
Abstract
Pediatric Crohn’s disease (CD) is characterized by a severe disease course with frequent complications. We sought to apply machine learning-based models to predict risk of developing future complications in pediatric CD using ileal and colonic gene expression. Gene expression data was generated from 101 formalin-fixed, paraffin-embedded (FFPE) ileal and colonic biopsies obtained from treatment-naïve CD patients and controls. Clinical outcomes including development of strictures or fistulas and progression to surgery were analyzed using differential expression and modeled using machine learning. Differential expression analysis revealed downregulation of pathways related to inflammation and extra-cellular matrix production in patients with strictures. Machine learning-based models were able to incorporate colonic gene expression and clinical characteristics to predict outcomes with high accuracy. Models showed an area under the receiver operating characteristic curve (AUROC) of 0.84 for strictures, 0.83 for remission, and 0.75 for surgery. Genes with potential prognostic importance for strictures (REG1A, MMP3, and DUOX2) were not identified in single gene differential analysis but were found to have strong contributions to predictive models. Our findings in FFPE tissue support the importance of colonic gene expression and the potential for machine learning-based models in predicting outcomes for pediatric CD.
Similar content being viewed by others


Introduction
Pediatric Crohn’s disease (CD) is the fastest growing age group for incidence of the disease with about 80,000 children in the US affected1,2,3. CD is characterized by a relapsing, remitting disease course with complications, such as strictures or perforation, affecting around 50% of patients within 5 years of diagnosis4,5. Pediatric CD follows a more severe disease course, more often involving strictures and fistulas6,7,8. These complications drive further morbidity and healthcare utilization associated with CD including growth failure, delayed puberty, hospitalizations, and surgery4,8.
Analysis of gene expression and identification of biological pathways which drive development of CD and CD complications may give insight into more precise treatment decision-making to prevent a complicated CD course. Genes associated with immune and cytokine pathways have been associated with CD development9,10,11,12,13. Further, specific genes including oncostatin M, IL1B, S100A8, and CXCL1 have been associated with response to anti-tumor necrosis factor therapy14,15,16. Genes controlling extracellular matrix production and inflammatory processes have been associated with strictures17,18,19. Predictive modeling which incorporates this genetic information to prognosticate disease course could assist with clinical decision-making.
Previous studies have developed predictive models for CD outcomes based on gene expression and other risk factors, most notably using the RISK cohort17. However, these studies relied on logistic regression models, which may fail to capture the multi-factorial, non-linear interactions between genes and clinical characteristics that predict increased risk for complications. Machine learning techniques, which have more capacity to capture these complex patterns, have been successfully applied to inflammatory bowel disease (IBD)-related topics including identification of risk genes, prediction of outcomes from serum proteins, and prediction of response to medication from multi-omic data20,21,22. However, they have not yet been applied specifically to prediction of complications for pediatric CD from gene expression.
The goals of our study are: (1) to identify genes which are differentially expressed in CD and complicated CD and (2) to apply machine learning techniques that use those genes to predict risk of complications. We hypothesize that machine learning techniques can incorporate the gene expression profiles of patients with complicated disease to outperform previous predictors.
Materials and methods
Study design and outcomes
This study included patient data from 120 patients that was collected at the University of North Carolina at Chapel Hill. This consisted of 101 colonic tissue specimens and 101 ileal tissue specimens of which 83 were matched. This included patients younger than 18 with suspected IBD, who underwent endoscopy between 2008 and 2012. Patients who were found to have no histologic evidence of gut inflammation were used as non-IBD controls. At the time of diagnosis, patients were selected based on non-penetrating, non-stricturing disease phenotype. This study was approved by the University of North Carolina Institutional Review Board (Study ID#: 15-0024). All experiments were performed in accordance with relevant guidelines and regulations and informed consent was obtained from patients’ guardians.
Disease behavior was defined according to the Montreal classification system. Disease complications included strictures (B2), fistulas (B3), progression to surgery, and experiencing remission. B2 and B3 disease were defined using endoscopy and/or imaging (fluoroscopy, CT, or MRI) and correlation with patient symptoms, in contrast to the non-stricturing, non-fistulizing phenotype (B1)23,24. Progression to surgery was defined as requiring an abdominal surgical procedure for resection of bowel. Remission was defined as experiencing a steroid-free interval of at least 6 months9. Outcomes were recorded with a mean follow-up period of 6 years.
