SpectraMax M2/M2E microplate reader (Molecular Devices). Cell viability was calculated relative to the average of DMSO control-treated cells.
Lentiviral Infections (shRNA)
shRNA plasmids were transfected into HEK293T cells using LipoD293 (SignaGen, Frederick, MD, USA) along with the pCMV-VSV-G vector (Addgene, Cambridge, MA, USA) and the pCMV-psPAX2 vector (a kind gift from Dr. Irwin Gelman, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA). pLKO-GFP vector was purchased from Sigma. All shRNA expressing plasmids were purchased from Sigma Aldrich: shAHR #1 TRCN0000245285; target sequence: 5’ GCAACAAGATGAGTCTATT 3’, shAHR #2 TRCN000021258; sequence: 5’ CGGCATAGAGACCGACTTA 3’, shSTING (TMEM173) TRCN0000134594; target sequence: 5’ GTTTACAGCAACAGCATCTAT 3’, shBRCA1 TRCN0000009823; target sequence: 5’ CTCTTAAAATATAAGACCTCTGGCATGAAT 3’. All lentiviral infections were performed as previously described41. Briefly, lentiviral supernatant was harvested at 48 h post transfection and transduced to cells in the presence of 8 \(\upmu\)g/mL hexadimethrine bromide (Sigma, St. Louis, MO, USA). Cells were incubated with virus for 12 h, followed by a 24-h incubation with non-virus growth media. Cells were then subjected to puromycin selection for 48–72 h. PURO-resistant surviving cells were aggregated and used for further experimentation.
Real time quantitative PCR (RT-qPCR)
Total cellular RNA was collected using the PureLink™ RNA Mini (ThermoFisher Scientific, Waltham, MA, USA cat. No. 12183025) with on-column DNase treatment, as per manufacturer’s recommendations. 1 μg of total RNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems Waltham, MA, USA cat. No. 4368814). Quantitative real time PCR was performed on a CFX Opus 96 Real-Time PCR System (Biorad, Hercules, CA, USA Cat. No. #12011319) using iTaq Universal SYBR® Green Supermix (Biorad, Hercules, CA, USA. Cat. No. 1725121) with the following human site-specific primers listed below. Data were analyzed using the Bio-Rad CFX Maestro software.
RPS20 FWD: AAGGATACCGGAAAAACACCC; RPS20 REV: TTTACGTTGCGGCTTGTTAGG
AHR FWD: GGTTGTGATGCCAAAGGA; AHR REV: GGGACTCGGCACAATAAAG
CYP1A1 FWD: GGAGCTAGACACAGTGATTG; CYP1A1 REV: AAGAGTGTCGGAAGGTCT
TIPARP FWD: CTCGTGTTTGAGCTGGTGAA; TIPARP REV: ACACGTTCATGGCATTCAAA
IFNB1 FWD: CTTCTCCACTACAGCTCTTTC; IFNB1 REV: CTGTCCTTGAGGCAGTATTC
CXCL10 FWD: CTCCCATCACTTCCCTACA; CXCL10 REV: GGAGTAGTAGCAGCTGATTTG
CCL5 FWD: ACACCCTGCTGCTTTGCCTACA; CCL5 REV: TCCCGAACCCATTCTTCTCTG
ISG15 FWD: CATCTTTGCCAGTACAGGAG; ISG15 REV: ACACCTGGAATTCGTTGC
IRF1 FWD: GACTCCAGCTACAACAGATG; IRF1 REV: CTTCCCATCCACGTTTGT
RNA-seq
Total cellular RNA was isolated using the PureLink RNA Mini kit with on-column DNase treatment (Thermo Fisher Sceintific) and RNAseq analysis was carried on as previously described73. Briefly, sequencing libraries were prepared with the TruSeq Stranded mRNA kit (Illumina) from 500 ng total RNA following manufacturer’s instructions and single-end sequenced on a NextSeq 500 (Illumina) following the manufacturer’s recommended protocol. Genome alignment of raw reads to the human reference genome (GRCh38.p13) using STAR74 and raw feature count normalization were performed at the Roswell Park Comprehensive Cancer Center’s Genomics Shared Resource. Differential expression analysis was carried out with DESeq275 and differential expression rank order was used for subsequent gene set enrichment analysis (GSEA)76, performed using the cluster profile package in R and collections available through the Molecular Signatures Database (MSigDB)77. Overlaps of DEG lists across companions were calculated by hypergeometric testing. All analyses were performed using R statistical software, version 4.1.1.
