
Abstract
Thrombocytopenia is a major and fatal complication in patients with acute myeloid leukemia (AML), which results from disrupted megakaryopoiesis by leukemic niche and blasts. Our previous research revealed that elevated interleukin-4 (IL-4) in AML bone marrow had adverse impact on multiple stages throughout megakaryopoiesis including hematopoietic stem cells (HSCs), but the specific mechanism remains unknown. In the present study, we performed single-cell transcriptome analysis and discovered activated oxidative stress pathway and apoptosis pathway in IL-4Rαhigh versus IL-4Rαlow HSCs. IL-4 stimulation in vitro led to apoptosis of HSCs and down-regulation of megakaryocyte-associated transcription factors. Functional assays displayed higher susceptibility of IL-4Rαhigh HSCs to tunicamycin and irradiation-induced apoptosis, demonstrating their vulnerability to endoplasmic reticulum (ER) stress injury. To clarify the downstream signaling of IL-4, we analyzed the transcriptomes of HSCs from AML bone marrow and found a remarkable down-regulation of the proteasome component Psmd13, whose expression was required for megakaryocytic-erythroid development but could be inhibited by IL-4 in vitro. We knocked down Psmd13 by shRNA in HSCs, and found their repopulating capacity and megakaryocytic differentiation were severely compromised, with increased apoptosis in vivo. In summary, our study uncovered a previous unrecognized regulatory role of IL-4-Psmd13 signaling in anti-stress and megakaryocytic differentiation capability of HSCs.
Similar content being viewed by others


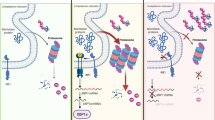
Introduction
The main causes of death, other than resistant disease or relapse, in patients with acute myeloid leukemia (AML) are hemorrhage and infection, stemming from severe thrombocytopenia and neutropenia1. Studies have shown that a variety of cells, cytokines, exosome contents are tampered in the leukemic microenvironment to support leukemia progression while inhibit proliferation and differentiation of residual hematopoietic stem and progenitor cells (HSPCs)2,3,4,5,6,7,8. Our previous work uncovered a link between hyperactive Interlukin-4 (IL-4) signaling and defective megakaryopoiesis in AML bone marrow. IL-4 exerted inhibitory effects on multiple stages along the path of megakaryocyte (MK) differentiation, from hematopoietic stem cells (HSCs) to MK progenitors. IL-4 antibody in combination with induction chemotherapy alleviated thrombocytopenia and prolonged overall survival of AML mice6. Yet the regulatory effect of IL-4 on HSCs and its mechanism remain unclear.
In the present study, we display that IL-4 renders HSCs susceptible to endoplasmic reticulum (ER) stress-induced apoptosis. IL-4 inhibits expression of the proteasome component Psmd13, whose down-regulation is detected in HSCs from AML bone marrow and impairs their repopulating capacity and megakaryocytic differentiation after transplantation.
Methods
Mice
C57BL/6-Ly5.1 (Ly5.1) and C57BL/6-Ly5.2 (Ly5.2) were purchased from the State Key Laboratory of Experimental Hematology (SKLEH). R26-tdTomato mice9 were purchased from Jackson Lab. β-actin-eGFP mice10 were kindly provided by Bing Liu (Academy of Military Medical Sciences, Beijing, China). Pf4-Cre mice11 were a gift from Junling Liu (Shanghai Key Laboratory of Tumor Microenvironment and Inflammation, China). Pf4-Cre mice were bred with R26-tdTomato mice to obtain Pf4-Tdtomato reporter mice. Mice experiments were approved by Institutional Animal Care and Use Committee of SKLEH and were carried out in accordance with ARRIVE guidelines12.
Flow cytometry
Detailed staining and enrichment procedures for flow cytometry have been previously described13.
Cell surface markers for phenotypical analyses of hematopoietic cells are listed as following: LT-HSC (Lin-cKit + Sca1 + Flt3-CD34-), MPP (Lin-cKit + Sca1 + Flt3 + CD34 +), MK (SSChighCD41high).