Specimen, mRNA, and data processing
Macroscopically uninflamed mucosal samples from the ascending colon and terminal ileum were obtained at the time of initial diagnosis, before therapy was started. These samples were preserved as formalin-fixed paraffin-embedded (FFPE) tissue.
RNA was isolated from FFPE tissue using the Quick-RNA FFPE MiniPrep (Zymo Research, Irvine, CA). This kit preserves mRNA content while using column-based DNase to eliminate DNA contamination. Total RNA was then purified using the MagMAX kit in the KingFisher system (ThermoFisher, Carlsbad, CA). RNA-seq libraries were prepared using TruSeq Stranded Total RNA with Ribo-Zero (Illumina, San Diego, CA). Paired-end (50 base pairs) sequencing was processed on the NovaSeq 6000 platform using default parameters (Illumina, San Diego, CA). Transcript expression was then quantified using Salmon with default parameters25.
Purity and integrity of the samples was assessed using a variety of quality control metrics. We first identified samples with a low number of transcripts counted (< 25,000). Further investigation of these samples confirmed low transcript integrity number (TIN)26, percentage of sequences aligned, and high duplication percentage. These samples (n = 2) were then discarded. Further, we used PCA (principal component analysis) plots to identify samples which did not cluster with their respective tissue (ileal or colonic) and discarded these samples as well (n = 5). Submission of raw and processed sequencing data to a public repository is pending.
Differential expression analysis
PCA showed that batch, sex, and TIN drove the greatest variation between samples that was unrelated to disease phenotype, so these variables were explicitly included as covariates. Additional factors of unwanted variation were identified using RUVSeq27. Control genes were selected by identifying the top 1000 genes with the lowest variance out of the top 5000 genes with the highest expression. Based on variation seen in relative log expression plots across samples, correlation between factors of unwanted variation and the desired outcomes, and the number of differentially expressed genes identified by DESeq2, we used one factor of unwanted variation for final analyses.
The filterbyExpression function from EdgeR was used to select genes with at least 10 read counts in 70% of samples28. Differential expression analysis was then performed using DESeq2 with false discovery rate (FDR) adjusted P-value (p-adj) of < 0.05 considered significant. Default settings, including Wald test with Benjamini–Hochberg correct for multiple tests were used. Final PCA plots were generated using the plotPCA function from DESeq2, based on the top 500 most variable genes, after applying the variance stabilizing transform (VST) and the removeBatchEffect function from limma29,30. Pathway analysis was performed using the Molecular Signatures Database hallmark gene set collection and fgsea31,32. Volcano plots were generated using EnhancedVolcano33. Exploratory data analysis and differential expression analysis was performed in R (v4.2)34.
Modeling
Predictive models were developed for the collected outcomes, including development of B2 phenotype, progression to surgery, and remission. Consecutive models were built including clinical variables alone (Table 1) and clinical variables with gene expression in order to evaluate the contribution of gene expression to overall predictions. Separate models were also built with and without rectosigmoid involvement, a clinical feature not previously reported in other predictive models for pediatric CD17,35. Based on the results of the differential expression analysis, colonic gene expression data was used. Models were trained based on normalized gene counts, processed as described above including filtering genes by expression, controlling for batch, sex, TIN, and 1 factor of variation, and normalizing using the variance stabilizing transformation27,28,29. Given the small sample size, leave-one-out cross-validation was used. With this approach, a unique model is trained for each sample in the dataset, that sample is excluded from training and used for evaluation, and model performance is calculated as an average across all samples. Genes were selected for inclusion within models using the least absolute shrinkage and selection operator (LASSO), a regularized linear model that identifies a concise set of predictive features. While many feature selection techniques exist, LASSO provides an efficient, multivariate method, which provides consistent, repeatable results36. Care was taken to apply gene selection within folds, with LASSO applied to only the training data for each fold.
