
Abstract
In goat breeds, the domestication followed by artificial selection for economically important traits have shaped genetic variation within populations, leading to the fixation of specific alleles for specific traits. This led to the formation and evolution of many different breeds specialised and raised for a particular purpose. However, and despite the intensity of artificial selection, natural selection continues acting, possibly leaving a more diluted contribution over time, whose traces may be more difficult to capture. In order to explore selection footprints as response of environmental adaptation, we analysed a total of 993 goats from four transboundary goats breeds (Angora, Boer, Nubian and Saanen) genotyped with the SNP chip 50 K using outlier detection, runs of homozygosity and haplotype-based detection methods. Our results showed that all methods identified footprints on chromosome 6 (from 30 to 49 Mb) for two specific populations of Nubian goats sampled in Egypt. In Angora and Saanen breeds, we detected two selective sweeps using HapFLK, on chromosome 21 (from 52 to 55 Mb) and chromosome 25 (from 1 to 5 Mb) respectively. The analysis of runs of homozygosity showed some hotspots in all breeds. The overall investigation of the selected regions detected combining the different approaches and the gene ontology exploration revealed both novel and well-known loci related to adaptation, especially for heat stress. Our findings can help to better understand the balance between the two selective pressures in commercial goat breeds providing new insights on the molecular mechanisms of adaptation.
Similar content being viewed by others

Introduction
Capra hircus is one of the most important worldwide farmed species and its domestication dates back to the early Neolithic era (ca. 11,000 YBP) in the Fertile Crescent1. Goats have been selected during centuries for different traits (milk, meat, wool or leather) show high resistance to stress and have a great ability to adapt to various agro-climatic conditions2. This quick adaptation implies several changes in physiology, morphology, behaviour, phenotype, and at the basis of all, in genetics. To analyse these genetic changes and the footprints that they left on the genome, the genome-wide scanning technology using SNP Arrays is a powerful and efficient tool widely used3 since several decades. For large transboundary breeds, genomic information has been used to identify candidate regions for traits of commercial interest and for application in breeding (genomic selection), investigating mainly the effect of artificial selection on the genome. In small local breed, genomic information has been exploited to investigate the ability to respond to environmental changes and challenges. This subdivision is generally based on the assumption that strong selection pressure applied to commercial breeds lead to negative impacts on their ability to adapt in comparison with traditional breeds, due to stronger connection to their original environments4. The role of the natural selection in shaping the genetic architecture of the highly selected, transboundary breeds has not yet been investigated. Little is known about how transboundary breeds have adapted to a wide range of different environments and management conditions. Indeed, natural selection continues acting, possibly leaving smaller but detectable contributions. To investigate this issue, it is important to first accurately choose an ideal model which go through strong anthropogenic selection over centuries and also to transboundary transport. Different methodologies that can be applied to detect selection signatures5, generally based on the comparison of statistics on genotypes at intra-populations versus inter-populations level. Two main categories of statistics have been developed at (1) intra-population (2) and inter-populations level. The first one is based on site frequency spectrum (SFS), linkage disequilibrium (LD) and reduced local variability. The second one focuses on single site differentiation and haplotype-based differentiation. Each of these approaches includes associated statistics and specific bioinformatic tools.
Objective of the study was to reveal breed-specific selection signatures linked to environmental variables and thus to identify loci potentially relevant for adaptation in commercial breeds. Our results will contribute to advancing knowledge on climate-driven adaptive evolution and to better understand the molecular mechanisms involved in this process. Moreover, results may find application in selective breeding and conservation management programs.
Results
Publicly available genotypes of four commercial goat breeds were used to reveal breed-specific selection signature linked to environmental variable. Combining three different statistical methods, we detected several polymorphisms that revealed loci potentially affecting adaptation to agro-climatic conditions.
Population relationships, clustering and outlier variant detection
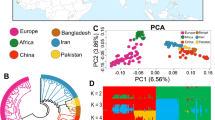
Figure 1 presents the PCA results where we can see that all populations clustered according to their geographic origin. The Nubian breed shows a clear separation not only between the two geographic areas (Argentina and Egypt) but also within the NBN_EGCH population (Fig. 1, A1) with the first two principal components that explain about 13% of genetic diversity (Fig. 1, A2). In Angora breed, all populations constitute well-defined clusters (Fig. 1, B1 and B2) except the Argentine population. Finally, in the Boer and Saanen breeds there are no well-defined clusters, even if it is possible to highlight a weak subdivision across them (Fig. 1, C1 and D1). The percentage of variance explained by the first two principal components indicates a low genetic diversity (Boer = 5% and Saanen = 8%, Fig. 1, C2 and D2).

Distribution of samples in relation to their scores on the first and second principal components obtained after principal component analysis for the four breeds. Each point represents a single individual and for each breed a different colour was assigned. The legend that explains the correspondence between breeds and colour is in the lower right corner. (A) In the Nubian breed, after separation due to a subpopulation from Egypt, the percentage of variance explained up to 13% (eigenvector 1, X-axis, 8% and eigenvector 2, Y-axis, 5%). (B) In the Angora breed, the two components explained 8.6% of total variation (eigenvector 1, X-axis, 5.1% of variation and eigenvector 2, Y-axis, 3.5%). (C) In the Boer breed, the total percentage of variance is 5.1% (eigenvector 1, X-axis, 3% and eigenvector 2, Y-axis, 2.1%). (D) In the Saanen breed, the variance was a total of 8% (eigenvector 1, X-axis, 4.6% and eigenvector 2, Y-axis, 3.4%).
The second step of the analysis with PCAdapt identified several polymorphisms as outliers and putative signs of local adaptation. The number of outliers as well as the corresponding chromosomes and the genes falling into the putative genomic region under selection are summarised in Table 1 for all breeds. We found a total of twelve outliers in Angora, and three in both Saanen and Boer breeds remaining after Bonferroni’s correction (Table 1, Fig. 2).

