
Abstract
Synthetic biology involves the engineering of logic circuit gates that process different inputs to produce specific outputs, enabling the creation or control of biological functions. While CRISPR has become the tool of choice in molecular biology due to its RNA-guided targetability to other nucleic acids, it has not been frequently applied to logic gates beyond those controlling the guide RNA (gRNA). In this study, we present an adaptation of split Cas9 to generate logic gates capable of sensing biological events, leveraging a Cas9 reporter (EGxxFP) to detect occurrences such as cancer cell origin, epithelial to mesenchymal transition (EMT), and cell–cell fusion. First, we positioned the complementing halves of split Cas9 under different promoters—one specific to cancer cells of epithelial origin (phCEA) and the other a universal promoter. The use of self-assembling inteins facilitated the reconstitution of the Cas9 halves. Consequently, only cancer cells with an epithelial origin activated the reporter, exhibiting green fluorescence. Subsequently, we explored whether this system could detect biological processes such as epithelial to mesenchymal transition (EMT). To achieve this, we designed a logic gate where one half of Cas9 is expressed under the phCEA, while the other is activated by TWIST1. The results showed that cells undergoing EMT effectively activated the reporter. Next, we combined the two inputs (epithelial origin and EMT) to create a new logic gate, where only cancer epithelial cells undergoing EMT activated the reporter. Lastly, we applied the split-Cas9 logic gate as a sensor of cell–cell fusion, both in induced and naturally occurring scenarios. Each cell type expressed one half of split Cas9, and the induction of fusion resulted in the appearance of multinucleated syncytia and the fluorescent reporter. The simplicity of the split Cas9 system presented here allows for its integration into various cellular processes, not only as a sensor but also as an actuator.
Similar content being viewed by others


Introduction
In synthetic biology, most of the logic control developed so far employ transcriptional regulators e.g., transcriptional activators or repressors to regulate gene expression1,2,3,4,5 to either increase reaction rates in metabolic pathways or respond to new input signals. While these genetic circuits work very predictably in prokaryotes, in eukaryotes they often require the use of genetic elements from prokaryotes or simple eukaryotes.
The domestication of CRISPR systems, the prokaryotic “immune system” that confers resistance and memory to phageal genetic elements, to eukaryotic cells has opened a new level of genetic engineering. CRISPR/Cas9 is composed by Cas9 endonuclease, responsible for executing the desired genetic modifications, and two small RNA molecules named CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA). While crRNA contains targeting sequence, the tracrRNA acts as a scaffold for Cas9 endonuclease6. In order to simplify the system, the crRNA and tracrRNA have been fused into a single guide-RNA (gRNA)7,8,9. Cas9/gRNA generates double-strand DNA breaks which can be repaired by either nonhomologous end-joining pathway (NHEJ), causing errors such as insertion/deletions (indels), or by homology-directed repair (HDR), which can be used to introduce specific DNA sequence changes. Such simplicity of CRISPR systems has made them the most adaptable system to manipulate endogenous genetic information, from gene deletion (knockout)10,11,12,13, gene insertion (knock-ins)14,15,16, to gene regulation (CRISPRi and CRISPR-TA)17, and genome editing18,19,20.
The Cas9 system has also been integrated into genetic logic gates whereas the input is provided by gRNAs that respond to a particular stimulus, such as the presence of a specific chemical21,22 or the activation of a specific signaling pathway or regulate multiplexed circuits to control transcriptional regulation (activation/inhibition) by modified Cas9 forms (CRISPRa and CRISPRi, respectively)23,24,25,26.
Yet, CRISPR/Cas9 has been rarely used as a sensor for biological events i.e., for identification of cancer cells27 or to control a complex phenotype such as cell fate28. In these cases, Cas9 and the gRNA have been positioned under specific promoters, each becoming one input to the AND logic gate. A split Cas9 system has previously been reported although not applied into logic circuits29, although it has been used to generate drug- and light-inducible systems by fusing halves of Cas9 to hetrodimerizers30,31.
The application of Cas9 into logic gates remains relatively unexplored, given its functionality both an actuator and a “memory” by permanent modifications on DNA. Here, we present another use of CRISPR/Cas9 by creating a synthetic (AND) logic gate using self-assembling split Cas9 (named Cas4.5N and Cas4,5C—as they sum up to Cas9), facilitated by DnaE inteins32, that need to be co-expressed (input 1 and 2) to activate a reporter, leading to a specific output (outcome). While studying cellular events can be detected by biochemical assays or biosensors, there are very few examples on the use of synthetic logic gates to record cellular events in eukaryotic cells.
Moreover, the presented 2-input system results in an active actuator that can be applied to many other biological outcomes. We show that detection of cell origin, phenotypical transition or cell–cell fusion can be specifically detected by split Cas9 logic gates.
Experimental section
Materials and reagents
Plasmid pSpCas9(BB)-2A-Puro (PX459) V2.0 was a gift from Feng Zhang (Addgene plasmid #6298833), pU6-sgGFP-NT1 was a gift from Stanley Qi & Jonathan Weissman (Addgene plasmid #46914; http://n2t.net/addgene:46914; RRID:Addgene_46914)8), pCAG-EGxxFP-Cetn1 was a gift from Masahito Ikawa (Addgene plasmid #50717; http://n2t.net/addgene:50717;RRID:Addgene_5071734), AmCyan-P2A-mCherry was previously created by us35 (Addgene plasmid #45350; http://n2t.net/addgene:45350; RRID:Addgene_4535035). The TWIST1 binding domain (TBD) promoter was created previously by us36, DnaE intein plasmids were a gift from Hideo Iwai (Addgene plasmid #34549; http://n2t.net/addgene:34549; RRID:Addgene_3454937 and #15335; http://n2t.net/addgene:15335; RRID:Addgene_1533538). Cell lines HEK-293, HSF, CCD 841 CoTr, H1299, H2170, SW480, A375, HeLa, U-2 OS, C2C12 and GC-2 were obtained from ATCC; H2170 stable cell line with vimentin reporter (VRCs) was created by us39. KOD-Xtreme hot-start DNA polymerase (Merck Millipore), DreamTaq™ Green PCR Master Mix (ThermoFisher Scientific), endonuclease DpnI and BbsI (Thermo Fisher Scientific), T7 Ligase (Thermo Fisher Scientific) and PNK (Thermo Fisher Scientific), Polyethylene Glycol Hybri-Max 1450 (PEG) (Sigma-Aldrich), TGFβ (human TGFβ1, Biorbyt, Cambridge, United Kingdom), Gibson Assembly® Master Mix (NEB), Ampicillin (BRAND), DNA Clean and Concentrator and Zyppy Plasmid Kits (Zymoresearch), Hoechst 33342 (Cayman), Dulbecco’s Modified Eagle Medium (DMEM/F-12), Dulbecco’s Modified Eagle Medium—high glucose (DMEM) (Sigma Aldrich), Penicillin and Streptomycin (Sigma Aldrich), fetal bovine serum (Sigma Aldrich), horse serum (Sigma Aldrich), and geneticin G418 (ThermoFisher Scientific). All primers were bought from Genomed (Warsaw, Poland).