Antibodies used are listed in Supplementary Table S1.
For apoptosis analysis, cells were cultured with mIL-4 (10 ng/mL) for 24 h and labeled with surface markers, then stained with an Annexin-V antibody and 7-AAD in binding buffer according to the manufacturer’s instructions (BD Bioscience).
Colony-forming assays
Cells (800 LKS + cells/well for multi-lineage colony formation in 24-well plates in a 0.5 mL volume) were directly pipetted into M3434 methylcellulose (Stem Cell Technologies) supplemented with 10 ng/mL mTPO and 10 ng/mL mFlt3L and incubated at 37 °C for 7–10 days in a humidified chamber.
Single cell liquid culture
For multi-lineage potential of HSCs, single-cell sorted HSCs were grown in IMDM (Gibco) supplemented with 10% FBS, L-Gln (2 mM), 1% β-mercaptoethanol (50 μM), mSCF (10 ng/mL), mTPO (10 ng/mL), mIL-3 (10 ng/mL), hEPO (1 U/mL), mGM-CSF (10 ng/mL). Colonies were evaluated after 9 days of culture. Unless otherwise indicated, cytokines were obtained from PeproTech.
Virus transduction
Production and packaging of lentivirus for stably knocking down Psmd13 (hU6-MCS-Ubiquitin-EGFP-IRES-puromycin) were accomplished by the company (Shanghai Genechem Co., LTD). LKS+ cells (CD45.2+) were flow sorted from donor mice. Cells were pre-cultured for 1 day, then transduced with viruses and incubated for another 3 days. The culture was maintained in StemSpan SFEM medium containing 100 ng/mL mSCF and 100 ng/mL mTPO. After transduction, GFP+ cells were sorted for assays.
Transplantation
For measurement of the reconstitution activity, IL4Rαhigh and IL4Rαlow HSCs were sorted from the BM of β-actin-GFP mice, and 1700 cells per group were then transplanted into lethally irradiated (9.5 Gy) B6-Ly5.2 recipient mice in competition with 3 × 105 CD45.2 + BM cells.
For Psmd13 knockdown assay, GFP + transduced cells were transplanted together with 3 × 105 CD45.2 + BM cells into lethally irradiated B6-Ly5.2 mice.
Single-cell RNA sequencing
LT-HSCs were purified by FACS sorting. Each single LT-HSC was placed into the lysis buffer by mouth pipette. Library preparation and processing of single cell RNA-seq data followed the detailed procedure described in Li et al.’s study14. Unique molecular identifiers (UMIs) was used to directly measure the number of transcripts for each gene. Cells with at least 2000 genes and more than 100,000 transcripts detected, and more than 40% reads mapping to genome were remained for further analysis. For all 34 single cells, 30 single cells passed quality control.
Differential expression genes between IL4Rαhigh and IL4Rαlow HSCs were identified by FindMarkers function of Seurat (Version 2.0)15 with default parameters. Log2 (TPM/10 + 1) was used as expression values. Genes with log2(fold-change) ≥ 0.59 or ≤ − 0.59 and p value < 0.05 were selected as differential expressed genes15. Canonical pathway analyses were performed by Ingenuity Pathway Analysis (QIAGEN Bioinformatics).
UMI-mRNA sequencing
LKS + cells were purified by FACS sorting. Total RNA was used as the input material for the library preparation. mRNA was firstly captured using mRNA Capture Beads with Oligo(dT), and purified with Binding Buffer and Washing Buffer. mRNA was then randomly fragmented into 100–200 nt in the Fragmentation Buffer and reverse-transcribed into cDNA subsequently. Synthesized cDNA was reclaimed using DNA Clean Beads and subjected to the adaptor ligation step. Purified cDNA was ligated to the pre-mixed adaptors with UMI using Ligase and Ligase Buffer in a one-step PCR reaction. The ligation products were again purified using DNA Clean Beads and then amplified using PCR. PCR products were finally purified using DNA Clean Beads and reclaimed with nuclease-free H2O. After the library preparation and pooling of different samples, the samples were subjected for Illumina sequencing. The libraries were sequenced on Illumina novaseq 6000 Platform for 6G raw data, and generated 150 nt pair-end reads.
