
Abstract
Detection of viruses by RNA and DNA sequencing has improved the understanding of the human virome. We sought to identify blood viral signatures through secondary use of RNA-sequencing (RNA-seq) data in a large study cohort. The ability to reveal undiagnosed infections with public health implications among study subjects with available sequencing data could enable epidemiologic surveys and may lead to diagnosis and therapeutic interventions, leveraging existing research data in a clinical context. We detected viral RNA in peripheral blood RNA-seq data from a COPD-enriched population of current and former smokers. Correlation between viral detection and both reported infections and relevant disease outcomes was evaluated. We identified Hepatitis C virus RNA in 228 subjects and HIV RNA in 30 subjects. Overall, we observed 31 viral species, including Epstein-Barr virus and Cytomegalovirus. We observed an enrichment of Hepatitis C and HIV infections among subjects reporting liver disease and HIV infections, respectively. Higher interferon expression scores were observed in the subjects with Hepatitis C and HIV infections. Through secondary use of RNA-seq from a cohort of current and former smokers, we detected peripheral blood viral signatures. We identified HIV and Hepatitis C virus (HCV), highlighting potential public health implications for the approach described this study. We observed correlations with reported infections, chronic infection outcomes and the host transcriptomic response, providing evidence to support the validity of the approach.
Similar content being viewed by others
Introduction
Studies of viruses in human tissues have helped to better understand the human virome and its role in human disease1,2. Through secondary use of human RNA and DNA sequencing to detect viruses, evidence has emerged regarding a healthy human blood virome3,4. In a study of the peripheral blood DNA virome in 8,240 individuals, Moustafa and colleagues3 repurposed human WGS data and were able to map the WGS data to sequences of 94 different viruses, including 19 human viruses, and observed differences in virus profiles across age, sex and ancestry3. Human Cytomegalovirus (CMV), HHV-6A, HHV-6B, Epstein–Barr virus (EBV) and Torque Teno Virus (TTV, Anellovirus) were among those detected. In a multi-tissue study repurposing RNA sequencing (RNA-seq) data by Kumata and colleagues4, EBV, CMV, TTV and human papillomavirus were among the viruses observed in blood.
We hypothesize that detection of viruses known to cause long-term latent infections within existing study populations would be of particular interest, as this could enable epidemiologic surveys and perhaps lead to diagnosis and therapeutic intervention for subjects from study populations of various diseases5. To assess the potential impact of this question in existing study populations, we sought to detect peripheral blood viral signatures by repurposing RNA-seq data in a population of current and former smokers with and without chronic obstructive pulmonary disease (COPD).
Results
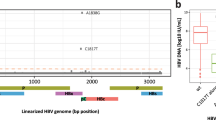
After quality control procedures, peripheral blood RNA-seq data were available for 3,984 samples, including 1601 COPD cases and 1609 controls (Table S1 in the online supplement). Approximately a third of subjects were current smokers and twenty-five percent were African American. Also included were 218 never-smokers. Using reads not mapped to the human genome during the gene expression analysis, we detected viral RNA in the blood RNA-seq data using PathSeq (see “Methods”). Focusing on viruses with public health implications, we detected Hepatitis C virus (HCV) RNA in 228 subjects and HIV RNA in 30 subjects, defined as the mapping of at least one read to the viral genome in at least two subjects. The relaxed threshold was implemented to reduce false negatives. In total we observed 31 viral species eclipsing the detection threshold, including Epstein-Barr virus and Cytomegalovirus (Table S2 in the online supplement).