Immunoblotting
Whole cell lysates were collected using RIPA buffer (50 mM Tris–HCl, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10% glycerol, 2 mM EDTA) and separated by SDS-PAGE on polyacrylamide gel (8–15% acrylamide depending on protein targets). Proteins were transferred to nitrocellulose membranes (Biorad, Hercules, CA, USA) and membranes were blocked in Blocking buffer (5% milk in TBS containing 0.1% Tween-20) for 1 h at room temperature. Membranes were incubated with primary antibody diluted in blocking buffer overnight at 4 °C. Appropriate HRP-conjugated secondary antibodies (Bio-Rad, Hercules, CA, USA) were used at 1:5,000 dilution in blocking buffer for 1 h at RT. Signals were visualized with BioRad chemiluminescence reagents and either xray films developed with a Konica SRX-101A (Konica Minolta, Wayne, NJ, USA) film processor or with a GeneGnome XRQ NPC system (Syngene, Frederick, MD, USA). Densitometric quantifications were performed with ImageJ (National Institutes of Health, Bethesda, MD, USA). Membranes were cut prior to hybridization with antibodies.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request. All sequencing data reported here are available in the NCBI Gene Expression Omnibus (GEO) database under GSE252119.
References
Aysola, K., et al. Triple negative breast cancer: An overview. Hereditary Genet. 2013(Suppl 2) (2013).
Waks, A. G. & Winer, E. P. Breast cancer treatment: A review. Jama 321(3), 288–300 (2019).
Zagami, P. & Carey, L. A. Triple negative breast cancer: Pitfalls and progress. npj Breast Cancer 8(1), 95 (2022).
Tavares, D. F. et al. Immunotherapy using PD-1/PDL-1 inhibitors in metastatic triple-negative breast cancer: A systematic review. Oncol Rev 15(2), 497 (2021).
Li, L., et al. Immunotherapy for triple-negative breast cancer: Combination strategies to improve outcome. Cancers (Basel). 15(1) (2023).
Farmer, H. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434(7035), 917–921 (2005).
Mehrgou, A. & Akouchekian, M. The importance of BRCA1 and BRCA2 genes mutations in breast cancer development. Med J Islam Repub Iran 30, 369 (2016).
Cortesi, L., Rugo, H. S. & Jackisch, C. An overview of PARP inhibitors for the treatment of breast cancer. Target Oncol 16(3), 255–282 (2021).
Wang, L. et al. Advances in the treatment of Ovarian cancer using PARP inhibitors and the underlying mechanism of resistance. Curr Drug Targets 21(2), 167–178 (2020).
Dias, M. P. et al. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nature Rev. Clin. Oncol. 18(12), 773–791 (2021).
Pantelidou, C. et al. PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov 9(6), 722–737 (2019).
Ding, L. et al. PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep 25(11), 2972-2980.e5 (2018).
Wang, Q. et al. STING agonism reprograms tumor-associated macrophages and overcomes resistance to PARP inhibition in BRCA1-deficient models of breast cancer. Nature Commun. 13(1), 3022 (2022).
Jiang, M. et al. cGAS-STING, an important pathway in cancer immunotherapy. J. Hematol. Oncol. 13(1), 81 (2020).
Kwon, J. & Bakhoum, S. F. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov 10(1), 26–39 (2020).
Murray, I. A., Patterson, A. D. & Perdew, G. H. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer 14(12), 801–814 (2014).
Safe, S. et al. Aryl hydrocarbon receptor (AhR) ligands as selective AhR modulators: Genomic studies. Curr Opin Toxicol 11–12, 10–20 (2018).
Kou, Z. & Dai, W. Aryl hydrocarbon receptor: Its roles in physiology. Biochem Pharmacol 185, 114428 (2021).
Al-Dhfyan, A., Alhoshani, A. & Korashy, H. M. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and β-Catenin and Akt activation. Mol Cancer 16(1), 14 (2017).
D’Amato, N. C. et al. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res 75(21), 4651–4664 (2015).
Stanford, E. A. et al. The role of the aryl hydrocarbon receptor in the development of cells with the molecular and functional characteristics of cancer stem-like cells. BMC Biol. 14(1), 20 (2016).
Rothhammer, V. & Quintana, F. J. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 19(3), 184–197 (2019).
Androutsopoulos, V. P., Tsatsakis, A. M. & Spandidos, D. A. Cytochrome P450 CYP1A1: wider roles in cancer progression and prevention. BMC Cancer 9(1), 187 (2009).
Takenaka, M. C. et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nature Neurosci. 22(5), 729–740 (2019).
Bender, M. J. et al. Dietary tryptophan metabolite released by intratumoral Lactobacillus reuteri facilitates immune checkpoint inhibitor treatment. Cell 186(9), 1846-1862.e26 (2023).