Statistics
Unless otherwise stated, data are expressed as mean ± standard error of the mean (SD) as indicated. P values were generated using unpaired Student’s t-test and analysis of variance. GraphPad Prism 5.0 software was used for the statistical analysis.
Ethical approval
All mice were bred and maintained under specific pathogen–free conditions in the animal facility at the Institute of Hematology in accordance with Institutional Animal Care and Use Committee of the Institute of Hematology.
Results
IL-4 stimulation increased apoptosis of HSCs
To search for clues to the innate effect of endogenous IL-4 on HSCs in steady state, we performed single-cell RNA sequencing (scRNA-seq) and acquired transcriptomes of 30 long-term HSCs (LT-HSCs) finally after strict quality control. We divided these transcriptomes into IL-4Rαhigh and IL-4Rαlow groups based on their expression of IL-4Rα beyond or below detection threshold respectively (Fig. 1A), hypothesizing that IL-4Rαhigh HSCs might exhibit more remarkable IL-4-primed gene expression profiling compared with IL-4Rαlow HSCs during their development in vivo. Ingenuity Pathway Analysis (IPA) of differentially expressed genes between two groups revealed that four pathways were possibly activated (p < 0.05, z-score > 0) whereas a large majority of canonical pathways were possibly inhibited (z-score > 0) (Supplementary Table S1). Among the activated pathways, NRF2-mediated oxidative stress pathway and apoptosis pathway were top two enriched (Fig. 1B). As expected, we observed significantly increased apoptotic rate of Lin-c-Kit + Sca1+ (LKS+) cells 24 h after IL-4 exposure (Fig. 1C). And the pro-apoptotic effect of IL-4 could be completely abrogated by IL-4 neutralizing antibody and anti-IL4Rα antibody (Fig. 1C). Our previous results showed that HSC-enriched LKS+ cells exhibted higher IL-4Rα level and more prominent activation of pStat6 signaling in response to exogenous IL-4 stimulation, compared to myeloid progenitors17. Among LKS+ subpopulations, LT-HSCs (CD34−Flt3−) were more susceptible to IL4-induced apoptosis than MPPs (CD34 + Flt3+) (Fig. 1D). Single cell liquid culture of LT-HSCs yielded significantly fewer and smaller colonies with IL-4 than without (Fig. 1E). Similarly, cell culture of LKS+ cells showed that the absolute cell number with IL-4 decreased compared with those without IL-4 (Supplementary Fig. 1C). Then, we extended the cell cycle analysis to the LKS+ cells 24 h after IL-4 exposure to determine whether IL-4 changed the proliferation or cell cycle state. However, Ki67 staining showed no significant difference (Supplementary Fig. 1A,B). We detected prominent up-regulation of pro-apoptotic genes (Bim, Bax, Bak1, Bid, Apaf1, Caspase6), p53 signaling genes (Trp53, Puma, p21, Mdm2) and stress-response genes (Atf3, Gadd45b, Atf4) in LKS+ cells within 1 h after IL-4 exposure (Fig. 1F). Among them, Caspase6 and Atf3 had been reported to be directly regulated by IL-4/Stat6 in lymphocytes18 and to facilitate the process of apoptosis19,20,21. Besides, megakaryocyte-associated transcription factors except for Gata2 were universally inhibited in LKS+ cells after IL-4 treatment (Fig. 1G), indicating reduced megakaryocytic differentiation potential.