Circular Manhattan plot of outliers SNP detected with PCadapt analysis. One different colour is assigned to each breed: coral for Saanen, purple for Nubian, green for Boer and blue for Angora. The red dashed line indicates the threshold of significance of 0.05%. Every point is a SNP and with amplified the significant ones.
In the Nubian breed we observed the highest number of putative markers under selection even after the Bonferroni correction (Supplementary Table 2 and Fig. 3). Since we found a strong overlap across three different analyses of a large, potential genomic region under selection in the CH6, we decided to re-analyse the Nubian dataset excluding this chromosome. After comparisons with the modified analysis, the number of outliers confirmed by both analysis with PCAdapt in Nubian breed was eight (into the CH2, CH3, CH4 and CH5, Table 2).

Frequency of Runs of Homozygosity for each class of length. Histograms are built with different colours for each breed (A = Nubian, B = Angora, C = Boer and D = Saanen) and every population is indicated with a different shade of the same colour.
Supplementary Figs. 1–4 show the Admixture analysis and the Supplementary Fig. 5 the cross-validation errors for each breed. These results agreed with the PCA and, together with the pairwise FST values, are in agreement with1 (Supplementary Table 1). In particular, both Admixture and FST confirmed the quite strong genetic structure found in the Nubian breed (Supplementary Table 1 and Supplementary Fig. 3).
Runs of Homozygosity
The Supplementary Figs. 7, 8, 9 and 10 have the Manhattan plots for the four breeds and for each population. In the Nubian breed, based on estimation of the genomic inbreeding coefficient (FROH), it is evident that both populations from Egypt had a higher level of inbreeding compared with the population from Argentina (0.13 and 0.14 for NBN_EGCH and NBN_EGCH1, and 0.11 for NBN_ARCH, Supplementary Table 3). Looking at the distribution of ROHs per class (Fig. 3A) the NBN_EGCH group revealed a higher amount of longer ROH (8–16 and > 16 Mb) compared to NBN_EGCH1 (4–8 Mb) and NBN_ARCH (2–4 Mb).
The Nubian populations from Egypt showed a high incidence of variants in ROH creating a peak on CH6, with a percentage of variants that overcomes 75% (Supplementary Fig. 9). The NBN_EGCH also showed another remarkable peak on CH25 and NBN_EGCH1 on CH18. We found similar patterns of homozygosity considering the FROH (Fig. 4A) and the percentage of ROH per chromosome (Fig. 5A).

Distribution of genomic inbreeding coefficient (FROH) or ROH-based inbreeding per chromosome and for each breed. Every bar represents a chromosome and a different colour is associated with a population for every breed. A = Nubian, B = Angora, C = Boer and D = Saanen.

Percentage of Runs Of Homozygosity. The bars exhibit the frequency of ROH per chromosome identified in every population for the four breeds. A different colour is assigned to each population within every breed. A = Nubian, B = Angora, C = Boer and D = Saanen.
Finally, we observed the presence of several hotspots of homozygosity (ROH islands) occurring mainly in both Egyptian populations but also one in the NBN_ARCH population (Fig. 6, Supplementary Table 4).

Graphical representation of the proportion of the genome covered by ROH Islands. One different colour is assigned to each breed and the order of breeds is based on the abundance of ROH Islands for a better visualization. From the most external to the centre: Boer (light green), Angora (light blue), Saanen (light orange) and Nubian (light pink).
In the Angora breed, both African populations showed the same pattern of homozygosity, where the sum and the mean of ROH for each individual were relatively high (Supplementary Figs. 11 and 12). On the contrary, the population from Argentina had very short ROH whereas the population from France displayed an intermediate situation. Interestingly, the France population exhibited the highest FROH value on CH21 in respect to the rest of populations (Fig. 4B) and ANG_ZACH had the highest value of genomic inbreeding coefficient (FROH = 0.22, Supplementary Table 3 and Fig. 4B). The analysis by classes of length revealed an interesting result for ANG_ARCH and ANG_ZACH populations which show several homozygous segments within the 4–8 and 8–16 Mb classes (Fig. 3B). Only the ANG_MGCH population had some hotspots characterised by homozygous segments of about 2 Mb (Fig. 6 and Supplementary Table 3). The percentage of ROH per chromosome (Fig. 5B) presented a similar pattern for all Angora populations. In the Boer breed, the genomic inbreeding coefficient ranged from 0.22 to 0.1 (Supplementary Table 3) where BOE_AUCH, BOE_NZCH, BOE_CHCH and BOE_USCH had the highest values. These results were confirmed when we look at the sum and mean of ROH (Supplementary Figs. 11 and 12) and the distribution per length class, where it is evident that several segments exceed the 16 Mb in length especially in the BOE_CHCH population (Fig. 3C). In general, similar patterns of homozygosity are found in all Boer populations considering the FROH (Fig. 4C) and the percentage of ROH per chromosome (Fig. 5C). In the Saanen breed, the populations from Switzerland, Tanzania and Argentina showed the highest value of FROH, ranging from 0.12 to 0.14 (Supplementary Table 3) and in general we found few ROH in all groups (Supplementary Table 4, Fig. 6) and only few hotspots in SAA_KECH population (Supplementary Table 4). The distribution by length class showed several segments < 2 Mb in the SAA_ARCH, SAA_CHCH and SAA_FRCH populations, while segments that exceed 16 MB are found in the SAA_TZCH population (Fig. 3D). A similar pattern of homozygosity considering the FROH (Fig. 4D) was found within all Saanen populations, however SAA_TZCH had the highest value of genomic inbreeding coefficient (FROH > 0.2) on CH23, CH24, CH25 and CH28. Regarding the percentage of ROH per chromosome (Fig. 5D), SAA_KECH showed a different pattern compared with the other Saanen populations. Another interesting finding is related to the abundance of hotspots present in the genome of Boer populations. In particular, we found the same genomic region in the CH6, ranging from 85 to 86 Mb in all groups and an additional region ranging from 80 to 82 Mb was absent only in BOE_UGCH and BOE_ZWCH. Furthermore, we discovered several long ROH islands in other chromosomes shared by some populations or exclusive of a particular population that were not discovered in previous studies6 (Fig. 6 and Supplementary Table 4).