HCV detection
A medical history of Hepatitis C infection was not obtained in COPDGene. However, 171 subjects with RNA-seq data had a self-reported history of liver disease at the 5-year follow-up and an additional 18 subjects reported liver disease at the 10-year follow-up. Of the 228 subjects with detectable HCV RNA, 77 were among the 189 total subjects with liver disease (Table 1), representing a significant enrichment (p < 0.0001) and suggesting high specificity. Although the 112 subjects self-reporting liver disease and lacking HCV RNA detection indicates the sensitivity may not be high, reduced HCV RNA loads would be observed in subjects undergoing treatment. Subjects with detected HCV RNA were younger with fewer comorbidities, and tended to be male, current smokers and African-American (Table 1). We did not observe a significant difference between the mapped read counts for the 77 subjects with self-reported liver disease compared with the remaining 151 (p value = 0.5; Mann–Whitney test).
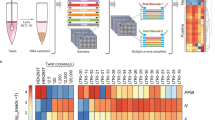
We created interferon scores using the RNA-seq gene expression data to observe correlation between detection of infections and host response (see “Methods”)6. We integrated the viral detection data with the scores for the interferon alpha and interferon gamma pathways to observe score values in the subjects with HCV detection compared with the subjects lacking viral detection. We observed higher expression scores for interferon alpha and interferon gamma pathways (p value < 0.0001; t-test) in the subjects with HCV RNA detection (Fig. 1). We also observed significantly (p value < 0.0001; t-test) lower levels of a published HCV down expression score and higher levels of an HCV up expression score in the subjects with HCV detection (Fig. S1 in the online supplement), suggesting correlation between RNA detection and HCV infections.

HIV detection
Among the subjects with RNA-seq data, 105 self-reported HIV infections from questionnaires at any COPDGene study visit. Of the 30 subjects with detectable HIV RNA, 22 were among the 105 subjects who self-reported an HIV infection (Table 1), representing a significant enrichment (p < 0.0001). The eight remaining subjects (Table 1) with detected HIV RNA could be either false-positive findings or undiagnosed individuals. The 83 subjects self-reporting HIV infections and not identified via RNA-seq analysis indicates the sensitivity may not be high, potentially due to therapies reducing HIV RNA loads. Although medication use data are available for 3820 of the 3984 subjects included in this study, of the 66 total subjects reporting use of medication to treat HIV 65 had also self-reported an HIV infection. The one additional subject treated with HIV medication was not among those with detectable HIV RNA. Subjects with detected HIV RNA tended to be younger, current smokers and African-American (Table 1). We did not observe a significant difference between the mapped read counts for the 22 subjects with self-reported infections compared with the remaining eight (p value = 0.7; Mann–Whitney test). We observed higher expression scores for interferon alpha and interferon gamma pathways (p value < 0.0001; t-test) in the subjects with HIV RNA detection (Fig. 1). We also observed significantly (p value < 0.0001; t-test) lower levels of a published HIV down expression score and higher levels of IFI27 in the subjects with HIV detection (Fig. S1 in the online supplement), suggesting correlation between RNA detection and HIV infections. Relevant to the temporal relationship, 15 of the 22 with detectable HIV RNA first self-reported infections at the 5-year follow-up (Table 1), the blood sample collection visit. HCV RNA was detected in 20 of the 105 subjects who self-reported HIV infections and was detected in seven of the 30 subjects with detectable HIV RNA. High rates of co-occurrence of HIV and HCV are consistent with known shared risk factors for these infections7.
Discussion
Through repurposing of blood RNA-seq data, we detected RNA of 31 viral species including HCV and HIV. The protocols used in the creation of the RNA-seq data in this study did not include polyA enrichment. A previous study demonstrated difficulty detecting viral RNA in polyA enriched RNA-seq data compared with non-polyA enriched RNA-seq data8. Although Kumata and colleagues4 detected some of the top viruses identified in this study, including EBV and CMV, their exclusion of HIV based on mapping specificity concerns and use of polyA enriched data from the Genotype-Tissue Expression (GTEx) project9 prevents a more complete comparison of findings, particularly with respect to HCV. The study of human viruses by Moustafa and colleagues3 involved the use of whole-genome DNA sequencing. Although we detected viral gene expression mRNA from infections caused by some DNA viruses identified in that study, such as EBV, CMV and TTV, the RNA from HCV and HIV infections would not be represented in their results to allow a direct comparison.