Increased apoptosis of HSCs in response to IL-4. (A) Violin plots showing distribution of expression levels of IL-4Ra across single cells of LT-HSCs. N = 5 for IL-4Rahigh group and n = 25 for IL4Ralow group. (B) Predicted activation and inhibition of canonical pathways in IL-4Rahigh vs IL-4Ralow HSCs by Ingenuity Pathway Analysis (IPA). (C) Left, representative FACS plots for apoptosis analysis of LKS+ cells with and without mIL-4 (10 ng/mL) treatment for 24 h. Right, the apoptotic rate of LKS+ cells upon indicated treatments for 24 h. Anti-IL-4 and anti-IL-4Rα antibody were used both at a concentration of 10 μg/mL. N = 3 per group. 3 independent experiments. (D) Apoptosis analysis of LT-HSCs and MPPs in the absence or presence of mIL-4 (10 ng/mL) for 24 h. (E) Single-cell colony number and diameter of LT-HSCs after 9 days liquid culture in the absence or presence of mIL-4 (10 ng/mL) (n = 60 cells per group). (F) Expression of apoptosis and stress response associated genes quantified by qRT-PCR (normalized to Gapdh) in LKS+ cells with (red bar) or without (black bar) IL-4 treatment for 1 h. N = 3 per group. (G) Megakaryocytic transcription factors expression quantified by qRT-PCR (normalized to Gapdh) in LKS + cells cultured in vitro with or without IL-4 (10 ng/mL) for 24 h. N = 3 per group. Data are shown as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. ns no significance.
IL-4Rαhigh HSCs were more susceptible to endoplasmic reticulum stress induced apoptosis
As the transcriptomes of IL-4Rαhigh HSCs displayed apoptosis signature and reduced response to growth stimulatory signaling such as IL-3 and IL-6 (Fig. 1B), we expected them to exhibit a growth disadvantage compared with IL-4Rαlow HSCs. However, single cell colony assay did not demonstrate significant difference in colony number or size between IL-4Rαhigh and IL-4Rαlow HSCs (Fig. 2A). Besides, IL-4Rαhigh and IL-4Rαlow HSCs did not show significant difference in their reconstitution capacity of all five lineages including erythrocytes and platelets in transplantation assays using β-actin-GFP reporter mice as donors (Fig. 2B). We then investigated their response to stress conditions by treating them in vitro with tunicamycin, an inhibitor of protein glycosylation that induced unfolded protein response (UPR) and endoplasmic reticulum (ER) stress22,23. As a result, significantly higher apoptotic rate was detected in IL-4Rαhigh group than IL-4Rαlow group, indicating that IL-4Rαhigh HSCs were more susceptible to ER stress induced apoptosis (Supplementary Fig. 2A; Fig. 2D).

Susceptibility of IL4Rαhigh HSCs to unfolded protein response (UPR) and irradiation induced apoptosis. (A) Single-cell colony number and diameter of IL4Rαhigh and IL4Rαlow LT-HSCs after 9 days liquid culture (n = 50 cells per group). (B) Mean contribution of 1700 IL-4Rαhigh (black bar) and IL-4Rαlow (grey bar) LT-HSCs to myeloid cells, T cells, B cells, platelets and erythrocytes in peripheral blood 12 weeks after transplantation. N = 7 mice per group. 2 independent experiments. (C) Mean fluorescence intensity (MFI) of IL-4Rα in LT-HSCs 16 h post 2 Gy radiation. N = 3 mice per group. 2 independent experiments. (D) The apoptotic rate of FACS-sorted IL-4Rαhigh and IL-4Rαlow LT-HSCs after treated with tunicamycin (0.35 μg/mL) for 24 h. N = 4–6 for each group. (E) The apoptotic rate of IL-4Rαhigh and IL-4Rαlow LT-HSCs 16 h post 2 Gy radiation. N = 3 mice per group. 2 independent experiments. Data are shown as mean ± SD. **p < 0.01, ***p < 0.001. ns no significance.