For instance, a ROH ranging from 21 to 25 Mb on CH13 is shared only by the BOE_NZCH and BOE_CHCH populations, and another ROH of 12 Mb on the same chromosome is exclusive to the BOE_AUCH population. This stretched segment was found to partially overlap several shorter segments found in all population excluding the African ones. Other regions on CH3, CH7 and CH8 (Supplementary Table 4 and Fig. 6) were shared or partially overlapped in BOE_AUCH, BOE_CHCH, BOE_USCH and BOE_NZCH, and some segments are partially overlapped with ROH islands found in a previous study6.
Selective sweeps with HapFLK
The HapFLK analysis detected two significant selective sweeps. The first one was a region of about 5 Mb mapping on CH25 and ranging from 1 and 5 Mb, in the Saanen breed (FDR < 0.01), whereas the second one was of about 3 Mb, spanning between 52 and 55 Mb on CH21 in the Angora breed (FDR < 0.02). For the remaining two breeds, there were no significant regions after FDR correction (Fig. 7).

Circular Manhattan plot of selective sweeps detected with HapFLK. One different colour is the same to each breed: coral for Saanen, purple for Nubian, green for Boer and blue for Angora. The red dashed line indicates the threshold of significance of 0.05%. The grey dashed line indicates the two significant peaks in the CH25 (Saanen) and CH21 (Angora).
However, it is necessary to remark that, while some of the peaks did not achieve the statistical threshold of 0.05, some of them showed a co-localisation with selective sweeps identified in previous studies using the same populations6. The corresponding putative selective sweeps and the genes falling into these genomic regions are summarised in Table 2 for the Angora and Saanen breeds. The analysis revealed several novel and well-known genes that are associated to adaptation.
Comparing the results of the three programs used in this study, we can highlight only one overlapping genomic region presumably under selection on CH6 of Nubian breed. It is worth highlighting that in the Nubian breed there were sixteen outlier SNPs falling into the putative genes under selection. These gene were HSP40 on CH4, PTPRB on CH5, and ZCCHC4, PPARGC1A, LGI2, SEPSECS, IBSP, CCSER1 (5 SNP), LCORL (2 SNP) and KCNIP4 (2 SNP) on CH6. However, we found positional coincidences between one outlier SNP and the ROH island on CH3 of Boer breed, spanning from 79 to 95 Mb and shared by AUCH, NZCH, USCH and TZCH populations.
Candidate gene identification and functional analysis
Candidate genes within the two Mb intervals of the putative selected regions were retrieved with the Ensembl BioMart tool. The obtained lists were further analysed using relevant literature for verifying if there were genes associated with environmental adaptation (Table 4). In all four breeds, several loci are involved in metabolism and adipogenesis, as well as feed intake, immune response and growth or which expression is affected by the availability of food. More genes indirectly related to adaptation are discovered when we checked the two large hotspots common amongst all Boer populations and summarised in Table 3.
All biological process terms with P values < 0.1, including the number of genes assigned to these terms are summarized in Table 4. The extensive analysis of the identified GO terms revealed that the identified candidate genes have been associated with diverse biological functions, such the transmission of nervous signals and metabolic processes, all of them playing a role in basic functions of the organism probably in response to environmental pressure.
Discussion
Environmental factors are one of the forces influencing agricultural and the livestock sectors. Animals exposed to stressful environments exhibit various adaptive mechanisms, such as behavioural, physiological, endocrine, cellular, metabolic and biochemical for minimising the stressful conditions. Thus, adaptation is the natural strategy to ensure both welfare and efficiency. The action of selection leaves signs along the genome as responses to environmental and anthropogenic pressures that can be revealed using specific methodologies and bioinformatic tools. We applied three complementary methods for detecting a wider range of candidate genes that can be further investigated. The analysis with PCAdapt revealed, in Boer and Saanen breeds, a few markers potentially under selection for environment adaptation, whereas Angora and Nubian breeds showed several outliers distributed along different chromosomes.
Our findings led to the identification of target genes related to adaptation and more specifically to response at the temperature stress, energy homeostasis, photoperiod, immune/inflammatory response, reproductive and production traits. The responses to stress include not only reactions to extreme cold and hot temperatures but also the ability to adapt to harsh environmental conditions, such as poor-quality forage or water scarcity. In African indigenous chickens, the TOGARAM1locus, involved in the assembly of non-motile cilia and thus essential for cellular signal transduction, was found affected by heat-shock7. In fact, heat can induce a rapid loss of these important organelles8, deducing that this gene may play an important adaptive role in alleviating this effect in high temperature conditions. PDIA3 regulates cell growth and death according to oxygen concentrations and this gene was implicated in the thermal acclimatisation process in ovine liver tissue9, and in sperm–egg fusion in sheep and cashmere goats10. These genes were detected in Angora goat populations on CH13 near to the SNP outlier and within the selective sweep on CH21. The heat-shock protein 40 (HSP40) and the heat shock protein family A (HSP70) member 14 (HSPA14) belong to the heat-shock proteins (HSP) family, involved in cellular responses and for protein homeostasis and survival under stress conditions. In particular, HSP70 gene has been linked with heat tolerance and higher milk production in cattle11. These findings indicate the putative effects of selective pressure on this gene family favouring animals with better thermotolerance, performance and stress resilience12. TRPA1 is a member of the transient receptor potential (TRP) superfamily of ion channels. Studies in mice suggested that TRPA1 channels mediate cold temperature sensing in mammalian vagal sensory neurons, evoking major protective reflexes and thermoregulatory responses13. This gene was found in the putative selective sweep on the CH25 of Saanen goat: in this breed, populations are from different climatic areas (continental and temperate, following the Köppen–Geiger Climate Classification; Table 5), indicating a plausible association with thermal stress and cold adaptation. Another interesting candidate gene is TRHDE, a gene implicated in energy homeostasis, body temperature regulation14, in particular adaptation to hot arid environments in goats15 and high-altitude in Ethiopian sheep16.
