
Abstract
Wellcomia compar (Spirurina: Oxyuridae) is a pinworm that infects wild and captive porcupines. Despite clear records of its morphological structure, its genetics, systematics, and biology are poorly understood. This study aimed to determine the complete mitochondrial (mt) genome of W. compar and reconstruct its phylogenetic relationship with other nematodes. We sequenced the complete mt genome of W. comparand conducted phylogenetic analyses using concatenated coding sequences of 12 protein-coding genes (PCGs) by maximum likelihood and Bayesian inference. The complete mt genome is 14,373 bp in size and comprises 36 genes, including 12 protein-coding, two rRNA and 22 tRNA genes. Apart from 28 intergenic regions, one non-coding region and one overlapping region also occur. A comparison of the gene arrangements of Oxyuridomorpha revealed relatively similar features in W. compar and Wellcomia siamensis. Phylogenetic analysis also showed that W. compar and W. siamensis formed a sister group. In Oxyuridomorpha the genetic distance between W. compar and W. siamensis was 0.0805. This study reports, for the first time, the complete W. compar mt genome sequence obtained from Chinese porcupines. It provides genetic markers for investigating the taxonomy, population genetics, and phylogenetics of pinworms from different hosts and has implications for the diagnosis, prevention, and control of parasitic diseases in porcupines and other animals.
Similar content being viewed by others


Introduction
The Chinese Porcupine (Hystrix brachyura) is a member of the rodent family Hystricidae. As a common terrestrial wild animal, it occurs in a wide range of habitats, including primary and secondary forest, cultivated areas and plantations1. Artificial breeding of porcupines used to be very popular in China for its high medicinal and ornamental value2. Porcupines are relatively well-protected from predators but are subject to a degree of parasitic infestation. Thus, clarifying the species of parasites that porcupines are infected with is important for the control of parasitic diseases. Wellcomia compar, the so-called porcupine pinworm that belongs to Oxyuridae nematode, was first reported and the detail of its morphological structure was recorded by Leidy3. Similar to that of other pinworms, the life cycle of W. compar is simple and typically entails the ingestion of infective eggs orally, and then the larvae infiltrating the glandular fossa of the cecum mature into adult worms. The pinworm can adversely impact porcupine hosts by causing indigestion, weight loss, and poor coat condition. However, so far, few studies have reported the genetics, systematics, and biology of this pinworm.
The phylogeny and classification of nematodes have remained subjects of debate, and the poor resolution conferred by morphologic characteristics has resulted in incongruent classification systems. 18S rRNA was the first genetic marker used for phylogenetic classification of nematodes4. This and subsequent analyses based on 18S rRNA have led to the recognition of major lineages of nematodes5,6,7. Recent molecular analyses of mt genome sequences have provided selectable hypotheses for the phylogenetic relationships of nematodes based on 18S rRNA.
Mitochondria (mt) synthesise their own proteins and are involved in various physiological functions8. After a long period of evolution, the genetic composition of mt in most metazoans is now present consistently, suggesting that mt genes are important for maintaining the basic functions of mt9. The mt genome accumulates mutation rates at a faster rate than the nuclear genome and is characterised by fast evolution, maternal inheritance, and a simple structure. Therefore, it is considered a reliable marker in cladistic systematics10,11 and has been widely employed as a genetic marker for identifying and distinguishing organisms, particularly for population genetics among nematode species12,13,14. The 12 protein-coding genes (PCGs) of the mt genome are also often used to elucidate the phylogenetic relationships between species15,16,17,18,19,20,21.
Despite the availability of numerous advanced sequencing and bioinformatic methods, the 18S rRNA gene remains the primary genetic marker used for phylogenetic construction and molecular identification of Oxyuridomorpha. Additionally, other genes such as Cox1, ITS1, ITS2, and 28S rRNA are also employed for this purpose22,23,24,25,26. However, the use of complete genome mt for constructing and characterising Oxyuridomorpha phylogenies is infrequent. To date, only six Oxyuridomorpha species have had their complete mt genomes published17,18,19,20,21. Therefore, more mt genomes of Oxyuridomorpha nematodes are needed to provide useful molecular markers for population genetic and phylogenetic studies of pinworms.
