
Abstract
Augmentation of anaplerosis, or replenishment of carbon lost during intermediary metabolic transitions, is desirable in energy metabolism defects. Triheptanoin, the triglyceride of 7-carbon heptanoic acid, is anaplerotic via direct oxidation or 5-carbon ketone body generation. In this context, triheptanoin can be used to treat Glucose transporter type 1 deficiency encephalopathy (G1D). An oral triheptanoin dose of 1 g/Kg/day supplies near 35% of the total caloric intake and impacted epilepsy and cognition in G1D. This provided the motivation to establish a maximum, potentially greater dose. Using a 3 + 3 dose-finding approach useful in oncology, we studied three age groups: 4–6, 6.8–10 and 11–16 years old. This allowed us to arrive at a maximum tolerated dose of 45% of daily caloric intake for each group. Safety was ascertained via analytical blood measures. One dose-limiting toxicity, occurring in 1 of 6 subjects, was encountered in the middle age group in the context of frequently reduced gastrointestinal tolerance for all groups. Ketonemia following triheptanoin was determined in another group of G1D subjects. In them, β-ketopentanoate and β-hydroxypentanoate concentrations were robustly but variably increased. These results enable the rigorous clinical investigation of triheptanoin in G1D by providing dosing and initial tolerability, safety and ketonemic potential.
ClinicalTrials.gov registration: NCT03041363, first registration 02/02/2017.
Similar content being viewed by others


Introduction
In normal conditions, carbon enters the organism with the diet and exits via excretion as organic waste or respiration as CO2. The principal source of CO2 is cellular respiration, which comprises the full oxidation of glucose-derived pyruvate in the tricarboxylic acid (TCA) cycle. At completion, pyruvate oxidation via pyruvate dehydrogenase, together with other catabolic reactions, is associated with net carbon loss or negative carbon balance1. This is in part counteracted by the parallel carboxylation of a fraction of pyruvate, which replenishes TCA cycle intermediate substrates. This, and other carbon-refilling reactions, are collectively termed anaplerosis2. Both pyruvate oxidation and decarboxylation are essential to sustain the robust metabolic rate that characterizes neural tissue. Despite wide variation attributable to measurement methods, a significant fraction of brain glucose flux, perhaps amounting to 20%, is estimated to sustain anaplerosis2,3. Similarly to pyruvate dehydrogenase, whose deficiency leads preferentially to encephalopathy4, the relevance of pyruvate carboxylation and anaplerosis to brain function is underscored by the human disease pyruvate carboxylase deficiency, where even mild reductions in enzyme activity are associated with neural dysfunction5. Intermediary carbon depletion has also been documented in more common disorders such as epilepsy6. Thus, extending this reasoning to the preceding metabolic events, the availability of pyruvate-derived byproducts is also likely to be reduced in clinically relevant states associated with diminished brain glucose transport into and flux through the brain. An example of such a state is Glucose transporter deficiency type 1 (G1D), where decreased blood to brain and glial cell glucose transport7 is associated with encephalopathy8.
G1D constitutes primarily a neurological disorder without alteration of systemic metabolism9,10. Some of its manifestations prove fully or partially treatable with a ketogenic diet in a significant fraction of individuals11,12. Nevertheless, the diet’s main ketone byproducts, β-hydroxybutyrate and acetoacetate, are not anaplerotic. Further, the diet is also poor in carbohydrate and is thus associated with a reduced blood glucose concentration. This is counterintuitive in G1D because all individuals to date have been haploinsufficient (mouse null SLC2A1 mutations are embryonic lethal13) and thus possess substantial glucose transport capacity, and many respond favorably to elevated blood glucose7. Moreover, the ketogenic diet is associated with side effect risks14,15,16,17,18. These notions provide the motivation to study anaplerotic therapy for G1D.
One biochemical principle sustains this motivation, since an alternative anaplerotic route of entry into the TCA cycle is provided by the reaction catalyzed by propionyl coenzyme A carboxylase. This reaction enables the utilization of exogenously administered odd-carbon-number containing fatty acids such as heptanoate and its metabolic byproducts as substrates for anaplerosis. Used as a supplement to a regular diet, triheptanoin (C7), the triglyceride of heptanoate, is associated with favorable impact on several aspects of human G1D encephalopathy19.
Previous studies suggest that C7 dosed at up to 35% daily caloric intake causes no discernible abnormalities in normal individuals and may be beneficial in inherited lipid or carbohydrate metabolic disorders19,20,21,22,23. To discern if a greater dose is feasible in G1D children, we used a 3 + 3 dose-finding design adapted from oncological phase I clinical trials24,25. Our objectives were to determine the safety and tolerability of C7, its maximum tolerated dose (MTD) and its ketogenic potential via the formation of the C5 ketones β-ketopentanoate (BKP) and β-hydroxypentanoate (BHP).