PCR
All primers were designed in SnapGene Viewer and purchased from Genomed (Warsaw, Poland), Syncytin1 was recoded based on the amino acid sequence from the The Human Protein Atlas and ordered as a gBlock (ThermoFisher). PCR reactions were performed using a high-fidelity polymerase (KOD-Xtreme, Merck Millipore) according to manufacturer protocol. All products used for molecular cloning were then digested with fast digest DpnI restriction endonuclease (Thermo Scientific) to get rid of the methylated template plasmid. PCR products were column-purified before cloning using DNA Clean & Concentrator (Zymo research).
Molecular cloning
All vectors were created using Gibson Assembly40 according to manufacturer protocol (NEB). In brief, inserts and vectors were amplified by PCR, or digested using restriction enzymes in selected cases, followed by gel-purification and concentrations measured. Appropriate amounts were used for Gibson reaction (ratio insert:vector 3:1). Then, electroporation with 2ul of the reaction was performed by using prepared electrocompetent DH10B E. coli bacteria, and after 45 min, bacteria were plated onto agar plates for selection with an antibiotic. A screening colony was taken using PCR, and the sequence was verified. The EGxxFP-Cetn1 plasmid was modified by inserting the Puro-T2A cassette from pU6-sgGFP-NT1 (Addgene plasmid #46914) by recombination resulting in the Puro-T2A-EgxxFP plasmid. Cas4.5N was amplified by PCR from Cas9 plasmid and then fused at its C-terminus to DnaEN intein, while Cas4.5C was fused at its N-terminus to DnaEC intein. In the next step, the CMV general promotor (pCMV) in Cas4.5N-DnaEN was replaced by hCEA or the TWIST1 binding domain (TBD) specific promoters/genetic elements. Resulting vectors: phCEA-Cas4.5N-DnaEN, TBD-Cas4.5N-DnaEN, TBD-mCherry and pCMV-Cas4.5C-DnaEC. The gRNAs against EGxxFP-Cetn1 was cloned into both the Cas9 and the Cas4.5C plasmids, following the Zhang’s lab protocol33 using T7 ligase and BbsI (both from ThermoFisher).
Cell culture and transfection
Human embryonic kidney 293 (HEK293) cells, human skin fibroblast cells (HSF), human normal colon epithelial cells (CCD 841 CoTr), lung cancer cell lines (H1299, H2170, H2170 VRCs), malignant melanoma cell line (A375), cervical carcinoma (HeLa), colon cancer cell line (SW480), human bone osteosarcoma cell line (U-2 OS), murine myoblasts (C2C12), and murine spermatocyte cell line (GC-2), were cultured under standard conditions in the incubator (37 °C/5% CO2). Media DMEM/F12 (Sigma Aldrich) were supplemented with 10% Fetal Bovine Serum (PromoCell) and 1% Penicillin/Streptomycin solution. Transfections have been carried out using TurboFect reagent (Thermo Fisher), and Lipofectamine 3000®. Ratio DNA/reagent have been optimized for each cell line in order to obtain the best transfection efficiency.
One day before transfection H1299 and H2170 VRCs were seeded at density 5 × 104 cells/well into a 24-well plate, and co-transfected with one half of Cas9 protein (pCMV-Cas4.5C) driven by general promoter, construct with EGxxFP reporter plasmid, Cetn1 gRNA and second half of Cas9 (TBD-Cas4.5N) under a promoter activated during EMT—under the control of TWIST1, using Turbofect™ transfection reagent following the manufacturer’s protocol. Transfection with only one half of Cas9 protein was used as a negative control (NC). Next day after transfection cells were exposed to TGFβ (10 ng/mL) for 72 h to evaluate the expression level of EMT-related gene TWIST1. All experiments were performed in at least triplicate.
Equally, 24 h before transfection HSF, CCD 841 CoTr, C2C12, GC-2, A375, SW480, H1299, HeLa and U-2 OS cells were seeded at density 5 × 104 cells/well into a 24-well plate, and co-transfected with one half of Cas9 protein (pCMV-Cas4.5C) driven by general promoter, construct with EGxxFP reporter plasmid, Cetn1 gRNA and second half of Cas9 (phCEA-Cas4.5N) under an epithelial cancer-specific promoter, using Lipofectamine 3000® (CCD 841 CoTr, A375) or Turbofect™ (H1299, SW480 and HeLa) transfection reagents following the manufacturer’s protocol. Transfection with only one half of Cas9 protein was used as a negative control (NC), and transfection contains mCherry was used as a transfection control. All experiments were performed in at least triplicate.