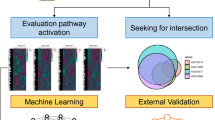
Multiple machine learning approaches were tested and compared, including LASSO, random forest (RF), gradient boosting (XGB), deep neural networks (NN)37. RF and XGB are decision tree-based methods, while NN, also known as deep learning, uses layers of non-linear functions to process data36. Each model was assessed using area under the receiver operating characteristic curve (AUROC) and area under the precision-recall curve (AUPRC). Feature importance was determined for the LASSO model using its coefficients. Coefficients were summarized across cross-validation folds by summing the absolute value for each fold. PCA plots were then generated using the genes with the highest coefficient values across all folds. Model training, evaluation, and interpretation was performed in Python (v3.8) using the Scikit-Learn and Tensorflow libraries37,38,39. The overall analysis strategy is summarized in Fig. 1. Code to reproduce differential expression analysis and model development is available at https://github.com/gomezlab/ped_ibd_rnaseq.

Flowsheet summarizing analysis strategy. CD Crohn’s disease, ML Machine learning.
Results
Study population characteristics
After applying quality control, 56 CD patients with colon samples and 56 CD patients with ileum samples were included in the study cohort, while 46 non-IBD patients with colon samples and 46 non-IBD patients with ileum samples were used as controls. For CD patients with colon samples, 33.9% of patients were female, the average age of diagnosis was 11.7, and 69.6% of patients had ileocolonic disease. 19.6% of patients developed B2 complications, 10.7% developed B3 complications, 32.1% required surgery, and 76.8% experienced a period of remission (Table 1). Of note, all 12 patients who developed B2 complications required surgery and 12 of 19 (63.1%) of patients who required surgery had B2 complications.
Differential expression analysis
PCA of CD compared with non-IBD samples showed some differences in disease status across the first principle component for both colonic and ileal tissues (Fig. 2A,B). We first identified differentially expressed genes (DEGs) between patients with CD compared with non-IBD controls, in both colonic and ileal tissue. In total, 10,973 DEGs were identified for colonic tissue and 8799 for ileal tissue (p-adj < 0.05) (Fig. 2C,D). Genes related to inflammatory response (CXCL8, AQP9, INHBA, IL1B, CXCL6, and IL6) were upregulated in CD compared with non-IBD, while genes related to DNA repair (MPC2, VPS28, EDF1, ALYREF, and PCNA) and oxidative phosphorylation (IDH3B, ATP5MC1, ATP5ME, MRPL11, COX7C, and PHB2) were downregulated. A complete list of all differential expression results is available in Supplementary Table 1 (colon) and 2 (ileum).

Differential gene expression analysis for pediatric patients with Crohn’s disease versus controls. (A) PCA plot based on colonic gene expression. (B) PCA plot based on ileal gene expression. (C) Volcano plot showing differentially expressed genes with p < 0.05 and log2 fold change > 1.5 based on colonic gene expression. (D) Volcano plot based on ileal gene expression (same criteria). (E) Gene set enrichment analysis based on Hallmark pathways for colonic gene expression. (F) Gene set enrichment analysis based on Hallmark pathways for ileal gene expression.
We then analyzed DEGs between patients experiencing specific outcomes (B2—stricturing, B3—fistulizing, progression to surgery, and remission) and those who did not. Of the four outcomes, B2 showed the clearest difference in gene expression (Fig. 3A,B). For colonic tissue, genes related to extracellular matrix (ECM) production (MMP3, MMP1, CHI3L1), as well as inflammatory processes (CXCL5, CXCL8, AQP9, INHBA) were downregulated in patients who experienced B2 complications. The Hallmark pathways interferon-gamma response, inflammatory response, and epithelial mesenchymal transition were notably downregulated (Fig. 3C). A full list of differential expression results for B2 in colonic tissue is available in Supplementary Table 3. For B2 in ileal tissue, no significant DEGs were identified. Analysis of DEGs for B3 showed 2 for colon and 1 for ileum, although these showed no specific pattern. For progression to surgery, 4 DEGs were identified for colon and 1 for ileum. This included upregulation of mitochondrial genes (MTCO1P12 and MTND1P23) and downregulation of UCN2 and CXCL5 in colonic tissue. For ileal tissue, MTCO1P12 was upregulated. Finally, analysis of remission showed no DEGs.