Moreno-Nieves, U. Y. et al. The aryl hydrocarbon receptor modulates the function of human CD56(bright) NK cells. Eur J Immunol 48(5), 771–776 (2018).
Hughes, T. et al. The transcription Factor AHR prevents the differentiation of a stage 3 innate lymphoid cell subset to natural killer cells. Cell Rep 8(1), 150–162 (2014).
Shin, J. H. et al. AHR regulates NK cell migration via ASB2-mediated ubiquitination of filamin A. Front Immunol 12, 624284 (2021).
Nguyen, N. T. et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. 107(46), 19961–19966 (2010).
Shinde, R. et al. Apoptotic cell–induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nature Immunol. 19(6), 571–582 (2018).
Hezaveh, K. et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 55(2), 324-340.e8 (2022).
Elson, D. J., Kolluri, S. K. Tumor-suppressive functions of the aryl hydrocarbon receptor (AhR) and AhR as a therapeutic target in cancer. Biology (Basel), 12(4) (2023).
Tarnow, P., Tralau, T. & Luch, A. Chemical activation of estrogen and aryl hydrocarbon receptor signaling pathways and their interaction in toxicology and metabolism. Expert Opin Drug Metab Toxicol 15(3), 219–229 (2019).
O’Donnell, E. F. et al. The aryl hydrocarbon receptor mediates raloxifene-induced apoptosis in estrogen receptor-negative hepatoma and breast cancer cells. Cell Death Dis 5(1), e1038 (2014).
Jang, H.S., et al. Identification of a Raloxifene analog that promotes AhR-mediated apoptosis in cancer cells. Biology (Basel) 6(4) (2017).
Elson, D. J. et al. Induction of aryl hydrocarbon receptor-mediated cancer cell-selective apoptosis in triple-negative breast cancer cells by a high-affinity benzimidazoisoquinoline. ACS Pharmacol Transl Sci 6(7), 1028–1042 (2023).
Goode, G. D. et al. Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB-231 human breast cancer cell line. Int J Cancer 133(12), 2769–2780 (2013).
Bekki, K. et al. The aryl hydrocarbon receptor (AhR) mediates resistance to apoptosis induced in breast cancer cells. Pestic Biochem Physiol 120, 5–13 (2015).
Gozgit, J. M. et al. PARP7 negatively regulates the type I interferon response in cancer cells and its inhibition triggers antitumor immunity. Cancer Cell 39(9), 1214-1226.e10 (2021).
Ma, Z. et al. AhR diminishes the efficacy of chemotherapy via suppressing STING dependent type-I interferon in bladder cancer. Nature Commun. 14(1), 5415 (2023).
Bianchi-Smiraglia, A. et al. Inhibition of the aryl hydrocarbon receptor/polyamine biosynthesis axis suppresses multiple myeloma. J Clin Invest 128(10), 4682–4696 (2018).
Zhao, C., et al. Polyamine metabolism controls B-to-Z DNA transition to orchestrate DNA sensor cGAS activity. Immunity (2023).
Hestermann, E. V., Stegeman, J. J. & Hahn, M. E. Relationships among the cell cycle, cell proliferation, and aryl hydrocarbon receptor expression in PLHC-1 cells. Aquatic Toxicology 58(3), 201–213 (2002).
Novikov, O. et al. An aryl hydrocarbon receptor-mediated amplification loop that enforces cell migration in ER-/PR-/Her2-human breast cancer cells. Mol Pharmacol 90(5), 674–688 (2016).
Chandrashekar, D. S. et al. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 19(8), 649–658 (2017).
Chen, F. et al. Global impact of somatic structural variation on the cancer proteome. Nature Commun. 14(1), 5637 (2023).
Zhang, Y. et al. Proteogenomic characterization of 2002 human cancers reveals pan-cancer molecular subtypes and associated pathways. Nat Commun 13(1), 2669 (2022).
Chen, F. et al. Pan-cancer molecular subtypes revealed by mass-spectrometry-based proteomic characterization of more than 500 human cancers. Nature Commun. 10(1), 5679 (2019).
Győrffy, B. Discovery and ranking of the most robust prognostic biomarkers in serous ovarian cancer. Geroscience 45(3), 1889–1898 (2023).
Opitz, C. A. et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478(7368), 197–203 (2011).
Grimaldi, G., Rajendra, S. & Matthews, J. The aryl hydrocarbon receptor regulates the expression of TIPARP and its cis long non-coding RNA, TIPARP-AS1. Biochem Biophys Res Commun 495(3), 2356–2362 (2018).