We observed significant enrichment of subjects with reported liver disease among those with detectable HCV RNA. We also observed a significant enrichment of subjects self-reporting HIV infections among those with detectable HIV RNA. These findings along with the elevated interferon pathway score in those with detectable RNA suggest we have identified HCV and HIV infections and a signature of the host response, given the role of interferons in response to HCV10,11 and HIV infections12,13. The concordance of virus detection and levels of the HCV and HIV host response scores from published transcriptomic studies also suggests we have observed chronic viral infections.
From 2013 to 2016, approximately 2.4 million people in the United States were living with a HCV infection14 and between 10 and 20% of HCV infections will lead to cirrhosis over 20–30 years15 with an increased risk of hepatocellular carcinoma. Direct-acting antiviral (DAA) therapy has been highly effective over the last decade14, highlighting the importance and benefits of HCV detection and diagnosis. In 2019, approximately 1 million people were living an HIV infection16. However, approximately 13% were not yet diagnosed16, highlighting the potential impact of HIV detection for both population surveillance and individual counseling within existing and future research study populations.
Limitations of this study include the lack of information regarding HCV infections, though an ongoing effort to return results to study subjects will provide clinical test results to allow further evaluation of the sensitivity and specificity of the RNA-seq approach. Clinically determined HIV-negatives were also lacking and prevented a full evaluation of HIV RNA detection accuracy. We also lack a validation cohort with infection-related questionnaire data. Although we are focused on secondary use of existing sequencing data, if these data do not exist for a particular study population, metagenomic approaches are available to provide more optimized viral detection17. The lack of control samples in this study would also be addressed in a properly designed metagenomic experiment. Although this study does not involve clinical diagnostic methods, we believe the public health implications of the viruses detected justifies the use of our approach, as it leverages existing data to enable targeted clinical interventions for study participants.
This study is the first to identify RNA signatures of HCV and HIV in the peripheral blood of a COPD-enriched population of current and former smokers through secondary use of RNA-seq data. In this study we were able to identify signatures of viral species with pathogenic and public health implications and observed correlations with reported infections, chronic infection outcomes and host transcriptomic response. Secondary mining of existing sequencing data in disease study populations using bioinformatic approaches may reveal chronic viral infections for cost-efficient epidemiologic studies and lead to additional clinical validation and therapeutic interventions where necessary.
Methods
Study subjects
The Genetic Epidemiology of COPD (COPDGene) study is a longitudinal cohort study that includes more than 10,000 non-Hispanic White and African American subjects enrolled at 21 centers across the United States18. The COPDGene cohort includes primarily current and former cigarette smokers. The five-year follow-up visit included questionnaires, spirometry, chest computed tomography scans, and collection of blood for complete blood cell counts and RNA sequencing. Subjects were at least one month removed from an acute respiratory infection. The RNA-sequencing and data processing methods were reported previously6,19. Briefly, paired end reads with nominal 75 bp length were generated on an Illumina HiSeq 2500 flow cell. Sequencing was performed to an average depth of 20 million reads. For the human gene expression analyses, STAR aligner20 was used to map the reads to GRCH38 and RSubreads produced gene-level counts21 with Ensembl gene annotation22. We confirmed concordance between sex-specific expression features and reported sex, and concordance between variants called from RNA sequencing reads and corresponding DNA genotyping.
Viral RNA detection
Using reads that were not mapped to the human genome during the gene expression analysis, we detected viral signatures in the COPDGene 5-year follow-up whole blood RNA-seq data using PathSeq, the microbial detection pipeline from the Genome Analysis Toolkit (GATK4)23, as described in a previous microbiome study6. Filtering of the unmapped reads was performed using PathSeq and the host reference in the GATK Resource Bundle23. The filtering step helps to addresses quality, host contamination or repetitive sequence issues. We then mapped the cleaned read data to viral reference genomes using PathSeq. The viral genomic reference was created using representative genomes (12,148 genomic entries) from the National Center for Biotechnology Information (NCBI). Taxonomy information for the viral genomic data was also obtained from NCBI (RefSeq-release95.catalog.gz). PathSeq output included the mapped read counts for each sample and viral species.