In this study, we identified several genes that are involved in the lipid metabolism, adipogenesis and feed intake, directly or indirectly related to energy balance. CLVS2 participates in regulation of foetal development in cattle that underlie the effects of early maternal nutrient restriction17. Interestingly, if we compare the Nubian populations from Egypt and Argentina, we can observe a clear discrepancy on the resources available in terms of food and water, since Egypt is a country characterised by a hot and dry climate. The same observation applies to Angora, as the populations from Argentina and South Africa, sampled in an arid zone, shared a common result in the analysis of ROH, with a greater number of medium-large homozygote segments (4–8 Mb), and thus suggesting a certain degree of selection that is occurred not recently. The CCSER1 locus was previously associated with the feed efficiency in beef cattle18 and in sheep19. It is worth highlighting that this gene lies close to well-known genes associated to body size, growth and height and it falls within the large genomic region we identified in Nubian. Our findings are confirmed by previous studies that reported a strong positive selection around the ABCG2, SPP1, LAP3, NCAPG, LCORL, PKD2, IBSP, and MEPE genes in domestic goats and sheep19.
In all breeds, we also pinpointed genes under selection for altitude adaptation, like DCLRE1C, FANCM and PPP1R12A in the Angora population, MCPH1 and ANGPT2 in the Boer group, TRHDE and IBSP in the Nubian and TRAP1 (as discussed above), CEBPZ, HMOX2, NMRAL1 in Saanen. HMOX2, involved in hypoxia response and the neighbouring NMRAL1, involved in synthesis of nitric oxide, are thought to be contributors to adaptation to high altitude in humans20,21. Edea and co-workers22 observed PPP1R12A to be associated with high altitude adaptation in Ethiopian sheep, and previous studies have already demonstrated that hypoxia increased phosphorylation of this gene23.
One of the most important and predictable environmental variations is seasonality in temperate zones, based on photoperiodism over the year24. Two out of four breeds studied here (Angora and Saanen) showed several candidate genes linked to physiological adjustments driven by photoperiodism. For example, CLOCK is one of the most important genes that controls circadian rhythms by regulating various physiological functions including sleep, body temperature, blood pressure, endocrine, cardiovascular and immune systems25. The CLOCK gene also has an impact on energy metabolism influencing the rhythms of feeding behaviour26. In the Angora breed, we found the KDR gene that is related to coat colour, and that falls into the same genomic segment that contains other genes like SRD5A3, TMEM165, PDCL2, EXOC1L, CEP135, SCFD2, FIP1L1, LNX1, PDGFRA, CLOCK, NMU and EXOC1 found under selection in Reggiana cattle27.
Stress can affect the immune system by inducing alteration of inflammatory processes and the animal’s inflammatory response is a survival mechanism to cope with pathogenic or non-pathogenic challenges28. Oxidative stress is considered an imbalance between oxidant and antioxidant status and considered one of the key factors causing the weakening of immune system in animals that have undergone heat stress. Macrophages and neutrophils play an important role in innate immunity by producing nitric oxide. NMRAL1, a candidate gene in Saanen breed, is related to the synthesis of nitric oxide and maybe could play a role in the activation of inflammatory processes. Several studies suggest that exposure to heat results in oxidative stress, thus promoting cytotoxicity29 and cellular damage30. It is remarkable how we detected, again in Saanen breed and as previously mentioned, the gene HMOX2 that is involved in the antioxidant response like its homologous HMOX1 gene that has been reported to play a role in "tissue tolerance"—the ability to resist pathogens, inflammation, or oxidative stress-mediated damage during infection or inflammation in humans31. This intersection amongst oxidative imbalance, immune, and physiological responses has been already described in sheep28.
In the selective sweep of CH21 (Angora breed), we found several interesting genes like FKBP and LRFN5. LRFN5 is involved in immune system in cattle32. It is worth noting that this gene maps inside a QTL region identified in sheep and involved in scrapie infection, a disease of the nervous system33. In the same genomic region of CH21, we also found MAP1A, that allows the maintenance and restructuring of adult neurons34 and maps inside a QTL affecting classical scrapie incubation time in a population of scrapie-infected35.
The thermal environment is the largest single stressor affecting the efficiency of animal production systems. Some evidence from field studies in sheep36 highlighted that the physiological and behavioural adaptations that allow animals to maintain homeothermy, negatively impact their growth, welfare and reproduction. Therefore, it is not surprising that our analysis revealed within the selected regions several genes that correlated to reproduction traits like fertility and productive performances, including growth and development. The expression of NR3C1 was explored in the ovine uterus37,38 discovering the crucial role of endometrial functions during early pregnancy in sheep. The effects of environmental stressors are also evident in male reproductive performances. Testicular thermoregulation is imperative to produce healthy viable spermatozoa39. We found in our study SCAPER, SEPTIN12, RODGI, selected in the Saanen group whereas HORMAD1, TDRKH, CELF3 and OAZ3 in the Nubian group, all genes related to spermatogenesis and fertility in mice40 and humans41.
It has been reported that a reduction in wool fiber diameter is a consequence to deteriorating food quality and availability42. Based on our results, we observed selection signatures in the FGF1 gene, a member of the fibroblast growth factor family and involved in the growth and development of various tissues and organs. FGF1 was also the target gene of a miRNA that had an effect on growth and development of hair follicles in sheep43.
Our results showed several novel and established genes that are correlated with milk, meat and growth traits (development, body size and height). Amongst them, the most important were retrieved in the selective sweep on the CH6 of Nubian populations, that includes CCSER1, LAP3, MED28, FAM184B, DCAF16, NCAPG, LCORL, SLIT2, PACRGL, KCNIP4, PPARGC1A, these loci were described in cattle44, sheep19,45 and goats46.