In this study, we aimed to sequence and analyse the complete mt genome of W. compar and compared it to the gene arrangement order of other pinworms. In addition, we used the concatenated coding sequences (CDS) of 12 PCGs from 33 Spirurina nematodes and an Enoplea nematode to reconstruct a more reliable phylogeny.
Methods
Ethics statement
No animal was harmed during the course of this research. This study was approved by the Animal Ethics Committee of the Zunyi Medical University (approval no. LVRIAEC [2016] 2-059).
Parasite isolates and DNA extraction
Adult W. compar parasites were collected from infected porcupines in Zunyi, Guizhou Province, China. The samples were washed with sterile saline, preserved in 100% ethanol and stored in a − 20 °C refrigerator until use. The isolation of total genomic DNA from the samples was carried out using the Wizard Genomic DNA Purification Kit (Promega, Madison, Wisconsin, USA), following the instructions provided by the manufacturer. The DNA concentration and quality of the extracted samples were assessed using a Nanodrop 2000 spectrophotometer. The total DNA was detected by agarose gel electrophoresis using an Agilent 2100 Bioanalyzer.
Mitochondrial genome sequencing and assembly
The whole genome shotgun strategy was used to construct libraries containing DNA fragments of 400–500 bp that were randomly interrupted by a covaris ultrasonic crusher. Paired-end genomic library (approximately 400 bp inserts) was constructed according to the manufacturer’s instructions (Illumina), and were sequenced by next-generation sequencing technology based on the Illumina HiSeq sequencing platform. A5-miseq v2015052227 and SPAdesv3.9.0 (http://cab.spbu.ru/software/spades/)28 were used to assemble high-quality second-generation sequencing data to construct contig and scaffold sequences. The contig was compared with the nt library on NCBI using BLAST v2.2.31+, and the contig from mt was screened. Then, collinearity analysis was carried out using the mummer v3.129 software to determine the positional relationship and to fill the gaps between contigs. Finally, A5-miseq software was selected for splicing results, and Pilon v1.1830 software was used to correct the results to obtain the final mt sequence.
Gene annotation and analysis
The complete mt genome sequence was uploaded to the MITOS web server (http://mitos.bioinf.uni-leipzig.de/) for functional annotation31. The genetic code was set to 05 invertebrate, and the remaining were set according to the default parameters set by MITOS. For an undetectable tRNA gene, the approximate position was determined by comparison with the corresponding tRNA sequence in the reference genome32. The secondary structure was manually drawn to determine the final sequence and structure. The base compositions (%) of the complete mt genome, PCGs, and rRNA genes were calculated using Mega X33. The CG View visualisation software was used to map the entire mt genome34. The figure of Gene rearrangement was drawn using PhyloSuite v1.2.2 (http://phylosuite.jushengwu.com/installation_packages/PhyloSuite_v1.2.2_Win64_with_plugins.rar)35.