Transcriptomic scores
To observe transcriptomic signatures of the host response to the detected infections, we projected the RNA-seq gene expression data onto the Hallmark pathway gene set collection from MSigDB24 using gene set variation analysis via the R package GSVA25, as previously described6. We focused on the Hallmark interferon alpha and interferon gamma pathway gene sets. This method creates a composite transcriptomic expression score for the set of genes within each of the pathways. We also created scores using genes observed to be differentially expressed in the blood of HCV and HIV infected individuals. In a study of peripheral blood mononuclear cells (PBMCs), Bolen and colleagues26 identified 109 genes differentially expressed in HCV patients relative to uninfected controls. Using GSVA and the 56 genes down-expressed in HCV and 53 genes up-expressed, we created HCV down expression and up expression scores. For HIV, we used the set of 10 genes identified by Ockenhouse and colleagues27, observed to be down expressed in the PBMCs of HIV seropositive individuals and differentiating seropositive from seronegative, to create an HIV down expression score. Previous studies showed higher expression of the gene IFI27 in the peripheral blood of HIV infected individuals compared with seronegative controls28,29; we also analyzed expression of this gene to provide evidence of HIV infections. For this analysis, RNA-seq data were retained for genes with variance in the upper 80th percentile and for genes with average read counts greater than five. GSVA output provides expression pathway scores for each subject and gene set.
Ethics statement
All subjects in this study provided written consent for study procedures, including genetic analysis. COPDGene was approved by the Institutional Review Boards at all participating centers. The research methods were carried out in accordance with the relevant guidelines.
Ethics approval and consent to participate
All subjects in this study provided written informed consent. COPDGene was approved by the Institutional Review Boards at all participating centers.
Clinical center | Institution title | Protocol number |
|---|---|---|
National Jewish Health | National Jewish IRB | HS-1883a |
Brigham and Women’s Hospital | Partners Human Research Committee | 2007-P-000554/2; BWH |
Baylor College of Medicine | Institutional Review Board for Baylor College of Medicine and Affiliated Hospitals | H-22209 |
Michael E. DeBakey VAMC | Institutional Review Board for Baylor College of Medicine and Affiliated Hospitals | H-22202 |
Columbia University Medical Center | Columbia University Medical Center IRB | IRB-AAAC9324 |
Duke University Medical Center | The Duke University Health System Institutional Review Board for Clinical Investigations (DUHS IRB) | Pro00004464 |
Johns Hopkins University | Johns Hopkins Medicine Institutional Review Boards (JHM IRB) | NA_00011524 |
Los Angeles Biomedical Research Institute | The John F. Wolf, MD Human Subjects Committee of Harbor-UCLA Medical Center | 12756-01 |
Morehouse School of Medicine | Morehouse School of Medicine Institutional Review Board | 07-1029 |
Temple University | Temple University Office for Human Subjects Protections Institutional Review Board | 11369 |
University of Alabama at Birmingham | The University of Alabama at Birmingham Institutional Review Board for Human Use | FO70712014 |
University of California, San Diego | University of California, San Diego Human Research Protections Program | 070876 |
University of Iowa | The University of Iowa Human Subjects Office | 200710717 |
Ann Arbor VA | VA Ann Arbor Healthcare System IRB | PCC 2008-110732 |
University of Minnesota | University of Minnesota Research Subjects’ Protection Programs (RSPP) | 0801M24949 |
University of Pittsburgh | University of Pittsburgh Institutional Review Board | PRO07120059 |
University of Texas Health Sciences Center at San Antonio | UT Health Science Center San Antonio Institutional Review Board | HSC20070644H |
Health Partners Research Foundation | Health Partners Research Foundation Institutional Review Board | 07-127 |
University of Michigan | Medical School Institutional Review Board (IRBMED) | HUM00014973 |
Minneapolis VA Medical Center | Minneapolis VAMC IRB | 4128-A |
Reliant Medical Group | Institutional Review Board/Research Review Committee Saint Vincent Hospital – Reliant Medical Group – Fallon Community Health Plan | 1143 |
Data availability
Phenotype and the RNA sequencing data are available in dbGaP, accessions phs000179 and phs000765. https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000179.v6.p2. https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000765.v3.p2.