Materials and methods
We followed the ethical standards of the Helsinki Declaration of 1975 (as revised in 1983) and was approved by the Institutional Review Board approval of UT Southwestern Medical Center. Informed consent was obtained in writing from all the subjects or legally authorized representatives. Assent was also equally documented for cognitively-capable children between 10 and 17 years of age.
We utilized sets of data from three independent groups of G1D individuals prospectively studied at the Rare Brain Disorders Program of the University of Texas Southwestern Medical Center: (1) 14 subjects (ages 2–27 years, group A) who consumed C7 at a 35% caloric dose and were previously reported as part of the first study of triheptanoin in G1D19 (ClinicalTrials.gov Identifier NCT02018315, first registration 23/12/2013). No dose-finding was part of that study, since the 35% dose was chosen based only on the dose used in other unrelated disorders. However, a new analysis of these data is used here for initial dose tolerability estimation and the data are therefore presented as newly separated by age group. This was not previously reported. As such, only the information relevant to the present work is contained in a table and the age dependence of tolerability discussed; (2) 12 subjects (ages 4–16 years, group B) enrolled in a 3 + 3 dose-finding phase I clinical trial (NCT03041363, first registration 02/02/2017). These subjects were studied between March and December of 2017 and their data were intended solely for the determination of the MTD and associated safety and tolerability; (3) 7 individuals (ages 2–11 years, group C) who received analytical testing at the MTD, once it was determined using the knowledge gained from group A and group B. These subjects were studied between January 2018 and December 2020 (ClinicalTrials.gov Identifier NCT03181399, first registration 08/06/2017). They received evaluation of ketonemia and additional safety assessments and these data are used in the present work. These subjects then proceeded to a still ongoing, different study unrelated to the goals of the present work, and this is therefore not further described here.
Participant characteristics
The diagnosis was ascertained via DNA analysis of the SLC2A1 gene, which encodes Glut1, or via fluoro-deoxyglucose positron emission tomography (PET) of the brain when DNA results did not reveal a probable pathogenic variant7,9,26. The DNA variants cited here refer to transcript NM_006516.2 and were deemed pathogenic as per each clinical DNA testing report. Enrolment followed the order of contact made by eligible subjects. Eligible contacts spontaneously exceeded enrolment targets. All individuals were consuming a regular or a modified Atkins diet. The latter type of diet was selected as it represents the most common alternative to a standard ketogenic diet12, thus facilitating enrolment in this study. There was no consideration of geographic location (U.S. or abroad) or disease severity. The phenotypic features of all subjects included a variable combination of intellectual disability, epilepsy, ataxia, or episodic apraxia. For the 3 + 3 study, medications, including antiseizure drugs, were not allowed to change 30 days prior to or during the study. Supplementary Table S1 lists ages, clinically significant DNA variants, phenotypes and previous treatments for the 14 group A subjects previously studied 19. Tables 1 and 2 indicate the same characteristics for groups A and B. Supplementary Table S2 lists the complete inclusion and exclusion criteria for group B.
Treatment protocol
MTD study
A standard 3 + 3 phase I dose de-escalation design was used in group B subjects. The MTD was defined as the dose at which fewer than one third of patients experienced a dose-limiting toxicity (DLT). As part of the study design, if dose 1 exceeded the MTD, the dose would be de-escalated in subsequent subjects to dose 2. If dose 2 proved intolerable, then 35% would be considered as the MTD based on the dose tolerated by group B19. Intra-patient dose escalation was permitted. The dose range under consideration (35–45%) was informed by nutritional standards27,28. Figure 1 illustrates the division of subjects by age: group BI (4–6 years old), group BII (6.8 to 10 years old) and group BIII (11–16 years old). Following a standard physical (including neurological) examination and a nutritional assessment to estimate average daily calories consumed, the subjects received baseline blood measurements (comprehensive metabolic panel, complete blood count, lipid panel, lactate, and β-hydroxybutyrate). C7 was then dosed for each subject as a percentage of the daily caloric intake (dose 1 = 45%; dose 2 = 40%), divided into 4 doses per day. To minimize insulin-mediated suppression of ketogenesis and potential competition of dietary octanoate with C729, each dose was consumed 45–60 min before meals. A reduction in calories from other dietary sources compensated for the extra C7 calories. All subjects ingested C7 daily for 7 days. We measured tolerance over 7 days because we previously reported acceptable tolerance of the 35% dose after 2 days30. They received physical examination, tolerability and toxicity assessments every 2–3 days until exit, with additional telephone contact on non-physical examination days to assess side effects. Blood studies were repeated at physical examination visits. Upon discontinuation of C7 on day 8, the subjects received a physical examination, repeat blood studies and tolerability and toxicity assessment, all of which was again repeated on day 10. Side effects were again assessed one-month post-exit via telephone. Toxicity was assessed using the Common Toxicity Criteria (version 5.0) of the National Cancer Institute, National Institutes of Health. A dose limiting toxicity (DLT) was defined as any toxicity grade ≥ 3 (except for grade 3 nausea, vomiting, or diarrhea that could be controlled within 24 h with supportive care measures) or any unacceptable grade 2 toxicity resulting in treatment intolerance despite maximal medical treatment.