Phylogenetic analysis and genetic distance
The 33 mt genomes of nematode parasites were downloaded from NCBI (supplementary material Table S1), including 32 of Spirurina nematodes (mostly published papers) as ingroups and one of an Enoplea nematode, Trichuris suis, as an outgroup. Phylogenetic relationships among the 33 representative nematode species sequences and that of the W. compar mt DNA obtained in this study were reconstructed based on the CDS of 12 PCGs excluding the stop codon, and were concatenated. 12 PCGs were aligned in batches with MAFFT36 using the '–auto' strategy and codon alignment mode. Each gene was translated into amino acid sequences using the invertebrate mt genetic code in MAFFT and aligned based on its amino acid sequence using the default settings. The alignments were back translated into the corresponding nucleotide sequences, and refined using the codon-aware program MACSE v. 2.0337 with the invertebrate mt genetic code, which preserves the reading frame and allows the incorporation of sequencing errors or sequences with frameshifts. Ambiguously aligned fragments of 12 alignments were removed in batches using Gblocks38 with the following parameter settings: minimum number of sequences for a conserved/flank position (16/16), maximum number of contiguous non-conserved positions (8), minimum block length (10), and allowed gap positions (with half). The final sequences of the 12 PCGs were concatenated into single alignments for phylogenetic analyses using phylosuite35. ModelFinder39 was used to select the best-fit partition model (edge-linked) using Bayesian information criterion (BIC). Best-fit partition model for BI and ML trees are listed (supplementary material Tables S2, S3) according to BIC. Bayesian inference phylogenies were inferred using MrBayes v3.2.7a40 under the partition model (2 parallel runs, 4 independent Markov chains run for 3,000,000 metropolis-coupled MCMC generations, sampling a tree every 100 generations), in which the initial 25% of sampled data were discarded as burn-in. ML phylogeny was inferred using IQ-TREE v1.6.12 using an edge-linked partition model for 50,000 ultrafast bootstraps41,42. Phylograms were drawn using Interactive Tree Of Life (iTOL) v6 (https://itol.embl.de/itol.cgi)43. The calculation of pairwise genetic distances between phylogenetic trees was conducted using MEGA X33. Variance estimation was performed utilising concatenated PCGs sequences, employing the Bootstrao method with 1000 generations, and modeling the distances using the P-distance.
Ethics approval and consent to participate
No animal was harmed during the course of this research. We confirm that all experiments were performed in accordance with relevant guidelines and regulations. All experimental procedures involving animals were reviewed, approved and supervised by the Animal Ethics Committee of Zunyi Medical University (approval no. LVRIAEC [2016] 2-059).
Results and discussion
Genome structure, organisation and composition
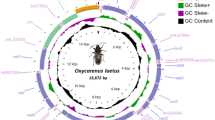
The complete mt genome of W. compar (GenBank accession No. MW059037) was a 14,373 bp closed-circular molecule (Fig. 1), encoding an entire set of 36 genes, which included 12 PCGs (cox1-3, nad1-6, nad4l, atp6, and cytb), 22 tRNA genes, two rRNA genes (s-rRNA and l-rRNA), and a non-coding region (NCR)) (Table 1). However, this was different from the Enoplea nematode (such as Trichuris_suis) gene set, which has an atp8 gene44. All genes were transcribed in the same direction on the N-strand. The distribution of genes in the mt genome was identical to those of Wellcomia siamensis17 and Syphacia obvelata18. The overall base composition of the mt genome of W. compar was as follows A: 25.7%; T: 53.0%; G: 16.9%; C: 4.4%; G + C: 21.3%; and A + T: 78.7%. The overall A-T and G-C skews values were -0.346 and 0.586, respectively (Table 2). The intergenic spacer sequence was 613 bp in total and included 28 regions ranging in size from 1 to 147 bp. The overlapping nucleotide fragments were scattered in one place, located between trn H and rrn S (-2 bp).

Circular map of the mt genome of W. compar. Scale is approximate. 12PCGs and 2 rRNA have standard nomenclature. The 22 tRNA genes are designated by a single-letter code for the corresponding amino acid, with numbers used to distinguish the two leucine and serine designated tRNAs. Grey region rerensents NCR. From the inside to the outside of the circle diagram, the first circle represents the scale, the second circle represents G-C skew, the third circle represents GC content, and the fourth circle represents the arrangement of PCGs, tRNA genes, and rRNA genes on the genome.
Gene annotation and analysis
The mt genome of W. compar encodes 12 PCGs, which contain 3420 codons with a total length of 10,260 bp. The content of A + T was 77.5%, which was far higher than G + C. The nucleotides in metazoan mt genomes are not randomly distributed, and this nucleotide bias is often linked to the unequal usage of codons45. The nucleotide usage of the 12 PCGs in the mt genome of W. compar is shown in Table 3. The codons TTT (phenylalanine, 19.2%), TTA (leucine-2, 8.2%), ATT (isoleucine, 7.5%) usage were most frequent. Therefore, the nucleotides in PCGs were biased towards A and T. Most of the PCGs had the start codon TTG, except for cox 1 that uses ATA, and the other three genes (nad 1, nad 4 and cox 2) that use ATT (Table 1). Three types of stop codons were used: TAG (nad 1, cob and nad 5), TAA (nad 6, nad 4 and nad 4l) and an abbreviated stop codon T (cox 1, atp 6, nad 2, cox 3, nad 3 and cox 2), which is an incomplete TAA stop codon and is completed by the addition of 3` A residues to the mRNA (Table 1).