The large hotspot retrieved on CH6 and shared by all populations investigated, contained genes associated to reproduction and immune resistance. GnRHR regulates the production of gametes and gonadal hormones and it is important for reproduction control in buffalo, cattle and goats47,48,49. Interestingly, in a review investigating the evolution of GnRHR family genes and its receptors, the following genes surrounding the mammalian GnRHR1 (STAP1, UBA6, GnRHR, TMPRSS11D, TMPRSS11A, TMPRSS11F, TMPRSS11E and YTHDC1) and retrieved in our analysis, are conserved in human, mouse and other vertebrates50 suggesting that they can affect the same trait.
In the Gene Ontology (GO) analysis, all biological processes are related with neurological functions and the nervous system in Angora breed. The functional annotation exacerbates neurological pathways involving behavioural acetylcholine-mediated responses. Acetylcholine (ACh) is the neurotransmitter used for muscular activation and all biological processes converge to cholinergic transmission. These chemical signals act on regulation of smooth muscle contraction (GO:0006940) and as a component of presynaptic (GO:0042734) and postsynaptic (GO:0045211) membranes. In fact, the key genes in these pathways are CHRNA3, CHRNB4, CHRNA5, which are nicotinic acetylcholine receptors. Researchers demonstrated that mild hypoxia decreased ACh synthesis and the amino acid metabolism51. In this breed, we found genes related to hypoxia and in general to adaptation to harsh environments, thus suggesting that the nervous system regulates many processes that can affect the efficiency in maintaining homeostasis. Three out of four populations included in the Angora dataset were sampled in arid cold and with desert or steppe (Argentina and South Africa) and hot (Madagascar) environments– thus exposed to extreme conditions, whereas the French population is the only one that comes from a temperate climate.
The Boer, Saanen and Nubian groups shared a GO associated with retinoic acid activity pathway. Retinol is vitamin A, a fat-soluble compound that is required for vision, cellular proliferation and differentiation. Studies in cattle demonstrated that it regulates intramuscular adipose tissue and muscle development52. Retinol metabolism pathway is also involved in feed efficiency in livestock53 and in normal immunologic function54. In the Boer breed, there is another interesting GO regarding angiogenesis. As we discussed above, angiogenesis is involved in some high-altitude adaptation responses.
Boer populations belonging to this dataset come from many different climatic zones, with a wide range of environmental variations; for example, the population from Switzerland originated from a sample site with continental climate that exposed individuals to different temperatures in winter and summer, whereas the population from Australia is exposed to hot weather. Intriguingly WNT2B, that is a potential target gene in wool follicle development, showed a footprint of selection in this breed that is not farmed for this purpose, suggesting that it could be related to the local adaptation of some populations to the temperature regime. If additional studies would verify that a selective pressure is acting on this locus in this breed, it could be a further confirmation that natural selection continues affecting and leaving detectable traces.
In Saanen populations, the GO revealed also photoreceptor outer segment/photoreceptor cell maintenance processes that together with retinol acid activity pathway can lead us to hypothesize that some part of the genome is triggering mechanisms to the protection/maintenance of cells belonging to the visual system, and maybe adapting it to a new and variable condition of light. Moreover, in this breed, we see several genes related to DNA repair and oxidative stress that are also related to solar radiation.
In Nubian breed, the GO results showed links with energetic metabolism, protein, and fatty acids synthesis regulation, but also catabolic processes (proteins and collagen) and cellular response to oxidative stress. The phosphatase inhibitor activity is a remarkable finding, because this impedes the target enzyme activity, avoiding the protein and cellular lysis. Consequently, as the protein phosphatases, it negatively regulates the HPSs proteins55. Inhibitors of this protein can prevent cell and protein damage in response to thermal stress. The two populations from Egypt are subjected to many stressful factors, in particular to thermal stress, since they were sampled in arid, desertic zones with very hot temperatures, whereas the population from Argentina comes from a temperate climate, thus the local adaptation to bio-climatic conditions is evident.
Although our objective was not to compare the effectiveness of each program used to carry out the analysis for discovering genomic regions under selection, it has been possible to see a general agreement on the evident clues of the adaptative processes that synergically activate a complex gene network. In our study, we found well known loci that have been identified in previous studies in goats as well as novel genes that showed implications for biogeographical adaptation described in other species, in particular on other ruminants. Most of these studies focused on local or indigenous populations, thus highlighting a probable population-specific selection footprint. Detecting regions under selection is a complex task, and this is reflected from the intricate connections amongst genes and biological processes. Taken together, our findings indicated that natural selection operated and continues acting in commercial goat breeds despite human intervention. Moreover, they provided evidence of selection that may be specific to one or few populations (local adaptation), and this information could be useful to identify both causal variants that are involved in a particular phenotype or important adaptive traits and the affected genes. Further investigating the detected genes will shed light on the complex mechanisms involved in the adaptation process, and provide information on putative favourable variants. Such information could be use in selection/conservation programs, also via new breeding technologies.
Materials and methods
Since the aim of this work was to detect loci that are under natural selection in artificially selected goat breeds, we addressed this issue choosing the follow four commercial and transboundary breeds: Angora, Boer, Saanen and Nubian. Each of them is known to be selected for a specific productive trait (wool, meet, milk and dual-purpose, respectively) and were transported over centuries in different countries, thus exposed to multiple environmental variables with respect to their original countries. Considering these characteristics, the four breeds studied meet our goal.
Sampling, genotyping and quality control
Figure 8 describes the workflow followed for detecting genetic signatures in our dataset. Genotypic data were gathered for goat breeds with a worldwide distribution. A total of 993 individuals belonging to four commercial breeds with a worldwide distribution were included in the analysis: Angora (n = 366), Boer (n = 332), Nubian (n = 99) and Saanen (n = 163) breeds from AdaptMap project (http://www.goatadaptmap.eu/56) and 33 genotypes of Russian Saanen goat57. All individuals were previously genotyped with the Illumina GoatSNP50 BeadChip3. The raw dataset was updated to the latest goat genome map (ARS1.2) and the quality control was carried out using Plink v1.911058 (Table 1) excluding SNPs unmapped or mapped into the sex chromosomes, SNPs with minor allele frequency < 0.05%, markers that failure the Hardy–Weinberg test at a specified significance threshold of 1 × 10−6, and SNP with call rate < 95%. Since we investigate breed-specific selection signatures related to adaptation, this procedure was repeated for all the four datasets, yielding a total of 44,655, 46,124, 44,800 and 47,325 for Angora, Boer, Nubian and Saanen, respectively. A first PCA analysis carried out with SNPrelate59 R package to explore the genetic structure Egyptian Nubian samples revealed a strong population divergence between individuals. Thus, we split the Nubian in two subpopulations: EGCH and EGCH1 (Table 1). Further analyses of this breed were done considering three populations in the Nubian dataset.