C7 dose-finding. Subjects are divided in 3 age groups as described in the methods. Each circle represents one subject. Left: 14 group A G1D individuals studied at the 35% dose19. One DLT was observed in age group AII. Right: Tolerance of the 45% dose in group B following the 3 + 3 design. The MTD was determined at 45% in groups BI and BIII after all 3 subjects in each group tolerated this dose. Due to one DTL in group BII, an additional 3 subjects were added to this group, following after tolerance, in a determination of the same MTD for this age group. DLT Dose limiting toxicity.
C7 ketonemia
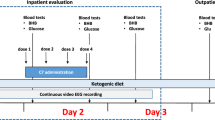
Group C subjects received baseline analytical measurements (comprehensive metabolic panel, complete blood count, lipid panel, lactate, β-hydroxybutyrate, β-ketopentanoate and β-hydroxypentanoate) and physical examination. They then observed an overnight fast. C7 was calculated at a daily dose of 45% of daily caloric intake. The following morning, two doses were administered at approximately 6 AM (before breakfast) and 10 AM (before lunch), each containing ¼ of the total daily dose. Before lunch, at 12–1 PM, blood was collected for determination of ketone bodies as described31, approximately 21 h after the baseline analysis. Figure 2 illustrates this experimental sequence.

Stimulation of C5 ketogenesis by C7. Plasma levels of C5 ketones before and after two C7 doses. Top and middle panels illustrate C5 ketone values before and after an overnight fasting period followed by two C7 doses. The baseline (pre-C7) values were obtained at 2–2:30 PM. Two C7 doses (each ¼ of the MTD) were administered approximately at 6 AM and 10 AM. The only morning meal was breakfast approximately at 8 AM. Each color symbol and time identifies one individual.
C7 dietary supplement
Triheptanoin (glycerol triheptanoate, Neobee 795) was provided by Stepan Lipid Nutrition from a supply used in the human food industry as an additive to dairy products or as an emollient in cosmetics. Participants and caretakers were instructed on its mode of administration. To facilitate consumption approximately one hour before meals, in some cases the C7 oil was mixed immediately before use with a small amount (less than 15 ml) of fat free, sugar free yogurt, pudding, or an equivalent low-calorie food.
Results
Subject characteristics
Group B subjects, summarized in Table 1, included 8 females and 4 males. Median age was 8.5 years (range 4–16). They were primary English and Spanish speakers, all of whom identified themselves as white. Eight (67%) participants were non-Hispanic and 4 (33%) were Hispanic. One subject who exhibited common disease features and decreased cerebrospinal fluid glucose (25 mg/dl) was enrolled using the findings of a PET study, consistent with the approximately 15% of G1D patients who do not harbor an identifiable a pathogenic variant in SLC2A110,
Group C subjects, studied for ketonemia, are summarized in Table 2. This group included 5 females and 2 males, with a median age of 8.5 years (range 4–16 years). It also included English and Spanish speakers. All of them identified themselves as white. Five participants identified as non-Hispanic, and two as Hispanic. One subject with no pathogenic mutation in SLC2A1 exhibited characteristic symptoms and a brain PET study indicative of G1D.
Adverse events
We previously collected safety data from group A subjects, comprising 265 G1D subject-months while receiving C7 treatment (19, with subsequent data collection until September 1 of 2015). These data originated from 14 G1D individuals (age 2–27 years) recruited from our program, who received C7 at a 35% dose. All received a regular diet adjusted to maintain the appropriate daily calories after the addition of C7. Of these, following study completion, 11 subjects opted to maintain longer term C7 consumption. There were no statistically significant differences in blood glucose, β-hydroxybutyrate, lactate, cholesterol (HDL, LDL, total), triglycerides, basic metabolic panel electrolytes, blood urea nitrogen, creatinine, creatine kinase, AST, ALT, GGT and blood cell count before and after C7 treatment. Three subjects experienced gastrointestinal discomfort within two days of initiation that subsided after decreasing the C7 dose, followed by a gradual increase to the desired dose over 7–14 days. One subject maintained pre-C7 levels of caloric consumption and gained 4 kg (baseline weight 52 kg) over 6 weeks, which were lost over another 6 weeks following nutritional counseling. There was no change in analytical parameters in this subject at maximum weight.