W. compar mt DNA contains 22 tRNA genes, which range from 53 bp (trnS1) to 64 bp (trnI and trnT). The two rRNA genes, rrnL and rrnS, were 959 bp and 732 bp in size, respectively (Table 1). rrnL is located between trnC and trnM, and rrnS is situated between trnH and trnA. The A + T content of rrnL and rrnS was 80.9% and 76.5%, respectively (Table 2).
The NCR, located between trnS2 and trnN, was 530 bp in length with a higher A + T content (97.2%) than of any other region of the mt genome.
Gene rearrangement in Oxyuridomorpha
The Oxyuridomorpha infraorder comprises seven families: Thelastomatoidae, Travassosinematidae, Hystrignathidae, Protrelloididae, Oxyuridae, Pharyngodonidae, and Heteroxynematidae5. To date, the mt genomes of many Oxyuridomorpha nematode infraorder lineages are still underrepresented or not represented, except for those of Oxyuridae and Heteroxynematidae. Oxyuridae includes seven species (including W. compar) and Heteroxynematidae, and we thus compared the gene rearrangement between the seven species of Oxyuridomorpha.
Gene rearrangement is mainly caused by mutations in the mt46, and for which the TDRL model is possibly the most widely accepted explanation hypothesis47,48 The mt gene arrangements in the seven species were not the same (Fig. 2), with W. compar being consistent with W. siamensis and S. obvelata but different from Oxyuris equi, Enterobius vermicularis, Aspiculuris tetraptera, and Passalurus ambiguus. The main difference was the occurrence of a transposition event (a position change of trnI for O. equi and E. vermicularis; a position change of trnY for A. tetraptera) or the number of NCR. trnI was inserted between NCR and trnN, and trnY was inserted between trnQ and trnC. P. ambiguus has two NCRs, consistent with other nematodes44,49,50,51, and an extra NCR in P. ambiguus was found between trnA and trnS2. Based on the order of gene arrangement, it can be inferred that W. compare has a closer evolutionary relationship with W. siamensis and S. obvelata.

Gene orders in the seven species in Oxyuridomorpha. All genes are transcribed in the same direction on the N-strand.
Phylogenetic analyses and genetic distance
Pairwise genetic distances in the phylogenetic tree are displayed in Data S1 of the Supplementary Material, where we find that the genetic distances between species are smaller within the same superfamily than the distances with species from other families. In Oxyuridomorpha, the genetic distance between W. compar and W. siamensis was 0.0805, which was lower than that between W. compar and other pinworms. Phylogenetic analyses of W. compar and the selected Spirurina nematodes were performed using maximum likelihood (ML) and Bayesian inference (BI) methods based on concatenated mitochondrial CDS of 12 PCGs (BI/ML [Fig. 3]). The mt genome sequences may provide reliable genetic markers for examining the taxonomic status of nematodes, particularly when PCG sequences are used as markers for comparative analyses15,16,17,18,19,20,21. Because some superfamilies were represented by a single species, this topology should be interpreted with caution. Phylogenetic analysis showed that the BI and ML trees both divided Spirurina into two clades: Spiruromorpha formed one separate clade, and Oxyuridomorpha + Rhigonematomorpha + Gnathostomatomorpha + Dracunculoidea + Ascaridomorpha formed another clade. The second clade was further sub-divided into two clades, Rhigonematomorpha + Gnathostomatomorpha + Dracunculoidea + Ascaridomorpha and Oxyuridomorpha. These results are consistent with a recent study in Spirurina47.

A phylogenetic tree of Spirurina inferred by BI and ML using Trichuris suis as the outgroup. Bayesian posterior probability (left) and bootstrap support (right) values of each clade are displayed. Wellcomia compar has been bolded. Inconsistent branches in ML were listed separately(right).