Hong, Z., et al. STING inhibitors target the cyclic dinucleotide binding pocket. Proc. Natl. Acad. Sci. USA 118(24) (2021).
Zhang, M. et al. Inhibitory targeting cGAS-STING-TBK1 axis: Emerging strategies for autoimmune diseases therapy. Front Immunol 13, 954129 (2022).
Elstrodt, F. et al. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Res 66(1), 41–45 (2006).
Rose, M. et al. PARP inhibitors: Clinical relevance, mechanisms of action and tumor resistance. Front Cell Dev Biol 8, 564601 (2020).
Wu, Y. T. et al. Tumor-targeted delivery of a STING agonist improvescancer immunotherapy. Proc Natl Acad Sci USA 119(49), e2214278119 (2022).
Zhu, C. et al. Potential for treatment benefit of STING agonists plus immune checkpoint inhibitors in oral squamous cell carcinoma. BMC Oral Health 21(1), 506 (2021).
Ghaffari, A. et al. STING agonist therapy in combination with PD-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br J Cancer 119(4), 440–449 (2018).
Reisländer, T., Groelly, F. J. & Tarsounas, M. DNA damage and cancer immunotherapy: A STING in the tale. Mol Cell 80(1), 21–28 (2020).
Liu, H. et al. Pharmacological boosting of cGAS activation sensitizes chemotherapy by enhancing antitumor immunity. Cell Rep 42(3), 112275 (2023).
Lehmann, B. D. et al. Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nature Commun. 12(1), 6276 (2021).
Thomas, A. et al. Tumor mutational burden is a determinant of immune-mediated survival in breast cancer. Oncoimmunology 7(10), e1490854 (2018).
Abdou, Y. et al. Immunotherapy in triple negative breast cancer: beyond checkpoint inhibitors. NPJ Breast Cancer 8(1), 121 (2022).
Loizides, S. & Constantinidou, A. Triple negative breast cancer: Immunogenicity, tumor microenvironment, and immunotherapy. Front Genet 13, 1095839 (2022).
Shen, R. et al. DNA damage and activation of cGAS/STING pathway induce tumor microenvironment remodeling. Front Cell Dev Biol 9, 828657 (2021).
Dai, G., Chen, X. & He, Y. The gut microbiota activates AhR through the tryptophan metabolite Kyn to mediate renal cell carcinoma metastasis. Front Nutr 8, 712327 (2021).
Ivashkiv, L. B. & Donlin, L. T. Regulation of type I interferon responses. Nat Rev Immunol 14(1), 36–49 (2014).
Cheon, H. et al. How cancer cells make and respond to interferon-I. Trends Cancer 9(1), 83–92 (2023).
Burstein, M. D. et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res 21(7), 1688–1698 (2015).
Araujo, J. M. et al. Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer. Sci Rep 8(1), 4899 (2018).
Nakamura, T. et al. STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. J ImmunoTherapy Cancer 9(7), e002852 (2021).
Ibrahim, N.E.-S. Targeted delivery of anti-tumour STING agonist. Nature Rev Bioeng 1(8), 544–544 (2023).
Chaudhry, K. A., et al. Aryl hydrocarbon receptor is a tumor promoter in MYCN-amplified neuroblastoma cells through suppression of differentiation. iScience (2023).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1), 15–21 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12), 550 (2014).
Reimand, J. et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat Protoc 14(2), 482–517 (2019).
Liberzon, A., et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27(12): 1739–40 (2011).
Acknowledgements
We are grateful to Dr. Dominic Smiraglia (Roswell Park) for insightful scientific discussions. We are grateful to Ms. Denisha Robinson for technical assistance. This work has been partly supported by NIH grant CA248018 (A.B-S.); the Roswell Park Alliance Foundation (A.B-S.), and the NCI Cancer Center Support Grant P30CA16056 (to the Roswell Park Comprehensive Cancer Center).
Author information
Authors and Affiliations
Contributions
J.C.M and A. B-S. designed the study; J.C.M, T.dSF., and K.A.C. performed experiments and analyzed data; M.O. and S.R.R. performed bioinformatics analyses; T.K, S.R.R. and A. B-S. supervised the study and analyzed data. The manuscript was written by J.C. M. and A.B.-S. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martin, J.C., da Silva Fernandes, T., Chaudhry, K.A. et al. Aryl hydrocarbon receptor suppresses STING-mediated type I IFN expression in triple-negative breast cancer. Sci Rep 14, 5731 (2024). https://doi.org/10.1038/s41598-024-54732-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54732-3
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