No subject in the current dose-finding and ketonemia studies experienced serious or unexpected adverse events. There were no appreciable physical examination changes in any of these subjects. Table 3 reflects the adverse effects and event noted in the dose-finding individuals. Eleven of the twelve subjects in the 3 + 3 study (group B) experienced mild adverse events. Seven (58%) had mild diarrhea or abdominal discomfort within 4 days of treatment initiation. Five (42%) experienced mild vomiting within 3 days of treatment initiation. In four of the five (33%), the digestive discomfort, diarrhea or vomiting symptoms resolved by reducing the amount of triheptanoin by one half and gradually titrating up the maximum target level over 3 days such that the 45% dose was considered tolerable. One subject exhibited persistent gastrointestinal intolerance. Therefore, following the 3 + 3 design, the middle age group was expanded by 3 more subjects to a total of 6, with the latter three exhibiting tolerance. One subject had a grand mal seizure during the study. The occurrence of this event was judged consistent with the previous frequency and severity of epilepsy in this subject. Group C subjects manifested no adverse events. All the participants reported baseline wellbeing one month after study completion.
Dose limiting toxicity and MTD
Figure 1 illustrates the tolerance of C7 at 35% dose19 and at 45% dose as determined with the 3 + 3 design (group B). For group B, no DLT was observed at dose level 1 in age groups 1 and 3. As mentioned above, there was one DLT (grade 3 with intolerable vomiting and abdominal discomfort) in the first cohort of age group BII. No additional DLT was observed in the subsequent cohort enrolled in this group. Therefore, since 1/12 subjects experienced a DLT at dose level 1 (0/3 in group BI, 1/6 in group BII and 0/3 in group BIII), the MTD was defined as dose 1. De-escalation to dose 2 was not necessary.
Safety
Analyses of all blood analytes revealed no significant change in the comprehensive metabolic panel, complete blood count, lipid panel, lactate, or β-hydroxybutyrate at the MTD (Tables 4 and 5).
C5 ketone body blood levels
Figure 2 illustrates the blood concentration of C5 ketone bodies following two doses of C7. The values are provided in Table 6. Although the purpose of these measurements was not to delineate the metabolokinetics of C7 derivatives, these results indicate that, while C5 ketosis was noted in all subjects, there was significant variability. In numerous cases there was no C5 ketosis measurable pre C7 treatment. In the rest of cases, where a measurable level of C5 ketones was detected before C7, the fold-change in ketone concentration in post-relative to pre-C7 conditions was 3- to 88-fold for BKP and 46-fold for BHP. No correlation was found between the levels of C5 ketones after C7 (R = 0.36), indicating that ketogenesis or circulating ketone body extraction is unequal for both ketone forms. There was also no correlation between pre-C7 β-hydroxybutyrate and post-C7 BKP levels (R = 0.32) or between pre-C7 β-hydroxybutyrate and post-C7 BHP levels (R = -0.5).
Discussion
To enable the rigorous clinical investigation of C7 as a treatment for G1D, we have determined the MTD, safety and tolerability of C7 in G1D. The selection of C7 was based on its insipid nature, suitable for use as a dietary supplement, in conjunction with its heptanoate content. C7 dosing was based on individualized caloric consumption calculated at the time of study entry rather than per-weight or other dosing methods because caloric requirements change during development and C7 is fully consumed in the course of metabolism. This contrasts with the dosing of interventions (such as drugs) that primarily modify receptors or other targets whose action depends on molecule-to-molecule binding and are not consumed for biological action.
The β-oxidation of carbons 1–4 of heptanoate generates two molecules of acetyl coenzyme A and one molecule of propionyl coenzyme A derived from carbons 5–7. The latter can enter the TCA cycle through propionyl coenzyme A carboxylase. In this context, triheptanoin (C7) contains three esterified heptanoic acid molecules. We previously used 13C-nuclear magnetic resonance (NMR) spectroscopy32, gas chromatography-mass spectrometry (GC–MS) and liquid chromatography-mass spectrometry (LC–MS) to elucidate the metabolism of infused [5,6,7-13C3]heptanoate in the G1D mouse3. This work revealed enrichment in heptanoate-derived plasma glucose via neoglucogenesis and increased cerebral acetyl coenzyme A and glutamine concentrations consistent with metabolism of heptanoate and or derivative C5 (i.e., containing five carbons) ketones in glia.
Previous C7 use in G1D and other disorders was based on doses that supply up to 35% of the daily caloric intake and are tolerable19,20,21,22,23. However, augmentation of metabolic flux via increase substrate or preserved enzyme activity is desirable to maximize potential neural performance benefits. This is exemplified by the effect of increased blood glucose level or availability on neural function, including the amelioration of seizures, in G1D7,33. A parallel metabolic enzyme activity-neural function correlate is illustrated by the absence of detectable metabolic deficits in the brain of mice mildly deficient in pyruvate dehydrogenase34, which become pronounced under a greater degree of deficiency4. Thus, we aimed for as high as tolerable a dose, with the limitation that all oils, including the form in which C7 is manufactured, can cause gastrointestinal intolerance. A second limitation to greater doses stems from a universal desire to retain a diet composition as common as possible35 and from the necessity to additionally consume essential fat not provided by C727,28.