Within Spiruromorpha, many nodes were well-supported. Of note, our results indicate that Tetrameres grusi is a sister to Spirocerca lupi. Although S. lupi and Thelazia callipaeda belong to the superfamily Thelazioidea, T. callipaeda did not cluster together with S. lupi, but instead showed an early diverging position to S. lupi. These results are identical to those previously reported50,52. Within Ascaridomorpha, the superfamily Ascaridoidea forms a well-supported clade, whereas Rhigonematoidea and Heterakoidea form a sister branch53,54, and they are the sister group of Seuratoidea. Notably, Ascaridoidea, Heterakoidea, and Seuratoidea belong to the common infraorder: Ascaridomorpha. The topology of this clade indicated that Rhigonematoidea is closely related to the superfamily Ascaridomorpha. In many early morphology-based classification systems, Rhigonematomorpha was considered to be related to Oxyuridomorpha (pinworms)55. Subsequent research based on 18S rRNA phylogeny separated Rhigonematomorpha from Oxyuridomorpha as a distinct group6. Our results are inconsistent with those of previous research, and we suggest that it may belong to Ascaridomorpha.
Our results show that Dracunculoidea is more closely related to Ascaridomorpha than Spiruromorpha, which is in agreement with recent studies42. In our study, Gnathostomatomorpha forms a sister branch with Rhigonematoidea + Ascaridomorpha, which is consistent with previous studies47,51,56. However, previous studies have also suggested that Gnathostomatomorpha is nested within Ascaridomorpha57, or that Gnathostomatomorpha forms a sister branch with Ascaridomorpha + Oxyuridomorpha + Dracunculoidea + Spiruromorpha58.
The phylogenetic trees derived from two analytical methods exhibited a high degree of similarity in their topological structure. Notably, the branching patterns of the trees within the Gnathostomatomorpha clade displayed confuseing characteristics. Furthermore, dissimilar topologies were observed in the Oxyuridomorpha clade. Our results revealed that W. compar was sister to W. siamensis, with high statistical support (Bayesian posterior probability = 1.00; bootstrap support = 100). In Oxyuridomorpha, W. compar and W. siamensis are clustered together and have closer relationships with S. obvelata than with E. vermicularis in both trees, which is further supported by the comparison of gene orders and the genetic distance (PCGs) in this study. In previous research, it was observed that A. tetraptera and S. obvelata exhibited clustering, while P. ambiguus was identified as the sister species to O. equi18. Another study confirmed that W. siamensis was the sister species to P. ambiguus, as mentioned in a prior investigation19. Additionally, a recent study47 found P. ambiuguus to be sister to A. tetraptera. However, our results differ from these findings, possibly due to the limited availability of the complete mt genome of pinworms. In the present study, preference was given to BI tree classification owing to its higher support values. Despite pinworms are nematodes of human and animal health significance, the sequencing and publication of mitochondrial genomes have been limited to only seven species. Consequently, there is a need to increase taxon sampling in order to facilitate future phylogenetic investigations of this infraorder using mt genomic datasets.
The systematics of the suborder Spiruromorpha (Spiruromorpha, Oxyuridomorpha, Rhigonematoidea, Gnathostomatomorpha and Ascaridomorpha) has been controversial for decades. However, sequence analysis based on mt genomes presents roughly similar phylogenetic trees, which differ from those constructed based on 18S rRNA and single mitochondrial genes. To date, phylogenetic analysis based on 18S rRNA genes has accumulated more than 2700 sequences59, covering many but not all nematode taxa. However, it lacks the resolution needed to elucidate the phylogenetic relationships of the entire nematode tree, as different taxa exhibit different gene mutation rates, meaning that any single marker is unlikely to perform equally well in inferring the phylogeny of all nematode taxa. A better strategy for analysing nematode phylogeny involves targeting multiple genes rather than one gene. A large number of genes can help offset the adverse effects of recalcitrant genes in the analysis60,61. An analysis based on mt genomes fits this trend better, perhaps representing a better future for systematics.