Cell lines HEK293, H1299, and H2170 were seeded at density 5 × 104 cells/well into a 24-well plate 24h before transfection. Using Turbofect™ transfection reagent following the manufacturer’s protocol, cells were transfected with either one half Cas4.5N with EGxxFP reporter plasmid, Cetn1 gRNA, or the second halve pCMV-Cas4.5C with Syncytin1 plasmid. Transfection with only one half of Cas9 protein was a negative control (NC). In PEG treatment, cells were transfected with either Cas4,5, gRNA and reporter plasmids, and next day after transfection cells were exposed to 50% solution of PEG for 3 min at RT to induce cell–cell fusion. Similarly, to create a stable cell lines HEK293, H1299, and H2170 cells were seeded at density 5 × 104 cells/well into a 24-well plate one day before transfection. Using Turbofect™ transfection reagent following the manufacturer’s protocol, cells were transfected with each halves (pCMV-Cas4.5N; pCMV-Cas4.5C). After 48 h, cells were selected with Puromycin (0.75–1 mg/ml) for over three weeks to generate stable lines. Then, stable cell lines were co-transfected—one half Cas4.5N with EGxxFP reporter plasmid, Cetn1 gRNA, and second halve pCMV-Cas4.5C with Syncytin1 plasmid.
C2C12 differentiation
For ddifferentiation of mouse myoblast cells (C2C12) general media was replaced with differentiation media (DMEM high glucose without sodium pyruvate, 2% horse serum, and 1% Penicillin/Streptomycin). Then, cells were seeded on 24-well plate at 90% confluency. Cells started to align to each other and fuse into multinucleated myotubes after 48 h. Medium was changed every 48h.
Wound-healing assay
For the Wound-healing assay cell lines of H1299 and H2170 VRCs (vimentin reporter cells)39 were seeded into 12-well plates at a density 5 × 104 cells/mL. One day later were co-transfected with one half of Cas9 protein (pCMV-Cas4.5C) driven by general promoter, construct with EGxxFP reporter plasmid, Cetn1 gRNA and second half of Cas9 TBD-Cas4.5N under the specific promoter activated during EMT—under the control of TWIST1, resulted in EGFP expression. 24 h after transfection cells were exposed to TGFβ (10 ng/mL) for 72 h to evaluate the expression level of EMT-related gene TWIST1. Next day the monolayer of cells was scratched to form a linear wound. The Wound-healing Assay images were performed up to 72h under Nikon Ti Eclipse confocal microscope.
Electrofusion
For cellular fusion was used the BTX’s ECM 380 ElectroCellManipulator. Condition were optimized, two settings of device voltage was used and of controlled electrical pulse: 700 V for 15 µs (3 pulses) and 300 V for 500 µs (1 pulse), acting on the cells. For cell electroporation was used hypotonic medium. After experiment cells were resuspended in nutrition medium.
Confocal microscopy
The cells were cultured on glass bottom plates suitable for confocal imaging. Visualization has been carried out using Nikon Ti Eclipse microscopy. The nuclei of cells before imaging have been Hoechst 33342 stained (concentration 10 µg/mL).
Results
Naturally split DnaE intein (DnaEN and DnaEC) has high self-affinity followed by efficient splicing and ligation of the flanking exteins38 but only when DnaEN ends with a Cysteine (Cys;C), while DnaEC begins with the following di-amino acids: Cys followed by either Tyr (CY), Trp (CW), Phe (CF), His (CH) or Met (CM)41. After analyzing the Cas9 amino acid sequence and we found only one CY [amino acids (aa) 119–120] and one CF (aa 613–614) (sequences in Supplementary material S1). Since the latter is close to the middle, it was selected, and the two resulting halves have been termed: Cas4.5N (aa 1–613) and Cas4.5C (aa 613–1423).
Inteins can spontaneously self-assemble and induce protein ligation of their fused cargoes—in this case, the complementary Cas4.5 fragments, generating the holo-Cas9 endonuclease.
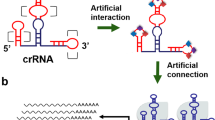
To detect Cas9 activity we adopted and modified the EGxxFP reporter34. This reporter is constructed on the basis of truncated but overlapping regions of the enhanced green fluorescent protein (EGFP) gene, with an early stop codon after 2/3 of the coding sequence, followed by a genomic (non-coding) fragment from the Cetn1 gene (stuffer) and a second fragment of the fluorescent protein covering about 500 bp of homology and the missing 1/3 of the respective gene. In the presence of active Cas9 and a targeting gRNA to the stuffer region, the homology regions undergo homologous recombination, creating the full-length EGFP gene and the fluorescent protein being expressed (Fig. 1). Co-expression of the 2 halves of Cas9 lacking inteins do not form a holo-enzyme as no reporter activity is detected (not shown).

Scheme of the principle of split-Cas9 as an AND logic gate.
Expression of individual Cas4.5 halves in cells, together with the gRNA, showed no reporter activity, proving that they are innocuous on their own. However, when both halves, containing inteins, were expressed in the same cell, Cas9 was active as detected by recombination of the reporter (EGxxFP → EGFP).
We then created an AND logic gate where one input is under a conditional promoter, while the other halve is under the ubiquitous (pCMV) promoter (Fig. 1). Certain parts of the system depend on specific promoters and other genetic elements for well-defined subpopulations. Based on that, we are able to program in what kind of cells the system will be activated.
Cell origin sensing system
As a proof-of-concept, spilt Cas9 was designed to selectively sense cancer cells of epithelial origin, using an epithelial cancer-specific promoter (phCEA). Carcinoembryonic antigen (CEA) has been previously used as a cancer marker due to its selective expression42,43, and its promoter has been proposed as cancer-specific and therapeutically useful44. Based on that we cloned Cas4.5N-DnaEN under the phCEA. The logic gate (phCEA-Cas4.5N-DnaEN/pCMV-DnaEC-Cas4.5C and gRNA) was then tested on multiple cell lines, and based on their known origins, we expected expression of EGFP only in epithelial cancer cells. For this, we selected various non-epithelial cells such as human skin fibroblasts (HSF) and myoblasts (C2C12); also normal epithelial cells (colon CCD 841 CoTr and spermatocyte GC-2), and cancer cells with epithelial origin: namely H1299 human non-small cell lung carcinoma, colon cancer cell line SW480, malignant melanoma cell line A375, cervical carcinoma cell line HeLa, and U-2 OS human bone osteosarcoma cell line. The logic gate resulted in EGFP expression in a pattern matching the cell lines’ origins—where only cancer cell lines of an epithelial origin had all elements expressed and thus EGFP expression, which means that all elements have been expressed and once the holo-Cas9 is formed it efficiently activates the reporter. The results confirmed selective expression phCEA-driven construct (Fig. 2). Our results are consistent with the known origins of the studied cell lines. Neither the non-epithelial cells (HSF and C212) nor the non-cancer epithelial cells (CCD 841 CoTr and GC-2) expressed the reporter, despite their comparable transfection efficiency to other lines (mock control) (Fig. 2A). In contrast, all cancer cells with epithelial origin (H1299, SW480, A375, HeLa, and U-2 OS) expressed the reporter (EGFP) (Fig. 2B).