Our subject selection from three age groups was based on age-dependent biological considerations and clinical observations to be considered in future clinical trials. First, nutritional fat requirements are age-dependent, varying by as much as 15% from early childhood to adolescence27,28. Second, the response to C7 might depend on age, which itself influences the level of fat consumption36 and on brain metabolic rate37. The cerebral metabolic rate for glucose is low at birth and rises to adult values by 2 years of age. It then continues to rise until, by 6 years, it nearly doubles. This elevated rate is maintained until approximately 10 years, when it begins to decline, approaching again the adult rate again near the end of the second decade. This time course parallels changes in the number of neurons, synapses, and dendritic spines in the human brain38,39.
The results indicate that the C7 MTD is not age dependent, as tolerability was similar for all ages. Consistent with the normal variability of stimulated blood ketone body levels in children40, the concentrations of the two C5 ketone bodies were variable across individuals and exhibited no correlation in several particular individuals. There was also no correlation between C5 ketogenesis and prior (i.e., immediately pre-C7) β-hydroxybutyrate ketogenesis. This suggests that ketogenesis and/ or blood ketone extraction arising from standard dietary fat and from C7 do not stem from the same metabolic process. Factors other than blood level determine metabolic efficacy (or lack thereof), especially considering the avid uptake of C5 ketones by several tissues rich in 3-oxoacid-CoA transferase, including the brain41,42,43. Correlating blood level with efficacy will be the subject of separate work. Of note, there are several possible metabolic effects derived from heptanoate, or its byproducts, in the brain3. This is due to the brain fuel potential of C5 ketones, heptanoate itself, and glucose from neoglucogenesis, all of which would be difficult to mechanistically separate without co-infusion labeling or other complex studies given the uncertainties about the magnitude of some of the relevant metabolic reactions44.
C7 is advantageous over other dietary therapeutic modalities given the possibility of combination with a regular or modified Atkins diet. This allows for a balanced diet, whereas the ketogenic diet is carbohydrate- and protein-restricted in favor of fat. Additionally, non-epileptic forms of G1D (i.e., apraxic, choreic, dystonic) continue to emerge45,46. Thus, simpler, effective diets are anticipated to be more widely accepted. Finally, relatively unbiased tools such a whole-exome or genome DNA analyses, comprehensive genomic hybridization and Sanger sequencing gene panels47 are increasingly uncovering G1D in young infants, for whom the ketogenic diet has not been fully tested48,49 but has received significant attention as a general epilepsy treatment50. Further, this diet provides reduced anaplerotic potential during this period of rapid brain growth51. Further studies will elucidate the compatibility of C7 with the ketogenic diet.
Our standard 3 + 3 phase I design52,53 yielded the MTD. Over 1,200 phase I oncology studies utilized this approach from 1991 to 200653, with a large fraction of subsequent studies adhering to the same design54. To our knowledge, the 3 + 3 design has not been used with neurologic or pediatric drug or food supplement investigations. Our results suggest that, despite a variety of existing trial design alternatives, the conceptual and procedural simplicity of the 3 + 3 design allow for effective dose finding53, especially in disorders with low prevalence or when drug availability is limited.
Conclusions
We were primarily motivated by the lack of an effective treatment for G1D. This includes, with few exceptions antiseizure medications7, the ketogenic diet and the modified Atkins diet35,46. C7 provides an anaplerotic alternative to these treatments. At 45% of caloric intake, C7 is safe and, after accounting for a period of frequent but transient gastrointestinal disturbance, suitable for clinical investigation. Future work may characterize C7 efficacy, including long term safety and tolerability, compatibility with the ketogenic diet and sources of individual variation in C5 ketone body metabolism.
Data availability
All the data are publicly available from the corresponding author.
Abbreviations
- BKP:
-
β-Ketopentanoate
- BHP:
-
β-Hydroxypentanoate
- C7:
-
Triheptanoin
- DLT:
-
Dose limiting toxicity
- G1D:
-
Glut1 deficiency
- Glut1:
-
Glucose transporter protein type 1
- PET:
-
Positron emission tomography
References
Jakkamsetti, V. M.-V., I; Ma, Q; Pascual, JM. in Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease, Sixth Edition (ed R N Rosenberg, Pascual, JM) (Academic Press, 2020).
Brunengraber, H. & Roe, C. R. Anaplerotic molecules: Current and future. J. Inherit. Metab. Dis. 29, 327–331. https://doi.org/10.1007/s10545-006-0320-1 (2006).
Marin-Valencia, I., Good, L. B., Ma, Q., Malloy, C. R. & Pascual, J. M. Heptanoate as a neural fuel: Energetic and neurotransmitter precursors in normal and glucose transporter I-deficient (G1D) brain. J. Cereb. Blood Flow Metab. 33, 175–182. https://doi.org/10.1038/jcbfm.2012.151 (2013).