Differential gene expression analysis for pediatric Crohn’s disease patients experiencing stricturing versus non-stricturing disease based on colonic tissue (A), PCA plot of colonic gene expression. (B) Volcano plot showing differentially expressed genes with p < 0.05 and log2 fold change > 1.5. (C) Gene set enrichment analysis based on Hallmark pathways. (D) Boxplots for selected genes, 0; non-stricturing, 1; stricturing.
Predictive modeling
We first developed models for each of the recorded outcomes based on clinical variables alone (sex, diagnosis age, disease location, perianal disease, and family history of IBD). Overall, these showed poor accuracy with AUROC of < 0.6 for all models for all outcomes. Adding gene expression resulted in a significant improvement in predictive ability (Fig. 4). For B2, neural networks (NN) showed the highest performance, with an AUROC of 0.806 (95% CI 0.753–0.859) compared with 0.583 (95% CI 0.518–0.649) for clinical variables alone. For remission and surgery, NN was also the highest performing model, obtaining an AUROC of 0.834 (95% CI 0.784–0.883) and 0.732 (95% CI 0.673–0.792) for each outcome respectively. AUROC and AUPRC results for all models are available in Supplementary Table 4.

Receiver operating characteristic curves for all models predicting pediatric Crohn’s disease complications based on clinical variables and gene expression RF random forest, XGB gradient boosting, NN neural network, AUROC area under the receiver operating characteristic curve, CI confidence interval.
Addition of rectosigmoid involvement to the clinical model also resulted in significant improvements for all outcomes compared with the original clinical variables with AUROC 0.7–0.8. Finally, combining all variable types (clinical variables, rectosigmoid involvement, and gene expression) resulted in the highest accuracy for B2, with NN showing an AUROC of 0.836, and remission, with XGB showing an AUROC of 0.834 (Fig. 5). In contrast, for surgery, clinical variables with gene expression and clinical variables with rectosigmoid involvement showed the best performance, with an AUROC for XGB of 0.751. AUROC and AUPRC results for these models are available in Supplementary Table 4.

Receiver operating characteristic curves for all models predicting pediatric Crohn’s disease complications based on clinical variables, rectosigmoid involvement, and gene expression RF random forest, XGB gradient boosting, NN neural network, AUROC area under the receiver operating characteristic curve, CI confidence interval.
Analysis of the LASSO prediction model for B2 to determine which genes showed the strongest contributions to model predictions revealed differences compared with differential expression analysis. Of the 131 genes used across all folds, 33 were found to be significantly differentially expressed. Genes related to inflammatory/immune processes were highly important, including CXCL9, DUOX2, and FOXP3. ECM-related genes were also important, including MMP3, MMP1, and CHI3L1. Genes with the largest cumulative absolute values for coefficients are listed in Fig. 6A. Pathway enrichment analysis showed that the Hallmark pathways interferon-gamma response and IL-6/JAK/STAT signaling showed the strongest enrichment (Fig. 6B). PCA plots based only on the top 20 genes identified by the LASSO models showed strong clustering of the B2 samples (Fig. 6C). Interestingly, of the 5 genes used in > 50% of folds (REG1A, FGL2, DMBT1, MMP3, and DUOX2), only 1 (DMBT1) was found to be significantly differentially expressed (Fig. 6D). Two of these, FGL2 and DUOX2 trended towards significance, with adjusted p-values of 0.17 and 0.07 respectively. Boxplots of expression of these specific genes showed clear differences between the two groups, but significant heterogeneity between samples.

Analysis of model predicting stricturing (B2) complications for pediatric Crohn’s disease (A) Top genes based on LASSO coefficients across all cross-validation folds. (B) Pathway analysis based on top genes. (C) PCA plot based on top genes. (D) Boxplots of expression by B2 status for genes used in > 50% of folds, but not found to be differentially expressed.