Irradiation causes DNA damage and prolonged elevation of reactive oxygen species (ROS) in HSCs24,25,26 and triggers ER stress to restore homeostasis or initiate apoptosis27. We exposed mice to 2 Gy irradiation and examined the apoptotic rate of HSC subpopulations 16 h later. We detected the change in IL-4R expression on HSCs before and after irradiation, but there was no significant difference (Fig. 2C). As the result showed, greatly larger proportions of IL-4Rαlow HSCs remained viable than IL-4Rαhigh HSCs, suggesting that IL-4Rαlow HSCs were more resistance to irradiation-induced stress (Supplementary Fig. 1B; Fig. 2E).
To gain further insight into the role of IL-4 on HSC maintenance, we isolated LKS+ cells from healthy BM and treated with IL-4 or not for 24 h for RNA-seq assay (Supplementary Fig. 2A,B). Gene expression data showed downregulation of hematopoietic stem progenitor cell differentiation genes and megkaryocyte differentiation genes in LKS+ cells treated with IL-4 (Supplementary Fig. 2C), implying IL-4 inhibited megakaryocytic differentiation capability of HSCs. In addition, apoptosis-associated genes and response to endoplasmic reticulum stress genes were positively enriched in LKS+ cells treated with IL-4 (Supplementary Fig. 2C). These data indicated that IL-4 signaling is related to UPR and ER stress, suggesting that IL-4 may act possibly as a stress signaling in HSCs.
IL-4 decreases Psmd13 expression in HSCs
The compromised adaptation of IL-4Rαhigh HSCs to ER stress inspired us to consider the possibility that IL-4 exerted its effect on HSCs by influencing protein metabolism. Proteasome is a major organelle responsible for degradation of redundant and misfolded proteins. The function of proteasome to lighten protein load is pivotal in UPR and ER stress response. In LKS+ cells isolated from AML mice bone marrow, we detected a significant reduced expression of Psmd13, a component of proteasome (Fig. 3A). Accordingly, in LKS+ cells exposed to IL-4, we observed robust inhibition of Psmd13 expression, and the inhibition effect of IL-4 could be completely abrogated by anti-IL4Rα antibody (Fig. 3B). Interestingly, megakaryocytic-erythroid progenitors (MEPs) possessed remarkably higher expression of Psmd13 than their upstream HSPC subsets (Fig. 3C), suggesting its possibly essential role in regulating MEP development.

IL-4 induces Psmd13 down-regulation. (A) qRT-PCR of the relative expression of Psmd13 in LKS+ cells from AML and control mice (n = 3 mice per group). (B) Histograms showing Psmd13 mRNA expression in LKS + cells with or without mIL-4 (10 ng/mL) and anti-IL4 (10 μg/mL) treatment for 24 h. N = 3 per group. (C) The gene expression profile of Psmd13 in HSPC subsets in ‘‘Gene Expression Commons’’ (https://gexc.stanford.edu). Calculated low/high expression level was represented by a color-coded heatmap: red represents high expression, white represents threshold level expression, and blue represents low expression. Data are shown as mean ± SD. *p < 0.05, ***p < 0.001. ns no significance.
Psmd13 down-regulation impairs megakaryocytic development
According to the above results, we knocked down Psmd13 by shRNA lentivirus in LKS+ cells (Supplementary Fig. 4A,B) and found Psmd13 suppression inhibited clonogenic potential of LKS+ cells (Fig. 4A). We then transplanted them into lethally irradiated mice to see their capacity to reconstitute megakaryocytic lineage. After 4 weeks, Psmd13 knockdown (KD) LKS+ cells exhibited significantly reduced overall reconstitution rate than scramble control (Fig. 4B). Among donor-derived nucleated cells in peripheral blood, we observed inhibited reconstitution of B, T, myeloid lineages and platelets compared to scramble shRNA control (Fig. 4C, Supplementary Fig. 4C,D). In bone marrow, the megakaryocytic reconstitution rate of Psmd13 KD LKS+ cells was significantly reduced than that of scramble control (Fig. 4D,E). Collectively, Psmd13 down-regulation impaired the repopulating potential of HSCs, especially to myeloid and megakaryocytic lineages.