Logic gate to identify only cancer cells of epithelial origin using halves of split Cas9 under a universal (pCMV) and a cell type-specific (phCEA) promoters. The expression of the green fluorescent protein reporter is observed only in cancer cells of epithelial origin. All cells were co-transfected with phCEA-Cas4.5N, pCMV-Cas4.5C/gRNA, the EGxxFP reporter and pCMV-mCherry (transfection control) constructs. Co-expression of the split Cas9 halves resulted in holo-enzyme formation via inteins, followed by Cas9-induced homologous recombination of EGFP. Only cancer cells of epithelial origin expressed both halves since the expression of Cas4.5N is restricted to these cells. Neither non-epithelial cell lines: human skin fibroblasts (HSF), murine myoblasts (C2C12), nor normal (non-cancerous) epithelial cells: CCD 841 CoTr and GC-2, showed any expression of the reporter (EGFP) (A), while all cancer cells with epithelial origin tested (H1299, SW480, A375, HeLa, and U-2 OS cells) expressed EGFP (B).
Recognising that a cell type can be labelled based on its gene expression characteristics, we decided to create another logic gate where a biological process is sensed—in this particular case, epithelial-mesenchymal transition (EMT). For this, we exchanged the universal promoter (pCMV) before Cas4.5C-DnaEC to a promoter that is activated by one of the essential EMT factors, TWIST145,46. TWIST1 activity is closely associated with EMT, correlating with tumor growth, metastasis and drug resistance, thereby diminishing the survival of cancer patients47. Using an artificial promoter containing TWIST1 binding domains (TWIST1-BD)36, driving one half, while the other half under the pCMV. As a result, only cells undergoing EMT, or having a mesenchymal phenotype, will express both Cas4.5 halves. To further trigger EMT, cells were exposed to TGFβ. Using the wound healing assay, 48 h after the addition of TGFβ, we began to observe higher reporter activity in epithelial carcinoma cells, both partially mesenchymal, H1299 and H2170 VRCs cells39 (Fig. 3A,B), predominantly at the edge of the wound, as it is often reported48,49. The cell morphology, the number of protrusions, on cells that transitioned into a more mesenchymal phenotype are clearly visible thanks to the reporter (Fig. 3B). While both these lines (H1299 and H2170 VRCs) are known to exhibit partial mesenchymal characteristics, and thus some reporter cells being observed in the non-induced controls (-TGFβ), the number of reporter-expressing cells increased dramatically after EMT induction, accompanied by noticeable changes on phenotype—more protrusions (insets in Fig. 3A,B).

Logic gate for sensing cells that are undergoing EMT. Representative images of H1299 lung cancer cells (A) and H2170 VRCs cells (B), both partly mesenchymal. Both cell lines, when exposed to TGFβ (+ TGFβ) as an EMT enhancer, increased the number of EGFP expressing cells as a proof of undergoing EMT. Morphological changes were visible after activation of TGFβ showed features of cells of mesenchymal origin—more spindle-shaped, they had more protrusions facilitating migration in comparison to controls.
In order to create a true logic gate, where both inputs are conditional, we designed an AND logic gate consisting of one half of Cas9 under the pTWIST1-BD, while the other half under the phCEA promoter. This configuration should selectively function only in cancerous cells with epithelial origin that are undergoing EMT. Following transfection with all plasmids into cells, they were exposed to EMT activator TGFβ. In the negative control, where no TGFβ was added (-TGFβ), only cancer epithelial cells partly mesenchymal (H1299, H2170) resulted in a lower number of cells expressing the green fluorescent reporter protein, while phenotypical epithelial cells (HeLa and U-2 OS) showed almost no reporter signal. However, upon exposure to TGFβ (+ TGFβ) the number of EGFP-expressing cells increased in every cell line tested (Fig. 4).

A two level AND logic gate, where epithelial cancer cells undergoing EMT are detected. Four epithelial cancer cell lines were transfected with split Cas9/gRNA system and the EGxxFP reporter. Then cells were stimulated to undergo EMT by the addition of TGFβ. While all these epithelial cancer cells were known to express the phCEA (Fig. 2), only cells with partial-EMT expressed the pTBD-Cas4,5C, without EMT stimulation (upper row, -TGFβ). But when EMT was induced (+ TGFβ), the number of positive cells increased dramatically in those partially mesenchymal (H1299 and H2170), and appeared in those more epithelial—HeLa and U-2 OS cells (lower row).
Detection of cell–cell fusion events
In addition of changes of gene expression in biological processes, there are other processes where two inputs could be detected by split Cas9 logic gate, such as cell fusion. This is both a physiological, e.g., placentation and skeletal muscle formation, and pathological as in cancer50,51, viral HIV52,53 or CoV2 infections54,55.
To detect cell–cell fusion, two groups of cells, were transfected separately with a different halve of Cas9, plus the gRNA and reporter. The logic gate here is that the inputs, N- or C-Cas4.5, can only be activated when two different cells fuse to each other, sharing a common cytoplasm.
To induce fusion, we applied two methods: the overexpression of a “fusogene” Syncytin1—a protein involved in syncytium formation in placenta56 that is known to promote the formation of multinucleated cells (syncytia)57,58; or a chemical-based method where polyethylene glycol (PEG), due to its known fusogenic properties59.