Discussion
Patients with pediatric CD who experienced stricturing complications showed a distinct colonic transcriptome at time of diagnosis compared with those who did not, with downregulation of inflammatory and extracellular matrix (ECM) production pathways. Patients who required surgery also showed downregulation of the ECM-related pathways. In contrast, there was no clear difference in the pattern of gene expression between patients who experienced fistulizing complications or those who experienced remission based on differential expression analysis. Machine learning-based models were able to incorporate information from gene expression to improve upon predictions based on clinical variables alone and predict with good accuracy which patients would develop stricturing complications, experience remission, or require surgery. This was despite limited changes in individual genes for the remission and surgery outcomes, suggesting improved predictions based on combinations of genes.
Previous studies have established a link between gene expression, particularly in the ECM and inflammatory pathways, and pediatric CD outcomes40. Haberman et al. identified DUOX2, MMP3, AQP9, and IL8 as highly upregulated and APOA1, NAT8, and AGXT2 as highly downregulated in ileal tissue for pediatric CD. These gene signatures were then used to predict steroid-free remission with an AUROC of 0.7219. Kugathasan et al. identified upregulation of several ECM-related gene ontology pathways in the ileum of pediatric CD patients experiencing B2 complications and used an ECM gene signature to predict development of B2 complications with an AUROC of 0.7217. Ta et al. also identified inflammatory and ECM gene signatures as associated with transmural healing for pediatric CD patients with inflammatory small bowel disease41. Finally, Dovrolis et al. studied fibrotic disorders across 9 different organ types, including fibrotic CD, and similarly showed differential expression of the genes MMP1, AQP9, and CXCL5 in fibrotic disease42.
The results of our study broadly agree with previous work and confirm the importance of ECM and inflammatory pathways for pediatric CD outcomes. However, they also differ from previous work in pediatric CD in that our analysis focuses on colonic rather than ileal tissue and shows downregulation of the inflammatory response and epithelial mesenchymal transition pathways in this tissue type. Location-based studies have shown that colonic and ileal disease show stark differences at the transcriptomic level43. The current results agree with previous studies suggesting prognostic significance of colonic gene expression for predicting mainly ileal complications, as the ileal transcriptome may be completely dominated by current, active disease23,44. Similar results were recently demonstrated in a single-cell transcriptomic profiling of CD, with terminal ileal samples dominated by inflammation and a higher total number of differentially expressed genes identified in the colon. This study also similarly identified alteration of mucin gene expression as a signal of rewiring of mucosal barrier function45. In addition, Bai et al. showed that CD patients have increased CD4 + T cells and memory-activated CD4 + T cells in the rectum compared with controls, suggesting a cellular sequelae of this differential expression46.
Of note, these results relied on FFPE tissue, which allowed assembly of a broader cohort at lower cost, but showed broad agreement with results based on fresh tissue, especially in CD versus non-IBD comparisons9. FFPE has been previously used in multiple previous studies, including of cardiac, breast, and rectal tissue, with overall robust results47,48,49. In addition, despite using a smaller training set and rigorous cross-validation, our models show higher predictive accuracy (AUROC > 0.8) compared with previous studies, demonstrating the potential for more complex, machine learning-based models to outperform traditional logistic regression.
Analysis of the contributions of individual genes to our models reveals associations between genes and outcomes that may be overlooked by single gene differential expression techniques. Due to heterogeneity in gene expression, these associations may not appear when groups are considered in aggregate. In particular, the genes REG1A, MMP3, and DUOX2 strongly influenced model predictions and have been found to be associated with IBD and disease severity in previous studies, but were not identified as significantly differentially expressed9,50,51.
Another interesting finding from our study was the strong inverse relationship between rectosigmoid involvement and development of stricturing disease. Previous studies have identified young age, ileocolonic involvement, perianal involvement, and early response to initial therapy as predictive of CD complications5,35,52. However, few studies have specifically examined rectosigmoid disease52. This finding merits further study in other populations.
Our results join a growing body of research highlighting the potential for machine learning to predict outcomes related to IBD and support clinicians in providing therapies tailored to those predictions. Machine learning has been used to predict hospitalization and outpatient steroid use53, response to biologic therapy54, post-operative CD recurrence55, and identify novel serum markers21. Machine learning can identify relationships within multi-omic, high dimensional data and is particularly well-suited to assist the transition from a “trial and error” approach to precision medicine in IBD56.