Abbreviations
- COPD:
-
Chronic obstructive pulmonary disease
- COPDGene:
-
Genetic epidemiology of COPD
- GATK:
-
Genome analysis toolkit
- GOLD:
-
Global initiative for chronic obstructive lung disease
- GSVA:
-
Gene set variation analysis
- HIV:
-
Human immunodeficiency virus
- HCV:
-
Hepatitis C virus
- MSigDB:
-
Molecular signatures database
- NCBI:
-
National center for biotechnology information
References
Rascovan, N., Duraisamy, R. & Desnues, C. Metagenomics and the human virome in asymptomatic individuals. Annu. Rev. Microbiol. 70, 125–141 (2016).
Cadwell, K. The virome in host health and disease. Immunity 42, 805–813 (2015).
Moustafa, A. et al. The blood DNA virome in 8000 humans. PLOS Pathog. 13, e1006292 (2017).
Kumata, R., Ito, J., Takahashi, K., Suzuki, T. & Sato, K. A tissue level atlas of the healthy human virome. BMC Biol. 18, 55 (2020).
Magiorkinis, G. et al. Potential for diagnosis of infectious disease from the 100,000 genomes project metagenomic dataset: Recommendations for reporting results. Wellcome Open Res. 4, 155 (2019).
Morrow, J. D. et al. Peripheral blood microbial signatures in current and former smokers. Sci. Rep. 11, 19875 (2021).
Kim, A. Y., Onofrey, S. & Church, D. R. An epidemiologic update on hepatitis C infection in persons living with or at risk of HIV infection. J. Infect. Dis. 207, S1–S6 (2013).
Yin, Q. et al. Assessment of viral RNA in idiopathic pulmonary fibrosis using RNA-seq. BMC Pulm. Med. 20, 81 (2020).
Lonsdale, J. et al. The genotype-tissue expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Mihm, S. et al. Interferon type I gene expression in chronic hepatitis C. Lab. Invest. 84, 1148–1159 (2004).
Napoli, J., Bishop, G. A., McGuinness, P. H., Painter, D. M. & McCaughan, G. W. Progressive liver injury in chronic hepatitis C infection correlates with increased intrahepatic expression of Th1-associated cytokines. Hepatology 24, 759–765 (1996).
Roff, S. R., Noon-Song, E. N. & Yamamoto, J. K. The significance of interferon-γ in HIV-1 pathogenesis, therapy, and prophylaxis. Front. Immunol. 4, 498 (2014).
Utay, N. S. & Douek, D. C. Interferons and HIV Infection: The good, the bad, and the ugly. Pathog. Immun. 1, 107–116 (2016).
Hofmeister, M. G. et al. Estimating prevalence of hepatitis C virus infection in the United States, 2013–2016. Hepatol. Baltim. Md 69, 1020–1031 (2019).
Westbrook, R. H. & Dusheiko, G. Natural history of hepatitis C. J. Hepatol. 61, S58–S68 (2014).
Centers for Disease Control and Prevention. HIV Surveillance Report, 2019. vol. 32 http://www.cdc.gov/hiv/library/reports/hiv-surveillance.html (2021).
Chiu, C. Y. & Miller, S. A. Clinical metagenomics. Nat. Rev. Genet. 20, 341–355 (2019).
Regan, E. A. et al. Genetic epidemiology of COPD (COPDGene) study design. COPD J. Chronic Obstr. Pulm. Dis. 7, 32–43 (2011).