Jakkamsetti, V. et al. Brain metabolism modulates neuronal excitability in a mouse model of pyruvate dehydrogenase deficiency. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aan0457 (2019).
Marin-Valencia, I., Roe, C. R. & Pascual, J. M. Pyruvate carboxylase deficiency: Mechanisms, mimics and anaplerosis. Mol. Genet. Metab. 101, 9–17. https://doi.org/10.1016/j.ymgme.2010.05.004 (2010).
Willis, S., Stoll, J., Sweetman, L. & Borges, K. Anticonvulsant effects of a triheptanoin diet in two mouse chronic seizure models. Neurobiol. Dis. 40, 565–572. https://doi.org/10.1016/j.nbd.2010.07.017 (2010).
Rajasekaran, K. et al. Metabolic modulation of synaptic failure and thalamocortical hypersynchronization with preserved consciousness in Glut1 deficiency. Sci. Transl. Med. 14, 2956. https://doi.org/10.1126/scitranslmed.abn2956 (2022).
Pascual, J. M. & Ronen, G. M. Glucose transporter type I deficiency (G1D) at 25 (1990–2015): Presumptions, facts, and the lives of persons with this rare disease. Pediatr. Neurol. 53, 379–393. https://doi.org/10.1016/j.pediatrneurol.2015.08.001 (2015).
Pascual, J. M. et al. Brain glucose supply and the syndrome of infantile neuroglycopenia. Arch. Neurol. 64, 507–513. https://doi.org/10.1001/archneur.64.4.noc60165 (2007).
Wang, D. et al. Glut-1 deficiency syndrome: Clinical, genetic, and therapeutic aspects. Ann. Neurol. 57, 111–118. https://doi.org/10.1002/ana.20331 (2005).
Wang, D., Pascual, J. M. & De Vivo, D. in GeneReviews(®) (eds M. P. Adam et al.) (University of Washington, Seattle Copyright © 1993–2022, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved., 1993).
Hao, J., Kelly, D. I., Su, J. & Pascual, J. M. Clinical aspects of glucose transporter type 1 deficiency: Information from a global registry. JAMA Neurol. 74, 727–732. https://doi.org/10.1001/jamaneurol.2017.0298 (2017).
Marin-Valencia, I. et al. Glut1 deficiency (G1D): Epilepsy and metabolic dysfunction in a mouse model of the most common human phenotype. Neurobiol. Dis. 48, 92–101. https://doi.org/10.1016/j.nbd.2012.04.011 (2012).
Stewart, W. A., Gordon, K. & Camfield, P. Acute pancreatitis causing death in a child on the ketogenic diet. J. Child Neurol. 16, 682. https://doi.org/10.1177/088307380101600910 (2001).
Best, T. H., Franz, D. N., Gilbert, D. L., Nelson, D. P. & Epstein, M. R. Cardiac complications in pediatric patients on the ketogenic diet. Neurology 54, 2328–2330. https://doi.org/10.1212/wnl.54.12.2328 (2000).
Berry-Kravis, E., Booth, G., Taylor, A. & Valentino, L. A. Bruising and the ketogenic diet: Evidence for diet-induced changes in platelet function. Ann. Neurol. 49, 98–103. https://doi.org/10.1002/1531-8249(200101)49:1%3c98::aid-ana13%3e3.0.co;2-2 (2001).
Hoyt, C. S. & Billson, F. A. Optic neuropathy in ketogenic diet. Br. J. Ophthalmol. 63, 191–194. https://doi.org/10.1136/bjo.63.3.191 (1979).
Klepper, J. GLUT1 deficiency syndrome in clinical practice. Epilepsy Res. 100, 272–277. https://doi.org/10.1016/j.eplepsyres.2011.02.007 (2012).
Pascual, J. M. et al. Triheptanoin for glucose transporter type I deficiency (G1D): Modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol. 71, 1255–1265. https://doi.org/10.1001/jamaneurol.2014.1584 (2014).
Roe, C. R., Bottiglieri, T., Wallace, M., Arning, E. & Martin, A. Adult polyglucosan body disease (APBD): Anaplerotic diet therapy (Triheptanoin) and demonstration of defective methylation pathways. Mol. Genet. Metab. 101, 246–252. https://doi.org/10.1016/j.ymgme.2010.06.017 (2010).
Roe, C. R. & Mochel, F. Anaplerotic diet therapy in inherited metabolic disease: Therapeutic potential. J. Inherit. Metab. Dis. 29, 332–340. https://doi.org/10.1007/s10545-006-0290-3 (2006).
Roe, C. R., Sweetman, L., Roe, D. S., David, F. & Brunengraber, H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J. Clin. Invest. 110, 259–269. https://doi.org/10.1172/jci15311 (2002).
Roe, C. R. et al. Carnitine palmitoyltransferase II deficiency: Successful anaplerotic diet therapy. Neurology 71, 260–264. https://doi.org/10.1212/01.wnl.0000318283.42961.e9 (2008).