Impairment of megakaryocytic development by Psmd13 down-regulation. (A) In vitro colony forming ability of Psmd13 knockdown LKS+ cells. (B–E) Reconstitution status of Psmd13 shRNA (1#) or scramble control transduced LKS+ cells 6 weeks after transplantation. C, percentage of donor-derived EGFP+ cells in peripheral blood (PB). D, fractions of T, B, and myeloid cell in EGFP + PB nucleated cells. E–F, percentage of donor-derived EGFP+ cells in bone marrow megakaryocytes (MKs). N = 6–7 mice per group. 2 independent experiments. Data are shown as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. ns no significance.
Since IL-4 treatment caused apoptosis in LKS+ cells, we next investigated the status of apoptosis of Psmd13 KD cells, to check if it is consistent with IL-4 exposure. The results showed that there was no significant difference in the proportion of apoptosis between the two KD groups and the control group (Supplementary Fig. 5A,B), which might be caused by the limitation of cell culture conditions in vitro and the short time of gene transfection. Then, according to the above transplantation scheme, we analyzed the apoptosis of donor-derived LKS+ cells four weeks after the transplantation of Psmd13 KD LKS+ cells, and found that the apoptosis rate of KD group was significantly higher than that of the control group (Supplementary Fig. 5C,D), which was consistent with the effect of IL-4.
Discussion
Our previous study reported overproduction of IL-4 from AML bone marrow and its inhibitory effect throughout the process of megakaryopoiesis in vivo6. The results suggested that IL-4 could be a promising therapeutic target combined with chemotherapy in AML, but the specific influence of IL-4 on normal HSPCs has been barely understood. In this study, we showed a weak resistance of HSCs to IL-4-induced apoptosis. Among HSCs, IL-4Rahigh subset was more susceptible to UPR and irradiation-induced apoptosis than IL-4Ralow. Stress can lead to severe functional loss or the persistence of oncogenes in HSCs, thus increasing the risk of leukemia28,29. We hypothesize that the interaction between immune cells-derived IL-4 and IL-4Rα on HSCs, and its pro-apoptotic consequence, may be an approach to clear away damaged or senescent HSCs after stress. Whether up-regulated IL-4Rα on HSCs is a signal calling for elimination and the underlying mechanism warrants further investigation.
A recent study from Pena-Martinez P et al. suggested the pro-apoptotic effect of IL-4 also occurred in AML blasts and it might be a potential therapeutic approach for AML30,31. They observed the caspase signaling pathway were significantly enriched in leukemia cells treated with IL-4 compared with the untreated group. However, we found IL-4-induced apoptosis was independent of caspase cascade in that it could not be rescued by either caspase-6 specific inhibitor or pan-caspase inhibitor (data not shown). The interrupted HSC function by IL-4-induced Psmd13 down-regulation hinted that proteasome system might also participate in the molecular mechanism. Regulation of HSC function via ubiquitin proteasome system has been studied by deleting different members of this complicated system using genetically modified models, which yielded distinct phenotypes by influencing different substrates32,33. As many of these components are enzymes and relatively easy for artificial intervention as targets, more attention should be paid to the role of ubiquitin proteasome system in stress hematopoiesis. Our research is limited in the axis of IL-4-Psmd 13, and we lack direct evidence that IL-4 signaling induces ER stress and UPR in HSCs, which would be explored through Psmd 13 knockout mice in the future to continue our research.