Parker, M. M. et al. RNA sequencing identifies novel non-coding RNA and exon-specific effects associated with cigarette smoking. BMC Med. Genomics 10, 58 (2017).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. The subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucl. Acids Res. 41, e108–e108 (2013).
Kersey, P. J. et al. Ensembl genomes 2016: More genomes, more complexity. Nucl. Acids Res. 44, D574–D580 (2016).
Walker, M. A. et al. GATK PathSeq: A customizable computational tool for the discovery and identification of microbial sequences in libraries from eukaryotic hosts. Bioinformatics 34, 4287–4289 (2018).
Liberzon, A. et al. The molecular signatures database hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinf. 14, 7 (2013).
Bolen, C. R. et al. The Blood Transcriptional Signature of Chronic Hepatitis C Virus Is Consistent with an Ongoing Interferon-Mediated Antiviral Response. J. Interferon Cytokine Res. 33, 15–23 (2013).
Ockenhouse, C. F., Bernstein, W. B., Wang, Z. & Vahey, M. T. Functional genomic relationships in HIV-1 disease revealed by gene-expression profiling of primary human peripheral blood mononuclear cells. J. Infect. Dis. 191, 2064–2074 (2005).
Palm, A. A. et al. Interferon alpha-inducible protein 27 expression is linked to disease severity in chronic infection of both HIV-1 and HIV-2. Front. Virol. 2, https://doi.org/10.3389/fviro.2022.929053 (2022).
Huang, H. et al. IFI27 is a potential therapeutic target for HIV infection. Ann. Med. 54, 314–325 (2022).
Putcha, N. et al. A simplified score to quantify comorbidity in COPD. PLoS ONE 9, e114438 (2014).
Acknowledgements
COPDGene Phase 3: Grant Support and Disclaimer: The project described was supported by Award Number U01 HL089897 and Award Number U01 HL089856 from the National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health. COPD Foundation Funding: COPDGene is also supported by the COPD Foundation through contributions made to an Industry Advisory Board that has included AstraZeneca, Bayer Pharmaceuticals, Boehringer-Ingelheim, Genentech, GlaxoSmithKline, Novartis, Pfizer, and Sunovion. COPDGene® Investigators – Core Units: Administrative Center: James D. Crapo, MD (PI); Edwin K. Silverman, MD, PhD (PI); Barry J. Make, MD; Elizabeth A. Regan, MD, PhD. Genetic Analysis Center: Terri H. Beaty, PhD; Peter J. Castaldi, MD, MSc; Michael H. Cho, MD, MPH; Dawn L. DeMeo, MD, MPH; Adel El Boueiz, MD, MMSc; Marilyn G. Foreman, MD, MS; Auyon Ghosh, MD; Lystra P. Hayden, MD, MMSc; Craig P. Hersh, MD, MPH; Jacqueline Hetmanski, MS; Brian D. Hobbs, MD, MMSc; John E. Hokanson, MPH, PhD; Wonji Kim, PhD; Nan Laird, PhD; Christoph Lange, PhD; Sharon M. Lutz, PhD; Merry-Lynn McDonald, PhD; Dmitry Prokopenko, PhD; Matthew Moll, MD, MPH; Jarrett Morrow, PhD; Dandi Qiao, PhD; Elizabeth A. Regan, MD, PhD; Aabida Saferali, PhD; Phuwanat Sakornsakolpat, MD; Edwin K. Silverman, MD, PhD; Emily S. Wan, MD; Jeong Yun, MD, MPH. Imaging Center: Juan Pablo Centeno; Jean-Paul Charbonnier, PhD; Harvey O. Coxson, PhD; Craig J. Galban, PhD; MeiLan K. Han, MD, MS; Eric A. Hoffman, Stephen Humphries, PhD; Francine L. Jacobson, MD, MPH; Philip F. Judy, PhD; Ella