For this purpose, we used transiently transfected cell lines (HEK293, H1299, H2170), each line expressing only one of the halves of Cas9 at a time (Cas4.5C or Cas4.5N), and co-transfected with the reporter (EGxxFP), as control cells, or either co-transfected with Syncytin1 plasmid or PEG-treated to induce cell–cell fusion. 24 h after transfection, cells expressing one half were admixed with either the same lineage, but expressing the complementing Cas4.5, or with a different cell type but expressing the complementing Cas4.5. As expected, in control samples cell fusion was undetectable, while a number of fusion events occurred, in particular in mixed cells HEK293 + H1299 and HEK293 + H2170, in those either expressing Syncytin1 or PEG-treated (Fig. 5A,B). However, treatment with PEG showed toxic effects on cells and therefore we continued only with Syncytin1 overexpression for further experiments. We then tested electrofusion—the use of high voltage electrical pulses to induce fusions of cells in close proximity60—on an admixture of the cells expressing either halve. For this aim, different voltages were tested: 300 V 500 µs one pulse vs 700 V 15 µs three pulses. Compared to control samples, where cell fusion was undetectable (Fig. 5C,F), in cells expressing both halves, electrofusion resulted in a few fusion events (Fig. 5D,G). Thus, to enhance fusion, we then combined both approaches, electrofusion and either expression of Syncytin1 (Fig. 5E,H). As a result, 700 V stimulation together with Syncytin1 induced fusion more effectively than the 300 V, in combination of different cell types HEK293 + H1299 (Fig. 5E), and HEK293 + H2170 (Fig. 5G). We show that in a mix of cells HEK293 + H2170, Syncytin1 overexpression alone and in pair with electrofusion visibly increased fusion events (Fig. 5B,H).

Logic gate to detect cell–cell fusion and schematic diagram of cell–cell electrofusion and hybrid cell formation. Images of admixed transiently transfected cells with complementing Cas9 halves HEK 293 + H1299 and HEK 293 + H2170 after fusion induction by overexpression of Syncytin-1, alone or in pair with electrofusion, or by PEG treatment. Control cells (NC) were transfected with only one half of Cas9 plus the reporter and fusogenic vectors (A,B). As we expected admixed cells HEK293 + H1299 expressing either one halve (as a control samples) and electrofused using 300V and 700V do not show any fusion events (C), compared to electrofusion of cells expressing both halves where we got results of a few cell fusion (D). After enhancement of fusion by electric pulses in pair with Syncytin1 overexpression, we visibly increased occurrence of fusion events (E). In control samples of admixed HEK293 + H2170 cells after electrofusion using 300 V and 700 V, also the fusion was undetectable (F), in comparison to Syncytin1-induced fusion cells plus electrofusion, where fusion cell were clearly visible (H). Images of admixed stable cell lines with complementing Cas9 halves (HEK293, H1299, H2170, HEK293 + H1299, HEK293 + H2170, and H1299 + H2170) after fusion induction by overexpression of Syncytin1, and Syncytin1 plus electrofusion. Control cells (NC) were transfected with only one half of Cas9 plus the reporter and fusogenic vectors. In relation to cells only expressed fusogenic Syncytin1 (G), the combination of Syncytin1 and electrical pulses (H) greatly enhanced the detection of syncytia. When fusing HEK293 with H1299 or H2170, the syncytia visibly formed contained more nuclei, than lung cancer cells in a mix within the same line. In summary, H1299 cells overexpressing Syncytin1 in pair with electrofusion at 700 V did not show significant fusion events (H). Higher efficiency was achieved by expression of Syncytin1 and lower voltage (300 V 500 µs one pulse) in stable expressing complementing Cas9 halves H1299 cells (I). Representative large multinucleated syncytia after electrofusion cells overexpressed Syncytin1 H1299 (using 300 V), H2170 cell lines (using 700 V), and mixed HEK 293 with H1299 cell line and HEK293 with H2170 cells (using 700 V) (J).
For more in-depth inquiry, we created stable cell lines, and used optimized settings to induce fusion of different cell, each line stably expressing one half of Cas9 (Cas4.5C or Cas4.5N), and co-transfected with the reporter (EGxxFP) and Syncytin1 plasmids. 24 h after transfection, the different cell types were admixed within either the same lineage, but expressing the complementing Cas4.5, or to a different cell type also expressing the complementing Cas4.5 (HEK293 with H2170, HEK293 with H1299, and H2170 with H1299) and then cells were electrofused (700 V for 15 µs). Control cells were co-transfected with only one half of Cas9 plus the reporter and Syncytin1 plasmids (negative control, NC). Compared to cells only overexpressing Syncytin1 (Fig. 5G), the presence of Syncytin1 and electrical pulses (Fig. 5H) greatly enhanced the detection of syncytia. We show that in a mix of different type of cells (when fusing HEK293 with H1299 or H2170) the syncytia formed were larger and contained more nuclei, than either cell type fused with its same lineage (Fig. 5G,H). There were no major fusion events in the mix of H1299 cells, whether only Syncytin1 was overexpressed (Fig. 5G) or in pair with 700 V stimulation (Fig. 5H). In summary, cells survival was lower after 1 long vs 3 shorter pulses. Surprisingly, the H1299 cells generated more fusion events at lower voltage (300 V for 500 µs) and 1 pulse (Fig. 5I). The presence of Syncytin1 and electrical impulses contributed significantly for syncytia to occur in H1299 (using 300 V) and H2170 cell lines (using 700 V), and mixed HEK 293 with H1299 or HEK293 with H2170 cells (using 700 V) (Fig. 5J).
We then asked whether we could detect naturally occurring cell fusions. For this, we selected murine C2C12 myoblast cells (Fig. 6), which undergo spontaneous fusion during differentiation into myotubules. C2C12 cells were separately transfected with either Cas9 half (Cas4.5C or Cas4.5N), gRNA and EGxxFP reporter. 24 h hours after transfection, cells were trypsynised and co-plated at 90% confluence. Subsequently, differentiation medium was added, and cells were incubated for up to seven days.

Applying split Cas9 logic gate to detect naturally occurring cell fusions. Murine C2C12 myoblast cells were transfected separately with either Cas9 half (Cas4.5C or Cas4.5N), gRNA and EGxxFP reporter, and then co-cultured together in differentiation medium. (A) C2C12 cells have a triangular shape while undifferentiated; (B) then they align with each other and begin to fuse—for better visualization of the two nuclei this image is show at higher magnification; (C) forming myotubules. NC—Negative control (cells with Split Cas9 halves, and EGxxFP reporter without gRNA); PC—(cells transfected with Cas9/gRNA and EGxxFP reporter). Additional images in Supplementary Fig. S1.