Graphical representation of workflow followed for detecting genetic signatures for adaptation.
For better understand the genetic background of the four breeds, we used Arlequin 3.5.2 program60 to calculate the pairwise FST and the Admixture 1.361 for the clusters analysis testing a number of clusters (K) equal to the number of populations composing each breed plus 3.
Data analysis
With the goal of leveraging the potential of the methods in capturing signals for regions under selection, we combined three complementary statistics with specific programs that can be used to calculate them62: Runs Of Homozygosity (ROH), FST-outliers detection and HapFLK methods were applied. ROH analysis compares genomic data within populations, and it is based on the detection of reduced local variability. The last two methods rely on the degree of differentiation due to locus-specific allele frequencies among populations and can be grouped into Single site (FST-outliers) and haplotype-based differentiation (HapFLK), respectively. To carry out these analyses DetectRUNS 0.9.4 package (R core 4.1), PCAdapt63 and HapFLK program v1.364 were used.
Runs of homozygosity
ROH are defined as two contiguous identical by descent (IBD) stretches of homozygous genotypes of a common ancestor present in an individual and inherited from both of its parents. The identification and characterization of ROH allow to reveal the population structure as well as footprints of natural and/or anthropogenic selection62. This analysis was carried out by using the R package DetectRUNS 0.9.4 package (R core v4.1) applying the “sliding windows” function and with the following setting: windowSize = 15, threshold = 0.1, minSNP = 15, ROHet = FALSE, maxOppWindow = 1, maxMissWindow = 1, maxGap = 106, minLengthBps = 1,000,000, minDensity = 1/10,000. We identified both ROH (length per class of ROH, total length per chromosome and sum at individual level, the frequency of SNP in each segment and visualising the homozygous segments per classes of length) and ROH islands (frequency of ROH at population level) ROH islands were plotted for all breeds using Biocircus65 package in R v.1.3.1073 (R core team 2020).
FST outliers detection
PCAdapt is a R package that uses statistical tools for outlier detection based on Principal Component Analysis (PCA). Briefly, this program tests how much each variant is associated with population structure, assuming that outlier variants are indicative of local adaptation. We determined the optimal number of PCs as recommended by Luu and co-workers63 using the graphical PCAdapt function and keeping PCs that correspond to eigenvalues to the left of the lower straight line in the screeplot (Supplementary Fig. 6) according to “Cattell’s rule”, that were 10 for all breeds. The P-values associated to the outlier variants were corrected with Bonferroni with a threshold of 0.05.
HapFLK analysis
With the HapFLK program v1.3 the loci under selection are revealed by comparing the genetic differentiation amongst the analysed populations with respect to the neutral drift model identifying genomic regions or loci showing deviations from neutrality (selective sweeps). The analysis was performed using the scripts available at https://forge-dga.jouy.inra.fr/projects/hapflk. The number of k (haplotype clusters) that better fits our data and estimated using the cross-validation procedure included in the fastPHASE software of66 was 35 for all breeds. The hapFLK statistic was computed as an average of 30 EM iterations to fit the Linkage Disequilibrium (LD) model. The P values obtained using the “Scaling_chi2_hapflk.py” script available at https://forge-dga.jouy.inra.fr/documents, were corrected for multiple comparisons using the false-discovery rate (FDR) method in R and SNPs (with a P value ≤ 0.05)were considered significant. Graphical representations of the Manhattan plots of the significant outliers and the selective sweeps retrieved with PCAdapt and HapFLK were done using CMplot package in R v.1.3.1073 (https://github.com/YinLiLin/R-CMplot).
Searching for candidate genes and pathways related to adaptation
The next step was to compare results from the three methodologies and to verify if identify genomic regions overlapped. Then, a screening within 1 Mb downstream and upstream of each significant marker was applied to pinpoint positional candidate genes, using Ensemble BioMart Capra hircus ARS1 data mining tool (https://m.ensembl.org/info/data/biomart/; Capra_hircus—Ensembl genome browser 108). Loci were investigated for each breed, focusing on previous studies about selection signatures mainly in goat, but also in other livestock species like sheep and cattle, since that the annotation of some genes in goat is still lacking or poor. Pathway enrichment analysis was performed to explore possible pathways involved in environmental adaptation. The genes identified from Ensemble BioMart were stored to perform a functional annotation using Ovis aries, Bos taurus and Homo sapiens databases by DAVID v6.867.
Data availability
The datasets analysed during the current study is available in the Dryad Repository (https://doi.org/10.5061/dryad.v8g21pt).
References
Colli, L. et al. Genome-wide SNP profiling of worldwide goat populations reveals strong partitioning of diversity and highlights post-domestication migration routes. Genet. Sel. Evol. 50, 58 (2018).
Zheng, Z. et al. The origin of domestication genes in goats. Sci. Adv. 6(21), eaaz5216 (2020).
Tosser-Klopp, G. et al. Design and characterization of a 52K SNP chip for goats. PLoS ONE 9, e86227 (2014).
Pogorevc, N. et al. Post-genotyping optimization of dataset formation could affect genetic diversity parameters: An example of analyses with alpine goat breeds. BMC Genomics 22, 546 (2021).
Islam, R. et al. Genome-wide runs of homozygosity, effective population size, and detection of positive selection signatures in six Chinese goat breeds. Genes (Basel) 10(11), 938 (2019).