A. Kazerooni, MD; Alex Kluiber; David A. Lynch, MB; Pietro Nardelli, PhD; John D. Newell, Jr., MD; Aleena Notary; Andrea Oh, MD; Elizabeth A. Regan, MD, PhD; James C. Ross, PhD; Raul San Jose Estepar, PhD; Joyce Schroeder, MD; Jered Sieren; Berend C. Stoel, PhD; Juerg Tschirren, PhD; Edwin Van Beek, MD, PhD; Bram van Ginneken, PhD; Eva van Rikxoort, PhD; Gonzalo Vegas Sanchez-Ferrero, PhD; Lucas Veitel; George R. Washko, MD; Carla G. Wilson, MS; PFT QA Center, Salt Lake City, UT: Robert Jensen, PhD. Data Coordinating Center and Biostatistics, National Jewish Health, Denver, CO: Douglas Everett, PhD; Jim Crooks, PhD; Katherine Pratte, PhD; Matt Strand, PhD; Carla G. Wilson, MS. Epidemiology Core, University of Colorado Anschutz Medical Campus, Aurora, CO: John E. Hokanson, MPH, PhD; Erin Austin, PhD; Gregory Kinney, MPH, PhD; Sharon M. Lutz, PhD; Kendra A. Young, PhD. Mortality Adjudication Core: Surya P. Bhatt, MD; Jessica Bon, MD; Alejandro A. Diaz, MD, MPH; MeiLan K. Han, MD, MS; Barry Make, MD; Susan Murray, ScD; Elizabeth Regan, MD; Xavier Soler, MD; Carla G. Wilson, MS. Biomarker Core: Russell P. Bowler, MD, PhD; Katerina Kechris, PhD; Farnoush Banaei-Kashani, PhD. COPDGene® Investigators – Clinical Centers: Ann Arbor VA: Jeffrey L. Curtis, MD; Perry G. Pernicano, MD. Baylor College of Medicine, Houston, TX: Nicola Hanania, MD, MS; Mustafa Atik, MD; Aladin Boriek, PhD; Kalpatha Guntupalli, MD; Elizabeth Guy, MD; Amit Parulekar, MD; Brigham and Women’s Hospital, Boston, MA: Dawn L. DeMeo, MD, MPH; Craig Hersh, MD, MPH; Francine L. Jacobson, MD, MPH; George Washko, MD. Columbia University, New York, NY: R. Graham Barr, MD, DrPH; John Austin, MD; Belinda D’Souza, MD; Byron Thomashow, MD. Duke University Medical Center, Durham, NC: Neil MacIntyre, Jr., MD; H. Page McAdams, MD; Lacey Washington, MD. HealthPartners Research Institute, Minneapolis, MN: Charlene McEvoy, MD, MPH; Joseph Tashjian, MD. Johns Hopkins University, Baltimore, MD: Robert Wise, MD; Robert Brown, MD; Nadia N. Hansel, MD, MPH; Karen Horton, MD; Allison Lambert, MD, MHS; Nirupama Putcha, MD, MHS. Lundquist Institute for Biomedical Innovationat Harbor UCLA Medical Center, Torrance, CA: Richard Casaburi, PhD, MD; Alessandra Adami, PhD; Matthew Budoff, MD; Hans Fischer, MD; Janos Porszasz, MD, PhD; Harry Rossiter, PhD; William Stringer, MD. Michael E. DeBakey VAMC, Houston, TX: Amir Sharafkhaneh, MD, PhD; Charlie Lan, DO. Minneapolis VA: Christine Wendt, MD; Brian Bell, MD; Ken M. Kunisaki, MD, MS. Morehouse School of Medicine, Atlanta, GA: Eric L. Flenaugh, MD; Hirut Gebrekristos, PhD; Mario Ponce, MD; Silanath Terpenning, MD; Gloria Westney, MD, MS. National Jewish Health, Denver, CO: Russell Bowler, MD, PhD; David A. Lynch, MB. Reliant Medical Group, Worcester, MA: Richard Rosiello, MD; David Pace, MD. Temple University, Philadelphia, PA: Gerard Criner, MD; David Ciccolella, MD; Francis Cordova, MD; Chandra Dass, MD; Gilbert D’Alonzo, DO; Parag Desai, MD; Michael Jacobs, PharmD; Steven Kelsen, MD, PhD; Victor Kim, MD; A. James Mamary, MD; Nathaniel Marchetti, DO; Aditi Satti, MD; Kartik Shenoy, MD; Robert M. Steiner, MD; Alex Swift, MD; Irene Swift, MD; Maria