As a positive control, we used cells transfected with Cas9, gRNA and EGxxFP reporter. As a negative control, we employed cells transfected with Split Cas9 halves and EGxxFP reporter without gRNA.
Undifferentiated C2C12 cells have a triangular shape (Fig. 6A), but as they differentiated they became spindle-shape, aligned to each other and fused to each other (Fig. 6B), eventually forming myotubules (Fig. 6C).
Discussion
Prokaryotes, which include bacteria and archaea, have simpler signaling processes than eukaryotic cells, which means that SynBio logic gates are easier to generate. In mammalian systems signaling is rather complex and therefore simple SynBio circuits are more demanding to design.
While biological processes or events can be detected using biochemical methods, the use of bioassays have become more and more commonplace. Yet, many bioassays contain single inputs and can hardly be adapted to other purposes. Here, we show that it is possible to generate logic gates using a split CRISPR/Cas9, which is an actuator with many downstream options61,62,63,64. Moreover, the activation of Cas9 also creates a memory so using this system we could detect biological processes after they occurred e.g. in vivo long after they occurred.
As CRISPR-Cas-mediated genome editing technologies have provided accessible and flexible means to alter, regulate and visualize genomes, they are considered a milestone for molecular biology in the twenty-first century. So far, CRISPR-Cas systems have found wide applications in the investigation of gene function, gene therapy, development of targeted drugs, and construction of animal models, which fully proves their great potential for further development10,65,66,67,68.
Together with other synthetic systems for detecting biological phenomena such as optogenetic, gene expression and cell-free sensors, CRISPR systems have shown incredible flexibility that can be easily translated to various research and medical contexts. For example, Liu et al. reported a modular AND logic gate based on the CRISPR-Cas9 system, contributing to a synthetic biology platform for targeting and controlling bladder cancer cells in vitro27.
Here, we introduced a new application of the CRISPR/Cas9, as a sensor for detection cancer cells with epithelial origin, using an epithelial cancer-specific promoter (phCEA), a biological process—cells undergoing EMT. The selection of the phCEA, a marker to detect the presence of cancer cells of epithelial origin is specific for various types of cancer including colorectal cancer44,69, supports the concept that CEA is one of the most promising target antigens for colorectal cancer immunotherapy in use. CEA-targeted immunotherapies are presently in the preclinical or clinical phase, including CEA vaccines and CEA-targeted CAR T cells or TCR-modified T cells70.
Finally, split Cas9 was used for the detection of cell–cell fusions. A process that has been reported to occur between tumor cells and surrounding cells71,72,73. Where normal cells surrounding cancer cells can acquire malignant characteristics, according to the proverb „lie down with dogs, rise up with fleas”. One of the mechanisms facilitating tumor progression as the tumor spreads to surrounding tissue is cell–cell fusion74. Cell–cell fusion has been suggested as one possible mechanism for tumor progression75,76. Given that EMT is closely related to tumor invasiveness, there is likely to be a link between cell fusion and tumor invasiveness. When cancer cells fuse with healthy differentiated cells, the resulting hybrid cell can develop the ability to divide uncontrollably and form tumours77,78,79. The most compelling evidence that cell–cell fusion drives cancerogenesis is the report by Lazova and collaborators80 where they found that a metastatic melanoma was a hybrid between the patient’s cells and the ones from his brother after an allogeneic bone-marrow transplantation. Since then, more reports of cell–cell fusion show that such events initiate or fuel tumorigenesis79,81.
The applications presented above are additional tools to the CRISPR palette. By modifying the genetic elements that control the expression of its individual elements, it can be programmed and created more complex logic gates. One of the possibilities offered by this system is the elimination of a selected cell subpopulation from a given cell mixture through the use of gRNAs directed against essential genes. Thus, the proposed method may have potential for cancer diagnosis, therapy and treatment monitoring.
In recent years, the potential of using Cas9 as an actuator has also become apparent, is still a relatively new approach, but it has the potential to enable new possibilities for precise control over biological systems. Moreover, the requirement of the gRNA allows a 3-input system that can target specific genomic loci or additional logic gates/circuits, and thus become a research tool for multiple applications (Fig. 7).

Engineering cells with CRISPR/Cas9 circuits.
In the last few years, SynBio has made evident that it can use engineering logic to provide insight into previously unrecognized mechanisms of natural phenomena6,82. As Cas9 has recently been shown to be associated with non-canonical RNAs in bacteria82, exploring the interplay among endogenous RNA and Cas9 in eukaryotes could point to new technology, including additional imaging reporters, to study gene activation and even enables therapy modalities in mammalian systems.
In the grand scheme, CRISPR provides new opportunities for constructing circuits that can control processes in living cells, with wide application in synthetic biology. Yet, there are still limitations that need to be overcome, such as improving delivery methods, what could be achieved by utilizing viral vectors or nanoparticles, in especially to primary cells and in vivo conditions. Another limitation lies in the characterisation of promoters or antigens that are only expressed in certain cell types or conditions, which would significantly improve specificity. Despite these challenges, our findings have expanded the use of CRISPR technologies, and we envision the development of more complex gates in the near future.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files]. The nucleotide (plasmid) data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB64624 (https://www.ebi.ac.uk/ena/browser/view/PRJEB64624.
References
Gardner, T. S., Cantor, C. R. & Collins, J. J. Construction of a genetic toggle switch in Escherichia coli. Nature 403, 339–342 (2000).
Elowitz, M. B. & Leibier, S. A synthetic oscillatory network of transcriptional regulators. Nature 403, 335–338 (2000).
Bikard, D. et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41, 7429–7437 (2013).
Dong, C., Fontana, J., Patel, A., Carothers, J. M. & Zalatan, J. G. Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nat. Commun. 9, (2018).
Kim, H., Bojar, D. & Fussenegger, M. A CRISPR/Cas9-based central processing unit to program complex logic computation in human cells. Proc. Natl. Acad. Sci. U.S.A. 116, 7214–7219 (2019).