Skolnik, J. M., Barrett, J. S., Jayaraman, B., Patel, D. & Adamson, P. C. Shortening the timeline of pediatric phase I trials: The rolling six design. J. Clin. Oncol. 26, 190–195. https://doi.org/10.1200/jco.2007.12.7712 (2008).
Sposto, R. & Groshen, S. A wide-spectrum paired comparison of the properties of the Rolling 6 and 3+3 Phase I study designs. Contemp. Clin. Trials 32, 694–703. https://doi.org/10.1016/j.cct.2011.04.009 (2011).
Pascual, J. M., Van Heertum, R. L., Wang, D., Engelstad, K. & De Vivo, D. C. Imaging the metabolic footprint of Glut1 deficiency on the brain. Ann. Neurol. 52, 458–464. https://doi.org/10.1002/ana.10311 (2002).
Gidding, S. S. et al. Dietary recommendations for children and adolescents: A guide for practitioners. Pediatrics 117, 544–559. https://doi.org/10.1542/peds.2005-2374 (2006).
Butte, N. F. Fat intake of children in relation to energy requirements. Am. J. Clin. Nutr. 72, 1246s–1252s. https://doi.org/10.1093/ajcn/72.5.1246s (2000).
Deng, S., Zhang, G. F., Kasumov, T., Roe, C. R. & Brunengraber, H. Interrelations between C4 ketogenesis, C5 ketogenesis, and anaplerosis in the perfused rat liver. J. Biol. Chem. 284, 27799–27807. https://doi.org/10.1074/jbc.M109.048744 (2009).
Roe, C. R. & Brunengraber, H. Anaplerotic treatment of long-chain fat oxidation disorders with triheptanoin: Review of 15 years Experience. Mol. Genet. Metab. 116, 260–268. https://doi.org/10.1016/j.ymgme.2015.10.005 (2015).
Kallem, R. R., Primeaux, S., Avila, A., Pascual, J. M. & Putnam, W. C. Development and validation of a LC-MS/MS method for quantitation of 3-hydroxypentanoic acid and 3-oxopentanoic acid in human plasma and its application to a clinical study of glucose transporter type I deficiency (G1D) syndrome. J. Pharm. Biomed. Anal. 205, 114335. https://doi.org/10.1016/j.jpba.2021.114335 (2021).
Marin-Valencia, I. et al. High-resolution detection of (13)C multiplets from the conscious mouse brain by ex vivo NMR spectroscopy. J. Neurosci. Methods https://doi.org/10.1016/j.jneumeth.2011.09.006 (2011).
Wang, R. C. et al. Red blood cells as glucose carriers to the human brain: Modulation of cerebral activity by erythrocyte exchange transfusion in Glut1 deficiency (G1D). J. Cereb. Blood Flow Metab. 43, 357–368. https://doi.org/10.1177/0271678X221146121 (2023).
Marin-Valencia, I. et al. Cortical metabolism in pyruvate dehydrogenase deficiency revealed by ex vivo multiplet (13)C NMR of the adult mouse brain. Neurochem. Int. 61, 1036–1043. https://doi.org/10.1016/j.neuint.2012.07.020 (2012).
Cervenka, M. et al. Metabolism-based therapies for epilepsy: New directions for future cures. Ann. Clin. Transl. Neurol. 8, 1730–1737. https://doi.org/10.1002/acn3.51423 (2021).
Chugani, H. T., Phelps, M. E. & Mazziotta, J. C. Positron emission tomography study of human brain functional development. Ann. Neurol. 22, 487–497. https://doi.org/10.1002/ana.410220408 (1987).
Chugani, H. T. & Phelps, M. E. Maturational changes in cerebral function in infants determined by 18FDG positron emission tomography. Science 231, 840–843. https://doi.org/10.1126/science.3945811 (1986).
Leen, W. G. et al. GLUT1 deficiency syndrome into adulthood: A follow-up study. J. Neurol. 261, 589–599. https://doi.org/10.1007/s00415-014-7240-z (2014).
Klepper, J. et al. Seizure control and acceptance of the ketogenic diet in GLUT1 deficiency syndrome: A 2- to 5-year follow-up of 15 children enrolled prospectively. Neuropediatrics 36, 302–308. https://doi.org/10.1055/s-2005-872843 (2005).
Saudubray, J. M. et al. Variation in plasma ketone bodies during a 24-hour fast in normal and in hypoglycemic children: Relationship to age. J. Pediatr. 98, 904–908. https://doi.org/10.1016/s0022-3476(81)80583-5 (1981).
Williamson, D. H., Bates, M. W., Page, M. A. & Krebs, H. A. Activities of enzymes involved in acetoacetate utilization in adult mammalian tissues. Biochem. J. 121, 41–47. https://doi.org/10.1042/bj1210041 (1971).