Our study has important limitations. First, it is based on a relatively small, single-institution dataset. While the exact models generated using this dataset may not be generalizable, the described methods for selecting and modeling on gene expression should be broadly applicable. Second, similar to previous studies, we were not able to consistently model B3 complications, likely due to the heterogeneity of the subtype17. Third, analyzing paired affected and unaffected regions for each patient may have captured the impact of inflammation on molecular phenotypes. Fourth, treatment in this study was left to the discretion of the primary pediatric gastroenterologist and differences in treatment selection had an unadjusted effect on outcomes. Finally, our analysis does not include other data types, such as small RNA, chromatin biology, serum markers, or microbial composition. Prediction of IBD outcomes by applying machine learning to these multi-omic data sources represents an exciting direction for future research22,57.
Conclusions
Pediatric CD patients who experience complications show a distinct colonic transcriptome at the time of diagnosis. Machine learning can use this information to predict future outcomes, including strictures, remission, or progression to surgery. Applied to larger, multi-institutional datasets, this approach can develop prognostic models to support clinicians in identifying which patients are at highest risk of CD-specific complications and tailor therapies to improve outcomes.
Data availability
Processed transcript counts are available at the Gene Expression Omnibus (GEO), accession # GSE221161. Raw sequences are available at the NIH database of Genomes and Phenotypes (dbGaP), accession # phs003156.v1.p1.
References
Kugathasan, S. & Hoffmann, R. The incidence and prevalence of pediatric inflammatory bowel disease (IBD) in the USA. J. Pediatr. Gastroenterol. Nutr. 39, S48–S49 (2004).
Benchimol, E. I. et al. Incidence, outcomes, and health services burden of very early onset inflammatory bowel disease. Gastroenterology 147, 803-813.e7 (2014).
Loftus, C. G. et al. Update on the incidence and prevalence of Crohn’s disease and ulcerative colitis in Olmsted County, Minnesota, 1940–2000. Inflamm. Bowel Dis. 13, 254–261 (2007).
Vernier-Massouille, G. et al. Natural history of pediatric Crohn’s disease: A population-based Cohort study. Gastroenterology 135, 1106–1113 (2008).
Thia, K. T., Sandborn, W. J., Harmsen, W. S., Zinsmeister, A. R. & Loftus, E. V. Risk factors associated with progression to intestinal complications of Crohn’s disease in a population-based cohort. Gastroenterology 139, 1147–1155 (2010).
Freeman, H. J. Age-dependent phenotypic clinical expression of Crohn’s disease. J. Clin. Gastroenterol. 39, 774–777 (2005).
Pigneur, B. et al. Natural history of Crohn’s disease: comparison between childhood- and adult-onset disease. Inflamm. Bowel Dis. 16, 953–961 (2010).
Abraham, B. P., Mehta, S. & El-Serag, H. B. Natural history of pediatric-onset inflammatory bowel disease: A systematic review. J. Clin. Gastroenterol. 46, 581–589 (2012).
Haberman, Y. et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Invest. 124, 3617–3633 (2014).
Neurath, M. F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 14, 329–342. https://doi.org/10.1038/nri3661 (2014).
Noble, C. L. et al. Characterization of intestinal gene expression profiles in Crohn’s disease by genome-wide microarray analysis. Inflamm. Bowel Dis. 16, 1717–1728 (2010).
Dovrolis, N. et al. The interplay between mucosal microbiota composition and host gene-expression is linked with infliximab response in inflammatory bowel diseases. Microorganisms 8, 438 (2020).
Gisbert, J. P. & Chaparro, M. Predictors of primary response to biologic treatment [Anti-TNF, Vedolizumab, and Ustekinumab] in patients with inflammatory bowel disease: From basic science to clinical practice. J. Crohn’s Colitis 14, 694–709 (2020).
West, N. R. et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 23, 579–589 (2017).
Leal, R. F. et al. Identification of inflammatory mediators in patients with Crohn’s disease unresponsive to anti-TNFα therapy. Gut 64, 233–242 (2015).