Liu, Y. et al. Reprogrammed tracrRNAs enable repurposing of RNAs as crRNAs and sequence-specific RNA biosensors. Nat. Commun. 13, 1–12 (2022).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Gilbert, L. A. et al. XCRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442 (2013).
Doudna, J. A. & Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1. https://doi.org/10.1126/science.1258096 (2014).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Jinek, M. et al. RNA-programmed genome editing in human cells. eLife 2013, (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/cas-mediated genome engineering. Cell 153, 910–918 (2013).
Li, M., Zhao, L., Page-McCaw, P. S. & Chen, W. Zebrafish genome engineering using the CRISPR–Cas9 system. Trends Genet. 32, 815–827. https://doi.org/10.1016/j.tig.2016.10.005 (2016).
Lee, S. H., Kim, S. & Hur, J. K. CRISPR and target-specific DNA endonucleases for efficient DNA knock-in in eukaryotic genomes. Mol. Cells 41, 943–952. https://doi.org/10.14348/molcells.2018.0408 (2018).
Bukhari, H. & Müller, T. Endogenous fluorescence tagging by CRISPR. Trends Cell Biol. 29, 912–928. https://doi.org/10.1016/j.tcb.2019.08.004 (2019).
Qi, L. S. et al. Repurposing CRISPR as an RNA-γuided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Cho, S. W., Kim, S., Kim, J. M. & Kim, J. S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232 (2013).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Chen, Y. Y., Jensen, M. C. & Smolke, C. D. Genetic control of mammalian T-cell proliferation with synthetic RNA regulatory systems. Proc. Natl. Acad. Sci. U.S.A. 107, 8531–8536 (2010).
Dominguez, A. A., Lim, W. A. & Qi, L. S. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 17, 5–15. https://doi.org/10.1038/nrm.2015.2 (2016).
Farzadfard, F. & Lu, T. K. Genomically encoded analog memory with precise in vivo dna writing in living cell populations. Science 346, (2014).
Gilbert, L. A. et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661 (2014).
Nissim, L., Perli, S. D., Fridkin, A., Perez-Pinera, P. & Lu, T. K. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol. Cell 54, 698–710 (2014).
Konermann, S. et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588 (2015).
Liu, Y. et al. Synthesizing and gate genetic circuits based on CRISPR-Cas9 for identification of bladder cancer cells. Nat. Commun. 5, (2014).
Chavez, A. et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12, 326–328 (2015).
Truong, D. J. J. et al. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 43, 6450–6458 (2015).
Nihongaki, Y., Kawano, F., Nakajima, T. & Sato, M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 33, 755–760 (2015).
Zetsche, B., Volz, S. E. & Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 33, 139–142. https://doi.org/10.1038/nbt.3149 (2015).
Iwai, H., Züger, S., Jin, J. & Tam, P. H. Highly efficient protein trans-splicing by a naturally split DnaE intein from Nostoc punctiforme. FEBS Lett 580, 1853–1858 (2006).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Mashiko, D. et al. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci. Rep. 3, (2013).
Potorac, I. et al. A vital region for human glycoprotein hormone trafficking revealed by an LHB mutation. J. Endocrinol. 231, 197–207 (2016).
Czapiński, J. et al. Development of a bioassay for continuous monitoring of TWIST1 activity (2021). https://doi.org/10.20944/PREPRINTS202103.0265.V1.
Muona, M., Aranko, A. S., Raulinaitis, V. & Iwaï, H. Segmental isotopic labeling of multi-domain and fusion proteins by protein trans-splicing in vivo and in vitro. Nat. Protoc. 5, 574–587 (2010).
Iwai, H., Züger, S., Jin, J. & Tam, P. H. Highly efficient protein trans-splicing by a naturally split DnaE intein from Nostoc punctiforme. FEBS Lett. 580, 1853–1858 (2006).
Kiełbus, M. et al. Genetically engineered lung cancer cells for analyzing epithelial–mesenchymal transition. Cells 8, (2019).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Shah, N. H. & Muir, T. W. Inteins: Nature’s gift to protein chemists. Chem. Sci. 5, 446–461. https://doi.org/10.1039/c3sc52951g (2014).
Grunnet, M. & Sorensen, J. B. Carcinoembryonic antigen (CEA) as tumor marker in lung cancer. Lung Cancer 76, 138–143. https://doi.org/10.1016/j.lungcan.2011.11.012 (2012).
Su, Y. T., Chen, J. W., Chang, S. C., Jiang, J. K. & Huang, S. C. The clinical experience of the prognosis in opposite CEA and image change after therapy in stage IV colorectal cancer. Sci. Rep. 12, (2022).
Cao, G. et al. Comparison of carcinoembryonic antigen promoter regions isolated from human colorectal carcinoma and normal adjacent mucosa to induce strong tumor- selective gene expression. Int. J. Cancer 78, 242–247 (1998).
Wei, S. C. et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 17, 678–688 (2015).
Zhu, Q. Q., Ma, C., Wang, Q., Song, Y. & Lv, T. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumor Biol. 37, 185–197. https://doi.org/10.1007/s13277-015-4450-7 (2016).
Qin, Q., Xu, Y., He, T., Qin, C. & Xu, J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 22, 90–106. https://doi.org/10.1038/cr.2011.144 (2012).
Rhim, A. D. et al. EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361 (2012).
Pastushenko, I. & Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 29, 212–226. https://doi.org/10.1016/j.tcb.2018.12.001 (2019).
Lu, X. & Kang, Y. Efficient acquisition of dual metastasis organotropism to bone and lung through stable spontaneous fusion between MDA-MB-231 variants. Proc. Natl. Acad. Sci. U.S.A. 106, 9385–9390 (2009).
Uygur, B. et al. Interactions with muscle cells boost fusion, stemness, and drug resistance of prostate cancer cells. Mol. Cancer Res. 17, 806–820 (2019).
Kondo, N., Marin, M., Kim, J. H., Desai, T. M. & Melikyan, G. B. Distinct requirements for HIV-cell fusion and HIV-mediated cell-cell fusion. J. Biol. Chem. 290, 6558–6573 (2015).