Fukao, T. et al. Enzymes of ketone body utilization in human tissues: Protein and messenger RNA levels of succinyl-coenzyme A (CoA):3-ketoacid CoA transferase and mitochondrial and cytosolic acetoacetyl-CoA thiolases. Pediatr. Res. 42, 498–502. https://doi.org/10.1203/00006450-199710000-00013 (1997).
Leclerc, J. et al. Metabolism of R-beta-hydroxypentanoate and of beta-ketopentanoate in conscious dogs. Am. J. Physiol. 268, E446-452. https://doi.org/10.1152/ajpendo.1995.268.3.E446 (1995).
Jeffrey, F. M. et al. Modeling of brain metabolism and pyruvate compartmentation using (13)C NMR in vivo: Caution required. J. Cereb. Blood Flow Metab. 33, 1160–1167. https://doi.org/10.1038/jcbfm.2013.67 (2013).
Pearson, T. S., Akman, C., Hinton, V. J., Engelstad, K. & De Vivo, D. C. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr. Neurol. Neurosci. Rep. 13, 342. https://doi.org/10.1007/s11910-013-0342-7 (2013).
Klepper, J. et al. Glut1 Deficiency Syndrome (Glut1DS): State of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open 5, 354–365. https://doi.org/10.1002/epi4.12414 (2020).
Wang, J., Gotway, G., Pascual, J. M. & Park, J. Y. Diagnostic yield of clinical next-generation sequencing panels for epilepsy. JAMA Neurol. 71, 650–651. https://doi.org/10.1001/jamaneurol.2014.405 (2014).
Klepper, J. et al. Introduction of a ketogenic diet in young infants. J. Inherit. Metab. Dis. 25, 449–460. https://doi.org/10.1023/a:1021238900470 (2002).
Dressler, A. & Trimmel-Schwahofer, P. The ketogenic diet for infants: How long can you go?. Epilepsy Res. 164, 106339. https://doi.org/10.1016/j.eplepsyres.2020.106339 (2020).
Treadwell, J. R. et al. Pharmacologic and dietary treatments for epilepsies in children aged 1–36 months: A systematic review. Neurology 100, e16–e27. https://doi.org/10.1212/WNL.0000000000201026 (2023).
Settergren, G., Lindblad, B. S. & Persson, B. Cerebral blood flow and exchange of oxygen, glucose, ketone bodies, lactate, pyruvate and amino acids in infants. Acta Paediatr. Scand. 65, 343–353. https://doi.org/10.1111/j.1651-2227.1976.tb04896.x (1976).
Kurzrock, R. et al. Moving beyond 3+3: The future of clinical trial design. Am. Soc. Clin. Oncol. Educ. Book 41, e133–e144. https://doi.org/10.1200/edbk_319783 (2021).
Hansen, A. R., Graham, D. M., Pond, G. R. & Siu, L. L. Phase 1 trial design: Is 3 + 3 the best?. Cancer Control 21, 200–208. https://doi.org/10.1177/107327481402100304 (2014).
Le Tourneau, C. et al. Heterogeneity in the definition of dose-limiting toxicity in phase I cancer clinical trials of molecularly targeted agents: A review of the literature. Eur. J. Cancer 47, 1468–1475. https://doi.org/10.1016/j.ejca.2011.03.016 (2011).
Acknowledgements
We thank the G1D individuals and their families for their participation. We are also grateful to Drs. Deepa Sirsi and Saima Kayani for assistance with group C subjects, and to Drs. Adam Hartman (NIH), Marc C. Patterson, Gabriel Ronen, Salvatore DiMauro, Eric H. Kossoff and Robert Rapaport (Glut1 Deficiency Foundation) for serving in the Data Safety Monitoring Committee of this study appointed in conjunction with NIH (subject groups B and C). We thank Jim Butterwick at Stepan Lipid Nutrition for a gift of triheptanoin.
Funding
Glut1 Deficiency Foundation (to J.M.P.), NIH grants NS094257, NS102588 and NS077015 (to J.M.P.) and Alicia Koplowitz Foundation (Ayuda de Estancia Corta to I.M). The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
Conception and design: J.M.P., C.A., C.R.R., S.S.; Acquisition, analysis, and interpretation of data: J.M.P., A.A., S.A.P., I.M., R.R.K., W.C.P., J.Y.P.; Drafting of the manuscript: J.M.P., A.A., I.M.; Critical revision of the manuscript for important intellectual content: All authors; Statistical analysis: C.A.; Obtaining funding: J.M.P.; Supervision: J.M.P.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Málaga, I., Avila, A., Primeaux, S. et al. Maximum dose, safety, tolerability and ketonemia after triheptanoin in glucose transporter type 1 deficiency (G1D). Sci Rep 13, 3465 (2023). https://doi.org/10.1038/s41598-023-30578-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-30578-z
This article is cited by
-
Combination of triheptanoin with the ketogenic diet in Glucose transporter type 1 deficiency (G1D)
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