Bank, S. et al. Polymorphisms in the NFkB, TNF-alpha, IL-1beta, and IL-18 pathways are associated with response to anti-TNF therapy in Danish patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 49, 890–903 (2019).
Kugathasan, S. et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: A multicentre inception cohort study. Lancet 389, 1710–1718 (2017).
Haberman, Y. et al. Mucosal inflammatory and wound healing gene programmes reveal targets for stricturing behaviour in paediatric Crohn’s disease. J. Crohn’s Colitis 15, 273–286 (2021).
Foster, J. D. et al. Application of objective clinical human reliability analysis (OCHRA) in assessment of technical performance in laparoscopic rectal cancer surgery. Tech. Coloproctol. 20, 361–367 (2016).
Isakov, O., Dotan, I. & Ben-Shachar, S. Machine learning-based gene prioritization identifies novel candidate risk genes for inflammatory bowel disease. Inflamm. Bowel Dis. 23, 1516–1523 (2017).
Ungaro, R. C. et al. Machine learning identifies novel blood protein predictors of penetrating and stricturing complications in newly diagnosed paediatric Crohn’s disease. Aliment. Pharmacol. Ther. 53, 281–290 (2021).
Gardiner, L. J. et al. Combining explainable machine learning, demographic and multi-omic data to inform precision medicine strategies for inflammatory bowel disease. PLoS One 17, e0263248 (2022).
Keith, B. P. et al. Colonic epithelial miR-31 associates with the development of Crohn’s phenotypes. JCI Insight 3, e122788 (2018).
Satsangi, J., Silverberg, M. S., Vermeire, S. & Colombel, J. F. The Montreal classification of inflammatory bowel disease: Controversies, consensus, and implications. Gut 55, 749–753 (2006).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Wang, L. et al. Measure transcript integrity using RNA-seq data. BMC Bioinform. 17, 1–16 (2016).
Risso, D., Ngai, J., Speed, T. P. & Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32(9), 896–902 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015).
Sergushichev, A. A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. (2016), bioRxiv 060012 https://doi.org/10.1101/060012.
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417 (2015).
Blighe, K., Rana, S. & Lewis, M. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. R package version 1.14.0. https://github.com/kevinblighe/EnhancedVolcano (2022).
R Core Team. R: A Language and Environment for Statistical Computing. https://www.r-project.org/ (2020).
Levine, A. et al. Complicated disease and response to initial therapy predicts early surgery in paediatric Crohn’s Disease: Results from the Porto group GROWTH study. J. Crohn’s Colitis 14, 71–78 (2020).
Géron, A. Hands-on machine learning with Scikit-Learn, Keras, and TensorFlow: Concepts, tools, and techniques to build intelligent systems (O’Reilly Media, 2019).
Pedregosa, F. et al. Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
scikit learn. https://scikit-learn.org/stable/modules/generated/sklearn.linear_model.LogisticRegression.html.
Chollet, F. & others. Keras. https://github.com/fchollet/keras (2015).
Alfredsson, J. & Wick, M. J. Mechanism of fibrosis and stricture formation in Crohn’s disease. Scand. J. Immunol. 92, e12990 (2020).
Ta, A. D. et al. Association of baseline luminal narrowing with ileal microbial shifts and gene expression programs and subsequent transmural healing in pediatric Crohn disease. Inflamm. Bowel Dis. 27, 1707–1718 (2021).
Dovrolis, N. et al. Co-expression of fibrotic genes in inflammatory bowel disease; A localized event?. Front. Immunol. 13, 133 (2022).
Gonzalez, C. G. et al. Location-specific signatures of Crohn’s disease at a multi-omics scale. Microbiome 10, (2022).
Toyonaga, T. et al. Increased colonic expression of ACE2 associates with poor prognosis in Crohn’s disease. Sci. Rep. 11, 13533 (2021).
Kong, L. et al. The landscape of immune dysregulation in Crohn’s disease revealed through single-cell transcriptomic profiling in the ileum and colon. Immunity 56, 444-458.e5 (2023).
Bai, X., Liu, W., Chen, H., Zuo, T. & Wu, X. Immune cell landscaping reveals distinct immune signatures of inflammatory bowel disease. Front. Immunol. 13, 861790 (2022).