Bertolini, F. et al. Genome-wide patterns of homozygosity provide clues about the population history and adaptation of goats. Genet. Sel. Evol. 50(1), 59 (2018).
Gheyas, A. A. et al. Integrated environmental and genomic analysis reveals the drivers of local adaptation in African indigenous chickens. Mol. Biol. Evol. 38, 4268–4285 (2021).
Prodromou, N. V. et al. Heat shock induces rapid resorption of primary cilia. J. Cell Sci. https://doi.org/10.1242/jcs.100545 (2012).
Li, Y. et al. Heat stress-responsive transcriptome analysis in the liver tissue of Hu sheep. Genes (Basel). 10, 395 (2019).
Lv, L., Ujisguleng, B., Orhontana, B., Lian, W. & Xing, W. Molecular cloning of sheep and cashmere goat Pdia3 and localization in sheep testis. Reprod. Domest. Anim. 46, 980–989 (2011).
Sodhi, M. et al. Novel polymorphisms in UTR and coding region of inducible heat shock protein 70.1 gene in tropically adapted Indian zebu cattle (Bos indicus) and riverine buffalo (Bubalus bubalis). Gene 527, 606–615 (2013).
Ur Rehman, S. et al. Genomic identification, evolution and sequence analysis of the heat-shock protein gene family in Buffalo. Genes (Basel) 11, 1–19 (2020).
Fajardo, O., Meseguer, V., Belmonte, C. & Viana, F. TRPA1 channels mediate cold temperature sensing in mammalian vagal sensory neurons: Pharmacological and genetic evidence. J. Neurosci. 28, 7863–7875 (2008).
Buga, A.-M. et al. Identification of new therapeutic targets by genome-wide analysis of gene expression in the ipsilateral cortex of aged rats after stroke. PLoS ONE 7, e50985 (2012).
Kim, E. S. et al. Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. Heredity (Edinb) 116, 255–264 (2016).
Edea, Z., Dadi, H., Dessie, T. & Kim, K. S. Genomic signatures of high-altitude adaptation in Ethiopian sheep populations. Genes Genomics 41, 973–981 (2019).
Diniz, W. J. S. et al. Cerebrum, liver, and muscle regulatory networks uncover maternal nutrition effects in developmental programming of beef cattle during early pregnancy. Sci. Rep. 11, 2771 (2021).
Abo-Ismail, M. K. et al. Development and validation of a small SNP panel for feed efficiency in beef cattle1. J. Anim. Sci. 96, 375–397 (2018).
Yurchenko, A. A. et al. High-density genotyping reveals signatures of selection related to acclimation and economically important traits in 15 local sheep breeds from Russia. BMC Genomics 20, 1–19 (2019).
Simonson, T. S. Altitude adaptation: A glimpse through various lenses. High Alt. Med. Biol. 16, 125–137 (2015).
Yang, D. et al. HMOX2 functions as a modifier gene for high-altitude adaptation in tibetans. Hum. Mutat. 37, 216–223 (2016).
Edea, Z. et al. Signatures of positive selection underlying beef production traits in Korean cattle breeds. J. Anim. Sci. Technol. 62, 293–305 (2020).
Zeng, Y. et al. MicroRNA-223 attenuates hypoxia-induced vascular remodeling by targeting RhoB/MLC2 in pulmonary arterial smooth muscle cells. Sci. Rep. 6, 24900 (2016).
Bradshaw, W. E. & Holzapfel, C. M. Evolution of Animal photoperiodism. Annu. Rev. Ecol. Evol. Syst. 38, 1–25 (2007).
Marcheva, B. et al. Circadian Clocks and Metabolism 127–155 (2013). https://doi.org/10.1007/978-3-642-25950-0_6.
Pickel, L. & Sung, H.-K. Feeding rhythms and the circadian regulation of metabolism. Front. Nutr. 7, 39 (2020).
Bertolini, F. et al. Comparative selection signature analyses identify genomic footprints in Reggiana cattle, the traditional breed of the Parmigiano-Reggiano cheese production system. Animal 14, 921–932 (2020).
Caroprese, M., Ciliberti, M. G., Albenzio, M. & Sevi, A. Climate Change Impact on Immune Response in Sheep. In Sheep Production Adapting to Climate Change 95–116 (Springer, Singapore, 2017). https://doi.org/10.1007/978-981-10-4714-5_4.
Mujahid, A., Yoshiki, Y., Akiba, Y. & Toyomizu, M. Superoxide radical production in chicken skeletal muscle induced by acute heat stress. Poult. Sci. 84, 307–314 (2005).
Beckman, K. B. & Ames, B. N. The free radical theory of aging matures. Physiol. Rev. 78, 547–581 (1998).
Soares, D. G., Basso, F. G., Scheffel, D. S., Hebling, J. & de Souza Costa, C. A. Responses of human dental pulp cells after application of a low-concentration bleaching gel to enamel. Arch. Oral Biol. 60, 1428–1436 (2015).
Wang, M. D., Dzama, K., Hefer, C. A. & Muchadeyi, F. C. Genomic population structure and prevalence of copy number variations in South African Nguni cattle. BMC Genomics 16, 894 (2015).
Cosseddu, G. M. et al. Gene expression profiling on sheep brain reveals differential transcripts in scrapie-affected/not-affected animals. Brain Res. 1142, 217–222 (2007).
Faller, E. M., Villeneuve, T. S. & Brown, D. L. MAP1a associated light chain 3 increases microtubule stability by suppressing microtubule dynamics. Mol. Cell. Neurosci. 41, 85–93 (2009).
Moreno, C. R. et al. Mapping of quantitative trait loci affecting classical scrapie incubation time in a population comprising several generations of scrapie-infected sheep. J. Gen. Virol. 91, 575–579 (2010).
van Wettere, W. H. E. J. et al. Review of the impact of heat stress on reproductive performance of sheep. J. Anim. Sci. Biotechnol. 12, 1–18 (2021).