Conclusions
The present study identified the complete mt genome sequence of W. compar, enriching the mt genome database of Oxyuridae. This is the first complete mt genome isolated from the body of Chinese porcupines and belongs to the same Wellcomia genus as W. siamensis previously isolated from Korea. We have compared the results with the mt genome of other Oxyuridomorpha pinworms. The gene sequence arrangement results show that W. compar is more closely related to W. siamensis and S.obvelata. Based on the mt genome sequence, we have determined the phylogenetic relationships within Spirurina, and the phylogenetic analysis indicates that W. compar is the sister group of W. siamensis, which is consistent with the gene arrangement results. In the phylogenetic analysis, we found that the species support of Oxyuridomorpha was low and inconsistent. Therefore, we believe that it is necessary to further investigate additional taxonomic groups and sequencing data to understand and document the evolution of this important group. This mt genome provides a new genetic marker for studying the molecular epidemiology, population genetics, and systematics of parasitic pinworms in animals and humans, and is of great significance for the diagnosis, prevention, and control of parasitic diseases in porcupines and other animals.
Data availability
All data supporting the conclusions of this article are included in this article. The data of W. compar in this study can be available from Genbank (MW059037).
Abbreviations
- mt:
-
Mitochondrial
- rRNA:
-
Ribosomal RNA
- tRNA:
-
Transfer RNA
- CDS:
-
Coding sequences
- PCG:
-
Protein-coding gene
- ML:
-
Maximum likelihood
- BI:
-
Bayesian inference
- NCR:
-
Non-coding region
References
Liu, S. Y. & Wu, Y. Handbook of the Mammals of China (The Straits Publishing & Distributing Group, 2018).
Tu, F., Huang, X., Zhang, Y. & Feng, Y. Complete mitochondrial genome of captive Chinese porcupine Hystrix hodgsoni (rodentia: hystricidae). Mitochondrial DNA Part B 5(2), 1945–1946 (2020).
Leidy, J. A synopsis of Entozoa and some of their ectocongeners observed by the author. Proc. Acad. Nat. Sci. Philad. 8(1), 42–58 (1856).
Blaxter, M. L. et al. A molecular evolutionary framework for the phylum Nematoda. Nature 392, 71–75 (1998).
De Ley, P. & Blaxter, M. L. Systematic position and phylogeny. In The Biology of Nematodes (ed. Lee, D. L.) 1–30 (Taylor & Francis, 2002).
De Ley, P. & Blaxter, M. L. A new system for Nematoda: Combining morphological characters with molecular trees, and translating clades into ranks and taxa. In Nematology Monographs and Perspectives (eds Cook, R. & Hunt, D. J.) 633–653 (Brill EJ, 2004).
Holterman, M. et al. Phylum-wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown Clades. Mol. Biol. Evol. 23(9), 1792–1800 (2006).
McBride, H. M., Neuspiel, M. & Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 16(14), R551–R560 (2006).
Brown, W. M., George, M. Jr. & Wilson, A. C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 76(4), 1967–1971 (1979).
Wolstenholme, D. R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 141, 173–216 (1992).
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Res. 27(8), 1767–1780 (1999).
Gao, J. F. et al. Comparative analyses of the complete mitochondrial genomes of the two ruminant hookworms Bunostomum trigonocephalum and Bunostomum phlebotomum. Gene 541(2), 92–100 (2014).
Liu, G. H. et al. Chabertia erschowi (Nematoda) is a distinct species based on nuclear ribosomal DNA sequences and mitochondrial DNA sequences. Parasit. Vectors 22(7), 44 (2014).
Liu, G. H., Li, J. Y. & Zhu, X. Q. Characterization of the complete mitochondrial genome of Setaria digitata (Nematoda: Setariidae) from China. J. Helminthol. 91(6), 772–776 (2017).
Hwang, U. W., Friedrich, M., Tautz, D., Park, C. J. & Kim, W. Mitochondrial protein phylogeny joins myriapods with chelicerates. Nature 413(6852), 154–157 (2001).
Liu, G. H. et al. Mitochondrial genome of the eyeworm, Thelazia callipaeda (Nematoda: Spirurida), as the first representative from the family Thelaziidae. PLoS Negl. Trop. Dis. 7(1), e2029 (2013).