Park, I. J. et al. A nine-gene signature for predicting the response to preoperative chemoradiotherapy in patients with locally advanced rectal cancer. Cancers 12, 800 (2020).
Jacobsen, S. B., Tfelt-Hansen, J., Smerup, M. H., Andersen, J. D. & Morling, N. Comparison of whole transcriptome sequencing of fresh, frozen, and formalin-fixed, paraffin-embedded cardiac tissue. PLoS One 18, e0283159 (2023).
Pennock, N. D. et al. RNA-seq from archival FFPE breast cancer samples: molecular pathway fidelity and novel discovery. BMC Med. Genom. 12, 1–18 (2019).
Kofla-Dlubacz, A., Matusiewicz, M., Krzystek-Korpacka, M. & Iwanczak, B. Correlation of MMP-3 and MMP-9 with Crohn’s Disease activity in children. Dig. Dis. Sci. 57, 706 (2012).
Van Beelen Granlund, A. et al. REG gene expression in inflamed and healthy colon mucosa explored by in situ hybridisation. Cell Tissue Res. 352, 639 (2013).
Torres, J. et al. Predicting outcomes to optimize disease management in inflammatory bowel diseases. J. Crohn’s Colitis 10, 1385–1394 (2016).
Waljee, A. K. et al. Predicting hospitalization and outpatient corticosteroid use in inflammatory bowel disease patients using machine learning. Inflamm. Bowel Dis. 24, 45 (2018).
Waljee, A. K. et al. Development and validation of machine learning models in prediction of remission in patients with moderate to severe crohn disease. JAMA Netw. Open 2, e193721–e193721 (2019).
Cushing, K. C. et al. Predicting risk of postoperative disease recurrence in Crohn’s disease: Patients with indolent Crohn’s disease have distinct whole transcriptome profiles at the time of first surgery. Inflamm. Bowel Dis. 25, 180–193 (2019).
Noor, N. M., Sousa, P., Paul, S. & Roblin, X. Early diagnosis, early stratification, and early intervention to deliver precision medicine in IBD. Inflamm. Bowel Dis. 28, 1254–1264 (2022).
Gubatan, J. et al. Artificial intelligence applications in inflammatory bowel disease: Emerging technologies and future directions. World J. Gastroenterol. 27, 1920–1935 (2021).
Acknowledgements
This study was supported by work from the University of North Carolina Pathology Services Core, High Throughput Sequencing Facility, and Translational Genomics Lab which are supported in part by an NCI Center Core Support Grant (5P30CA016080-42). We also appreciate the advice we received from Dr. Katherine Hoadley and Dr. Praveen Sethupathy. This study was supported by funding from the NIDDK (P01DK094779, 1R01DK104828, P30-DK034987) and the Helmsley Charitable Trust (SHARE Project 2). Kevin A Chen is supported by funding from the National Institutes of Health (UNC Integrated Translational Oncology Program T32-CA244125 to UNC/KAC).
Author information
Authors and Affiliations
Contributions
K.A.C.: study conceptualization, data curation, data analysis, model development, manuscript writing, N.N.: data curation, data analysis, manuscript editing, M.M.K.N.: data curation, data analysis, manuscript editing, A.S.: data curation, manuscript editing, C.U.J.: data analysis, model development, manuscript editing, M.R.S.: sample preparation, data curation, manuscript editing, G.L.: sample preparation, data curation, manuscript editing, C.B.: sample preparation, data curation, manuscript editing, L.-C.Z.: sample preparation, manuscript editing, S.B.: sample preparation, manuscript editing, M.R.: study conceptualization, data curation, manuscript editing, S.M.G.: study conceptualization, model development, manuscript editing, T.S.F.: study conceptualization, data curation, data analysis, model development, manuscript writing, S.Z.S.: study conceptualization, data curation, data analysis, model development, manuscript writing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, K.A., Nishiyama, N.C., Kennedy Ng, M.M. et al. Linking gene expression to clinical outcomes in pediatric Crohn’s disease using machine learning. Sci Rep 14, 2667 (2024). https://doi.org/10.1038/s41598-024-52678-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-52678-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