Simmons, R. M. et al. Insulin-like growth factor binding protein-1 in the ruminant uterus: Potential endometrial marker and regulator of conceptus elongation. Endocrinology 150, 4295–4305 (2009).
Dorniak, P., Welsh, T. H., Bazer, F. W. & Spencer, T. E. Endometrial HSD11B1 and cortisol regeneration in the ovine uterus: effects of pregnancy, interferon tau, and prostaglandins. Biol. Reprod. 86(4), 124 (2012).
Nichi, M. et al. Seasonal variation in semen quality in Bos indicus and Bos taurus bulls raised under tropical conditions. Theriogenology 66, 822–828 (2006).
Lin, Y.-H. et al. SEPTIN12 genetic variants confer susceptibility to teratozoospermia. PLoS ONE 7, e34011 (2012).
Dirami, T. et al. Assessment of the frequency of sperm annulus defects in a large cohort of patients presenting asthenozoospermia. Basic Clin. Androl. 25, 1–7 (2015).
Howden, S. M., Crimp, S. J. & Stokes, C. J. Climate change and Australian livestock systems: Impacts, research and policy issues. Aust. J. Exp. Agric. 48, 780 (2008).
Zhang, R., Wu, H. & Lian, Z. Bioinformatics analysis of evolutionary characteristics and biochemical structure of FGF5 Gene in sheep. Gene 702, 123–132 (2019).
Bongiorni, S., Mancini, G., Chillemi, G., Pariset, L. & Valentini, A. Identification of a Short region on chromosome 6 affecting direct calving ease in piedmontese cattle breed. PLoS ONE 7, 6–12 (2012).
Ruiz-Larrañaga, O. et al. Genomic selection signatures in sheep from the Western Pyrenees. Genet. Sel. Evol. 50, 9 (2018).
Guo, J. et al. Comparative genome analyses reveal the unique genetic composition and selection signals underlying the phenotypic characteristics of three Chinese domestic goat breeds. Genet. Sel. Evol. 51, 1–18 (2019).
Kadokawa, H., Pandey, K., Nahar, A., Nakamura, U. & Rudolf, F. O. Gonadotropin-releasing hormone (GnRH) receptors of cattle aggregate on the surface of gonadotrophs and are increased by elevated GnRH concentrations. Anim. Reprod. Sci. 150, 84–95 (2014).
Sosa, A. et al. Single nucleotide polymorphisms of GnRHR gene and its relationship with reproductive performance in Egyptian buffaloes. Egypt. J. Vet. Sci. 47, 41–50 (2016).
Bemji, M. N., Isa, A. M., Ibeagha-Awemu, E. M. & Wheto, M. Polymorphisms of caprine GnRHR gene and their association with litter size in West African Dwarf goats. Mol. Biol. Rep. 45, 63–69 (2018).
Kim, D.-K. et al. Revisiting the evolution of gonadotropin-releasing hormones and their receptors in vertebrates: Secrets hidden in genomes. Gen. Comp. Endocrinol. 170, 68–78 (2011).
Gibson, G. E., Peterson, C. & Sansone, J. Decreases in amino acid and acetylcholine metabolism during hypoxia. J. Neurochem. 37, 192–201 (1981).
Peng, D. Q., Smith, S. B. & Lee, H. G. Vitamin A regulates intramuscular adipose tissue and muscle development: Promoting high-quality beef production. J. Anim. Sci. Biotechnol. 12, 34 (2021).
Novais, F. J. et al. Identification of a metabolomic signature associated with feed efficiency in beef cattle. BMC Genomics 20, 1–10 (2019).
Xu, Q. et al. All-trans retinoic acid inhibits lipopolysaccharide-induced inflammatory responses in bovine adipocytes via TGFβ1/Smad3 signaling pathway. BMC Vet. Res. 15, 1–14 (2019).
Romero, R. D., Montero Pardo, A., Montaldo, H. H., Rodríguez, A. D. & Hernández Cerón, J. Differences in body temperature, cell viability, and HSP-70 concentrations between Pelibuey and Suffolk sheep under heat stress. Trop. Anim. Health Prod. 45, 1691–1696 (2013).
Stella, A. et al. AdaptMap: Exploring goat diversity and adaptation. Genet. Sel. Evol. 50, 61 (2018).
Deniskova, T. E. et al. SNP-based genotyping provides insight into the West Asian Origin of Russian Local Goats. Front. Genet. 12, 708–740 (2021).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Saravanan, K. A. et al. Selection signatures in livestock genome: A review of concepts, approaches and applications. Livest. Sci. 241, 104257 (2020).
Luu, K., Bazin, E. & Blum, M. G. B. pcadapt: An R package to perform genome scans for selection based on principal component analysis. Mol. Ecol. Resour. 17, 67–77 (2017).
Fariello, M. I., Boitard, S., Naya, H., SanCristobal, M. & Servin, B. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics 193, 929–941 (2013).
Cui, Y. et al. BioCircos.js: An interactive Circos JavaScript library for biological data visualization on web applications. Bioinformatics 32, 1740–1742 (2016).
Scheet, P. & Stephens, M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78, 629–644 (2006).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Acknowledgements
This research was funded by the European Union’s Horizon 2020 research and innovation program under grant agreement No 772787 (SMARTER).
Author information
Authors and Affiliations
Contributions
A.M., J.R.D., B.L.S., A.S.: Conceptualization. P.C., T.D., N.Z., A.D.: Data curation. A.M.: Formal analysis. A.M., J.R.D.: Investigation. A.M., T.B., T.D., A.D., N.Z.: Methodology. J.R.D., A.M.: prepared figures. A.S.: Project administration. A.M.: Writing – original draft. A.M., J.R.D., T.B., B.L.S., P.C., A.S., T.D., N.Z., A.D.: Writing – review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Manunza, A., Diaz, J.R., Sayre, B.L. et al. Discovering novel clues of natural selection on four worldwide goat breeds. Sci Rep 13, 2110 (2023). https://doi.org/10.1038/s41598-023-27490-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-27490-x
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