Park, J. K. et al. Monophyly of clade III nematodes is not supported by phylogenetic analysis of complete mitochondrial genome sequences. BMC Genom. 3(12), 392 (2011).
Wang, C. R. et al. Comparative analyses of the complete mitochondrial genomes of the two murine pinworms Aspiculuris tetraptera and Syphacia obvelata. Gene 585(1), 71–75 (2016).
Liu, G. H., Li, S., Zou, F. C., Wang, C. R. & Zhu, X. Q. The complete mitochondrial genome of rabbit pinworm Passalurus ambiguus: genome characterization and phylogenetic analysis. Parasitol. Res. 115(1), 423–429 (2016).
Kang, S. et al. The mitochondrial genome sequence of Enterobius vermicularis (Nematoda: Oxyurida)—An idiosyncratic gene order and phylogenetic information for chromadorean nematodes. Gene 429(1–2), 87–97 (2009).
Zhang, Y. et al. The complete mitochondrial genome of Oxyuris equi: Comparison with other closely related species and phylogenetic implications. Exp. Parasitol. 159, 215–221 (2015).
Nadler, S. A. et al. Molecular phylogeny of clade III nematodes reveals multiple origins of tissue parasitism. Parasitology 134(Pt 10), 1421–1442 (2007).
Canova, V., Del Rosario, R. M. & Abba, A. M. A new species of Wellcomia (Nematoda: Oxyuridae) in the plains viscacha (Rodentia: Chinchillidae) from Argentina, an emended diagnosis and an update of the genus Wellcomia. Parasitol. Res. 120(3), 929–940 (2021).
Okamoto, M., Urushima, H., Iwasa, M. & Hasegawa, H. Phylogenetic relationships of rodent pinworms (genus Syphacia) in Japan inferred from mitochondrial CO1 gene sequences. J. Vet. Med. Sci. 69(5), 545–547 (2007).
Qiu, J. H. et al. Sequence variability in internal transcribed spacers of nuclear ribosomal DNA among isolates of the oxyurid nematodes Syphacia obvelata and Aspiculuris tetraptera from mice reared in laboratories in China. J. Helminthol. 90(1), 81–85 (2016).
Carta, L. K., Thomas, W. K. & Meyer-Rochow, V. B. Two nematodes (Nematoda: Diplogastridae, Rhabditidae) from the invasive millipede Chamberlinius hualienensis Wang, 1956 (Diplopoda, Paradoxosomatidae) on Hachijojima Island in Japan. J. Nematol. 50(4), 479–486 (2018).
Coil, D., Jospin, G. & Darling, A. E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31(4), 587–589 (2015).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19(5), 455–477 (2012).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5(2), R12 (2004).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9(11), e112963 (2014).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69(2), 313–319 (2013).
Kearse, M. et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12), 1647–1649 (2012).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35(6), 1547–1549 (2018).
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L. & Beatson, S. A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 8(12), 402 (2011).
Zhang, D. et al. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20(1), 348–355 (2020).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30(4), 772–780 (2013).
Ranwez, V., Douzery, E. J. P., Cambon, C., Chantret, N. & Delsuc, F. MACSE v2: Toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 35(10), 2582–2584 (2018).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56(4), 564–577 (2007).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14(6), 587–589 (2017).
Ronquist, F. et al. MrBayes 32: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61(3), 539–542 (2012).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37(5), 1530–1534 (2020).
Minh, B. Q., Nguyen, M. A. & von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30(5), 1188–1195 (2013).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49(W1), W293–W296 (2021).
Liu, G. H. et al. Clear genetic distinctiveness between human- and pig-derived Trichuris based on analyses of mitochondrial datasets. PLoS Negl. Trop. Dis. 6(2), e1539 (2012).
Herbeck, J. T. & Novembre, J. Codon usage patterns in cytochrome oxidase I across multiple insect orders. J. Mol. Evol. 56(6), 691–701 (2003).
San Mauro, D., Gower, D. J., Zardoya, R. & Wilkinson, M. A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome. Mol. Biol. Evol. 23(1), 227–234 (2006).