By performing transplantation assays on LKS+ cells, Psmd13 KD cells exhibited significantly reduced overall reconstitution rate than scramble control. Among donor-derived nucleated cells in peripheral blood, we observed inhibited reconstitution of B, T, myeloid lineages and platelets compared to scramble shRNA control, which is not restricted to Mk-lineage. To figure this out, we will perform RNA-seq assay on Psmd13 KD LKS+ cells and scramble control in our future study, so as to clarify whether Psmd3 KD effect is restricted to Mk-lineage from the gene level. As for RBC chimerisms, we found Psmd13 suppression inhibited BFU-E clonogenic potential of LKS+ cells. However, in the transplantation assays, we used platelet-specific PF4-tdTomato reporter mouse LKS+ cells to infect shRNA, and indeed did not detect red RBC reconstruction, which will be perfected in our future study. Studies showed that platelet proteasome regulates platelet lifespan and viability only partially enhances platelet activation and aggregation by activating NF-κB34. More studies are, therefore, needed to explore how the proteasome system regulates the differentiation and development of Mk-lineage.
In summary, our study established a previous unrecognized link between IL-4-Psmd13 signaling and anti-stress as well as megakaryocytic differentiation potential of HSCs. Our findings provided evidence to counteract IL-4 signaling and to correct the disrupted proteasome function as a possible approach to alleviate thrombocytopenia in AML.
Data availability
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request. The RNA-seq data of this study have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE11653016.
References
Slats, A. M. et al. Causes of death—other than progressive leukemia—in childhood acute lymphoblastic (ALL) and myeloid leukemia (AML): The Dutch Childhood Oncology Group experience. Leukemia 19(4), 537–544 (2005).
Batsivari, A., Grey, W. & Bonnet, D. Understanding of the crosstalk between normal residual hematopoietic stem cells and the leukemic niche in acute myeloid leukemia. Exp. Hematol. 95, 23–30 (2021).
Cheng, H. & Cheng, T. “Waterloo”: When normal blood cells meet leukemia. Curr. Opin. Hematol. 23(4), 304–310 (2016).
Cheng, H. et al. Leukemic marrow infiltration reveals a novel role for Egr3 as a potent inhibitor of normal hematopoietic stem cell proliferation. Blood 126(11), 1302–1313 (2015).
Gong, Y. et al. Megakaryocyte-derived excessive transforming growth factor beta1 inhibits proliferation of normal hematopoietic stem cells in acute myeloid leukemia. Exp. Hematol. 60, 40–6.e2 (2018).
Gao, A. et al. Bone marrow endothelial cell-derived interleukin-4 contributes to thrombocytopenia in acute myeloid leukemia. Haematologica 104(10), 1950–1961 (2019).
Huang, D. et al. ANGPTL2-containing small extracellular vesicles from vascular endothelial cells accelerate leukemia progression. J. Clin. Investig. 131(1), e13898 (2021).
Wang, Y. et al. Leukemia cell infiltration causes defective erythropoiesis partially through MIP-1alpha/CCL3. Leukemia 30(9), 1897–1908 (2016).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13(1), 133–140 (2010).
Wright, D. E. et al. Cyclophosphamide/granulocyte colony-stimulating factor causes selective mobilization of bone marrow hematopoietic stem cells into the blood after M phase of the cell cycle. Blood 97(8), 2278–2285 (2001).
Chen, X. et al. PDK1 regulates platelet activation and arterial thrombosis. Blood 121(18), 3718–3726 (2013).
Percie du Sert, N. et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 18(7), e3000410 (2020).
Cheng, H., Liang, P. H. & Cheng, T. Mouse hematopoietic stem cell transplantation. Methods Mol. Biol. 976, 25–35 (2013).
Li, L. et al. Single-cell RNA-Seq Analysis maps development of human germline cells and gonadal niche interactions. Cell Stem Cell 20(6), 858–73.e4 (2017).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33(5), 495–502 (2015).
Dong, F. et al. Differentiation of transplanted haematopoietic stem cells tracked by single-cell transcriptomic analysis. Nat. Cell Biol. 22(6), 630–639 (2020).
Gao, A. et al. Bone marrow endothelial cell-derived interleukin-4 contributes to thrombocytopenia in acute myeloid leukemia. Haematologica 104, 1950–1961 (2019).