Elena Vega-Sanchez, MD. University of Alabama, Birmingham, AL: Mark Dransfield, MD; William Bailey, MD; Surya P. Bhatt, MD; Anand Iyer, MD; Hrudaya Nath, MD; J. Michael Wells, MD. University of California, San Diego, CA: Douglas Conrad, MD; Xavier Soler, MD, PhD; Andrew Yen, MD. University of Iowa, Iowa City, IA: Alejandro P. Comellas, MD; Karin F. Hoth, PhD; John Newell, Jr., MD; Brad Thompson, MD. University of Michigan, Ann Arbor, MI: MeiLan K. Han, MD MS; Ella Kazerooni, MD MS; Wassim Labaki, MD MS; Craig Galban, PhD; Dharshan Vummidi, MD. University of Minnesota, Minneapolis, MN: Joanne Billings, MD; Abbie Begnaud, MD; Tadashi Allen, MD. University of Pittsburgh, Pittsburgh, PA: Frank Sciurba, MD; Jessica Bon, MD; Divay Chandra, MD, MSc; Joel Weissfeld, MD, MPH. University of Texas Health, San Antonio, San Antonio, TX: Antonio Anzueto, MD; Sandra Adams, MD; Diego Maselli-Caceres, MD; Mario E. Ruiz, MD; Harjinder Singh.
Funding
Supported by NIH grants: K25 HL136846, R01HL130512, R01HL125583, U01HL089856, U01HL089897, R01HL124233, R01HL147326, and an Alpha-1 Foundation Research Grant. COPDGene is also supported by the COPD Foundation through contributions made to an Industry Advisory Board that has included AstraZeneca, Bayer Pharmaceuticals, Boehringer-Ingelheim, Genentech, GlaxoSmithKline, Novartis, Pfizer, and Sunovion.
Author information
Authors and Affiliations
Contributions
J.D.M.: analysis and interpretation of data, manuscript preparation and approval of the final version; P.J.C.: acquisition of data, review of manuscript and approval of the final version; R.P.C.: analysis of data, review of manuscript and approval of the final version; J.H.Y.: interpretation of data, review of manuscript and approval of the final version; G.L.K.: analysis of data, review of manuscript and approval of the final version; E.K.S.: acquisition of data, analysis and interpretation of data, manuscript preparation and approval of the final version; C.P.H.: acquisition of data, analysis and interpretation of data, manuscript preparation and approval of the final version.
Corresponding author
Ethics declarations
Competing interests
Dr. Castaldi has received consulting fees and grant support from GSK. Dr. Hersh has received grant support from Bayer, Boehringer-Ingelheim, Novartis and Vertex, and Consulting fees from Astra-Zeneca and Takeda. In the past three years, Edwin K. Silverman has received grant support from GSK and Bayer. All other authors declare that they have no competing interests related to this manuscript.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Morrow, J.D., Castaldi, P.J., Chase, R.P. et al. Hepatitis C and HIV detection by blood RNA-sequencing in cohort of smokers. Sci Rep 13, 1357 (2023). https://doi.org/10.1038/s41598-023-28156-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-28156-4
This article is cited by
-
Blood transcriptomics analysis offers insights into variant-specific immune response to SARS-CoV-2
Scientific Reports (2024)
-
Returning incidentally discovered Hepatitis C RNA-seq results to COPDGene study participants
npj Genomic Medicine (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