Chen, F. et al. Sequencing of the complete mitochondrial genome of Pingus sinensis (Spirurina: Quimperiidae): Gene arrangements and phylogenetic implications. Genes (Basel) 12(11), 1772 (2021).
Xu, X. D. et al. Novel tRNA gene rearrangements in the mitochondrial genomes of Praying mantises (Mantodea: Mantidae): Translocation, duplication and pseudogenization. Int. J. Biol. Macromol. 31(185), 403–411 (2021).
Liu, G. H. et al. Characterization of the complete mitochondrial genome of Spirocerca lupi: sequence, gene organization and phylogenetic implications. Parasit. Vectors 22(6), 45 (2013).
Gao, J. F. et al. Characterization of the mitochondrial genome of Tetrameres grusi and insights into the phylogeny of Spirurina. Int. J. Parasitol. Parasites Wildl. 6(17), 35–42 (2021).
Sun, M. M. et al. The complete mitochondrial genomes of Gnathostoma doloresi from China and Japan. Parasitol. Res. 115(10), 4013–4020 (2016).
Liu, G. H., Jia, Y. Q., Wang, Y. N., Zhao, G. H. & Zhu, X. Q. The complete mitochondrial genome of the gullet worm Gongylonema pulchrum: Gene content, arrangement, composition and phylogenetic implications. Parasit. Vectors 15(8), 100 (2015).
Kim, T. et al. Phylogeny of Rhigonematomorpha based on the complete mitochondrial genome of Rhigonema thysanophora (nematoda: chromadorea). Zool. Scr. 43(3), 289–303 (2014).
Du, H. et al. Complete mitogenome of Cruznema Tripartitum confirms highly conserved gene arrangement within family Rhabditidae. J. Nematol. 54(1), 20220029 (2022).
Skryabin, K. I. & Shikhobalova, N. P. A reconstruction of the classification of nematodes of the suborder Oxyurata Skrjabin, 1933. Trudi Gelmintologi Cheskoi Laboratoru Akademii Nauk. 5, 5–8 (1951).
Sun, M. M. et al. Complete mitochondrial genomes of Gnathostoma nipponicum and Gnathostoma sp., and their comparison with other Gnathostoma species. Infect. Genet. Evol. 48, 109–115 (2017).
Liu, G. H., Shao, R., Cai, X. Q., Li, W. W. & Zhu, X. Q. Gnathostoma spinigerum mitochondrial genome sequence: A novel gene arrangement and its phylogenetic position within the class chromadorea. Sci. Rep. 31(5), 12691 (2015).
Ahmed, M. et al. Phylogenomic analysis of the phylum Nematoda: Conflicts and congruences with morphology, 18S rRNA, and Mitogenomes. Front. Ecol. Evol. 9, 769565 (2022).
Holterman, M., Schratzberger, M. & Helder, J. Nematodes as evolutionary commuters between marine, freshwater and terrestrial habitats. Biol. J. Linn. Soc. 128, 756–767 (2019).
Smythe, A., Holovachov, O. & Kocot, K. Improved phylogenomic sampling of free-living nematodes enhances resolution of higher-level nematode phylogeny. BMC Evol. Biol. 19, 121 (2019).
Ahmed, M. & Holovachov, O. Twenty years after De Ley and Blaxter-how far did we progress in understanding the phylogeny of the phylum nematoda?. Animals (Basel). 11(12), 3479 (2021).
Funding
This research was funded by the National Natural Science Foundation of China (Grant No. 81660176).
Author information
Authors and Affiliations
Contributions
C.H. and S.Z.: Conceived of or designed study. C.H. and F.B.: Performed research, Writing—original draft. C.H., H.L. and T.Q.: Analyzed data, conducted the bioinformatics analyses. S.Z.: Supervision, project administration. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huo, C., Bao, F., Long, H. et al. The complete mitochondrial genome of Wellcomia compar (Spirurina: Oxyuridae) and its genome characterization and phylogenetic analysis. Sci Rep 13, 14426 (2023). https://doi.org/10.1038/s41598-023-41638-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-41638-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