Schroder, A. J., Pavlidis, P., Arimura, A., Capece, D. & Rothman, P. B. Cutting edge: STAT6 serves as a positive and negative regulator of gene expression in IL-4-stimulated B lymphocytes. J. Immunol. 168(3), 996–1000 (2002).
MacLachlan, T. K. & El-Deiry, W. S. Apoptotic threshold is lowered by p53 transactivation of caspase-6. Proc. Natl. Acad. Sci. U.S.A. 99(14), 9492–9497 (2002).
Mashima, T., Udagawa, S. & Tsuruo, T. Involvement of transcriptional repressor ATF3 in acceleration of caspase protease activation during DNA damaging agent-induced apoptosis. J. Cell. Physiol. 188(3), 352–358 (2001).
Liu, G. et al. Salermide up-regulates death receptor 5 expression through the ATF4-ATF3-CHOP axis and leads to apoptosis in human cancer cells. J. Cell Mol. Med. 16(7), 1618–1628 (2012).
DuRose, J. B., Tam, A. B. & Niwa, M. Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol. Biol. Cell 17(7), 3095–3107 (2006).
van Galen, P. et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 510(7504), 268–272 (2014).
Shen, H. et al. An acute negative bystander effect of gamma-irradiated recipients on transplanted hematopoietic stem cells. Blood 119(15), 3629–3637 (2012).
Hu, L. et al. Antioxidant N-acetyl-L-cysteine increases engraftment of human hematopoietic stem cells in immune-deficient mice. Blood 124(20), e45–e48 (2014).
Rodrigues-Moreira, S. et al. Low-dose irradiation promotes persistent oxidative stress and decreases self-renewal in hematopoietic stem cells. Cell Rep. 20(13), 3199–3211 (2017).
Kim, E. J., Lee, Y. J., Kang, S. & Lim, Y. B. Ionizing radiation activates PERK/eIF2alpha/ATF4 signaling via ER stress-independent pathway in human vascular endothelial cells. Int. J. Radiat. Biol. 90(4), 306–312 (2014).
Shlush, L. I. et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506(7488), 328–333 (2014).
Rossi, D. J., Jamieson, C. H. & Weissman, I. L. Stems cells and the pathways to aging and cancer. Cell 132(4), 681–696 (2008).
Pena-Martinez, P. et al. Interleukin 4 induces apoptosis of acute myeloid leukemia cells in a Stat6-dependent manner. Leukemia 32(3), 588–596 (2018).
Peña-Martínez, P. et al. Interleukin 4 promotes phagocytosis of murine leukemia cells counteracted by CD47 upregulation. Haematologica 107(4), 816–824 (2022).
Moran-Crusio, K., Reavie, L. B. & Aifantis, I. Regulation of hematopoietic stem cell fate by the ubiquitin proteasome system. Trends Immunol. 33(7), 357–363 (2012).
Sarodaya, N., Karapurkar, J., Kim, K. S., Hong, S. H. & Ramakrishna, S. The role of deubiquitinating enzymes in hematopoiesis and hematological malignancies. Cancers 12(5), 1103 (2020).
El-Kadiry, A. E. & Merhi, Y. The role of the proteasome in platelet function. Int. J. Mol. Sci. 22(8), 3999 (2021).
Funding
This work was supported by grants from the National Natural Science Foundation of China (82000113, 81900109, 82200124).
Author information
Authors and Affiliations
Contributions
A.G., S.X. and Y.G. designed and performed all the experiments, analyzed the data and wrote the manuscript. C.Z. and F.D. helped with transcriptomics analysis. Q.L. and F.W. helped with the mouse experiments. H.C. and S.H. helped with experimental design and data interpretation. T.C. conceived the study, designed the experiments, interpreted the results, wrote the paper and oversaw the research project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, A., Xu, S., Li, Q. et al. Interlukin-4 weakens resistance to stress injury and megakaryocytic differentiation of hematopoietic stem cells by inhibiting Psmd13 expression. Sci Rep 13, 14253 (2023). https://doi.org/10.1038/s41598-023-41479-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-41479-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