Yamamoto, M. et al. Cell-cell and virus-cell fusion assay-based analyses of alanine insertion mutants in the distal 9 portion of the JRFL gp41 subunit from HIV-1 Downloaded from. J. Biol. Chem 294, 5677–5687 (2019).
Papa, G. et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathogens 17, (2021).
Hörnich, B. F. et al. SARS-CoV-2 and SARS-CoV spike-mediated cell-cell fusion differ in their requirements for receptor expression and proteolytic activation. J. Virol. 95, (2021).
Sha, M. et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403, 785–789 (2000).
Chang, C., Chen, P. T., Chang, G. D., Huang, C. J. & Chen, H. Functional characterization of the placental fusogenic membrane protein syncytin. Biol. Reprod. 71, 1956–1962 (2004).
Strick, R. et al. Proliferation and cell-cell fusion of endometrial carcinoma are induced by the human endogenous retroviral Syncytin-1 and regulated by TGF-β. J. Mol. Med. 85, 23–38 (2007).
Pontecorvo, G. Production of mammalian somatic cell hybrids by means of polyethylene glycol treatment. Som. Cell Genet. 1, 397–400 (1975).
Trontelj, K., Ušaj, M. & Miklavčič, D. Cell electrofusion visualized with fluorescence microscopy. J. Vis. Exp. 1991. https://doi.org/10.3791/1991 (2010).
Moon, T. S., Lou, C., Tamsir, A., Stanton, B. C. & Voigt, C. A. Genetic programs constructed from layered logic gates in single cells. Nature 491, 249–253 (2012).
Siuti, P., Yazbek, J. & Lu, T. K. Synthetic circuits integrating logic and memory in living cells. Nat. Biotechnol. 31, 448–452 (2013).
Huang, T. P. et al. High-throughput continuous evolution of compact Cas9 variants targeting single-nucleotide-pyrimidine PAMs. Nat. Biotechnol. 41, 96–107 (2023).
Sheth, R. U., Yim, S. S., Wu, F. L. & Wang, H. H. Multiplex recording of cellular events over time on CRISPR biological tape. Science 358, 1457–1461 (2017).
Chen, S. et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160, 1246–1260 (2015).
Sánchez-Rivera, F. J. & Jacks, T. Applications of the CRISPR-Cas9 system in cancer biology. Nat. Rev. Cancer 15, 387–395 (2015).
Chow, R. D. et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat. Neurosci. 20, 1329–1341 (2017).
Lu, Y. et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 26, 732–740 (2020).
Baqar, A. R. et al. The role of preoperative CEA in the management of colorectal cancer: A cohort study from two cancer centres. Int. J. Surg. 64, 10–15 (2019).
Gonzalez-Exposito, R. et al. CEA expression heterogeneity and plasticity confer resistance to the CEA-targeting bispecific immunotherapy antibody cibisatamab (CEA-TCB) in patient-derived colorectal cancer organoids. J. ImmunoTher. Cancer 7, (2019).
Bhatia, B. et al. Evidence that senescent human prostate epithelial cells enhance tumorigenicity: Cell fusion as a potential mechanism and inhibition by p16INK4a and hTERT. Int. J. Cancer 122, 1483–1495 (2008).
Dittmar, T. et al. Characterization of hybrid cells derived from spontaneous fusion events between breast epithelial cells exhibiting stem-like characteristics and breast cancer cells. Clin. Exp. Metas. 28, 75–90 (2011).
Luo, F. et al. Bone marrow mesenchymal stem cells participate in prostate carcinogenesis and promote growth of prostate cancer by cell fusion in vivo. Oncotarget 7, 30924–30934 (2016).
Goldenberg, D. M. Horizontal transmission of malignancy by cellcell fusion. in Expert Opinion on Biological Therapy vol. 12 (Expert Opin Biol Ther, 2012).
Huang, C. M. et al. Hypoxia enhances fusion of oral squamous carcinoma cells and epithelial cells partly via the epithelial-mesenchymal transition of epithelial cells. BioMed Res. Int. 2018, (2018).
Lu, X. & Kang, Y. Cell fusion as a hidden force in tumor progression. Cancer Res. 69, 8536–8539. https://doi.org/10.1158/0008-5472.CAN-09-2159 (2009).
Pawelek, J. M. Tumour-cell fusion as a source of myeloid traits in cancer. Lancet Oncol. 6, 988–993. https://doi.org/10.1016/S1470-2045(05)70466-6 (2005).
Su, Y. et al. Somatic cell fusions reveal extensive heterogeneity in basal-like breast cancer. Cell Rep. 11, 1549–1563 (2015).
Gast, C. E. et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci. Adv. 4, (2018).
Lazova, R. et al. A Melanoma brain metastasis with a donor-patient hybrid genome following bone marrow transplantation: First evidence for fusion in human cancer. PLoS ONE 8, (2013).
Zhou, X. et al. Cell fusion connects oncogenesis with tumor evolution. Am. J. Pathol. 185, 2049–2060 (2015).
Jiao, C. et al. Noncanonical crRNAs derived from host transcripts enable multiplexable RNA detection by Cas9. Science 372, 941–948 (2021).
Acknowledgements
Authors would like to thank Addgene, and the colleagues depositing their plasmids there, for the support to the greater scientific community.
Funding
The study was supported by Medical University of Lublin DS442/2022-2023, and the Polish National Science Centre (NCN): DEC-2015/17/B/NZ1/01777 and DEC-2017/25/B/NZ4/02364 grants.
Author information
Authors and Affiliations
Contributions
A.P.-P. performed the experiments and analyses. J.C.z. assisted with specific promotors experiments and data analyses. JK assisted in the fusion experiment and helped with figures. A.P.-P., J.C.z., and A.R.-M. designed the experimental section of this research work and co-wrote the manuscript draft. A.R.-M. supervised the overall study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Przybyszewska-Podstawka, A., Czapiński, J., Kałafut, J. et al. Synthetic circuits based on split Cas9 to detect cellular events. Sci Rep 13, 14988 (2023). https://doi.org/10.1038/s41598-023-41367-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-41367-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
