
Abstract
Near-infrared organic light-emitting diodes (NIR OLEDs) with heavy metals are regularly reported due to the advantages of their various applications in healthcare services, veil authentication, and night vision displays. For commercial applications, it is necessary to look at radiance capacity (RC) instead of radiance because of power consumption. However, recent papers still reported only simple high radiance performance and do not look at device from the point of view of RC. To overcome this hurdle, we designed Ir(III)-based heteroleptic NIR materials with two types of auxiliary ligand. The proposed emitters achieve a highly oriented horizontal dipole ratio (Ir(mCPDTiq)2tmd, complex 1: 80%, Ir(mCPDTiq)2acac, complex 2: 81%) with a short radiative lifetime (1: 386 ns, 2: 323 ns). The device also shows an extremely low turn-on voltage (Von) of 2.2 V and a high RC of 720 mW/sr/m2/V. The results on the Von and RC of the device is demonstrated an outstanding performance among the Ir(III)-based NIR OLEDs with a similar emission peak.
Similar content being viewed by others

Introduction
OLEDs have been successfully established and are widely used as an important light source in conventional display and lighting markets1. In particular, OLEDs are pioneering a new market beyond the visible spectral range. Recently, NIR OLEDs are of great interest not only in the field of wearable healthcare systems such as photobiomodulation (PBM)2,3,4,5,6 and photodynamic therapies (PDT)7 but also for night-vision displays and optical signal processing. In particular, phototherapy studies using OLED have been steadily published in the past decade. It is known that the effect of phototherapy is maximized in the red or NIR wavelength, where the absorption of cytochrome c oxidase (CCO), a photoreceptor, occurs well8. Recently, it has been reported that there is a wound healing effect not only in the red region, but also by using NIR OLED2. Moreover, a low level light therapy (LLLT) technique, which heals using lower energy than that of PDT, is used in PBM such as wound healing. As a result, the device was driven in a low voltage area (6 V or less) for phototherapy, and a lower voltage is required through integration, such as a battery, to develop actual commercial products. Therefore, it is more important than anything else to realize NIR OLEDs from an RC point of view. Although new NIR emitters are continuously being designed for implementation of NIR OLEDs, the performance of NIR OLEDs is still far behind devices in the visible range due to energy gap law9,10. Many studies have been done to overcome the bottleneck of the intrinsic performance of NIR OLEDs, such as using thermally activated delayed fluorescence (TADF)11,12,13,14,15 or using transition heavy metals such as Pt(II)16,17,18,19,20, Os(II)21,22,23, and Ir(III)24,25,26,27,28,29,30 to obtain high efficiency. The method using TADF can transfer the non-radiative triplet exciton to the singlet space and obtain 100% of internal quantum efficiency (IQE). Moreover, Pt(II) and Os(II) complexes are extremely expensive to use in comparison to the Ir(III) complex. Conventionally, many transition metal-based NIR OLEDs have been simply reported in terms of high radiance16. However, the voltage at maximum radiance is so high that there are many restrictions on applying it to actual industries from the perspective of power consumption. Recently, it has been reported that a cyclopentadithiophene (CPDT) unit has many advantages, such as efficient charge transfer, high electron density and strong electron donor ability due to a structure with high planarity in which bithiophene is fused31,32,33. Therefore, it has been adopted as a unit in photoactive layers and hole transport layers of electronic materials such as organic solar cells and quantum-dot solar cells due to its optimal properties31,32,33,34,35.
In this study, CPDT-isoquinoline based Ir(III) complexes for NIR OLED application were synthesized and designed because the CPDT-based ligand has an electron-rich and significant extension of the conjugative structure that enables the emission peak to have a longer wavelength. Therefore, the Ir(III) complex with a CPDT-isoqinoline ligand has a small energy band gap (Eg, ≤ 2.1 eV) that is suitable for NIR emission. Furthermore, the substitution of the two methyl groups in sp3 carbon of CPDT can suppress the excessive aggregation between molecules34,36,37. In addition, we introduced two β-diketone ancillary ligands with long conjugation lengths to increase solubility and facilitate accurate wavelength control, as well as attempt to enhance the horizontal orientation24,38,39. Based on the above design concepts, the Ir(mCPDTiq)2tmd (complex 1), Ir(mCPDTiq)2acac (complex 2) were newly developed and chemical structures are shown in Fig. 1. The two compounds achieved maximum emission peaks of 760 nm and 755 nm in dichloromethane solution, respectively.

Chemical structures of (a) Ir(mCPDTiq)2tmd and (b) Ir(mCPDTiq)2acac.
Results and discussion
Synthesis and characterization
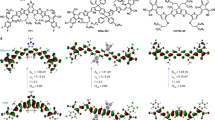
The newly developed iridium complexes were prepared using universal Stille cross-coupling reactions between (4,4-dimethyl-4H-cyclopenta[2,1-b:3,4-b']dithiophen-2-yl)trimethylstannane (4) and 1-chloroisoquinoline. They were formed into two new iridium complexes using different ancillary ligands, as shown in Fig. 2. The bridged dimer was synthesized by a common Nonoyama reaction and a base-catalyzed ligand exchange reaction method using iridium hydrate (IrCl3 nH2O) and a freshly prepared ligand(1-(4,4-dimethyl-4H-cyclopenta[2,1-b:3,4-b']dithiophen-2-yl)isoquinoline (5)). Then, complex 1 and complex 2 were synthesized from an iridium dimer [(mCPDTiq)2Ir(μ-Cl)]2 and 2,2,6,6-tetramethyl-3,5-heptanedione (tmd), acetylacetone (acac), respectively. The synthesized iridium complexes were characterized via 1H-NMR, 13C-NMR spectroscopy and high-resolution mass spectroscopy. All intermediates were also confirmed by 1H-NMR, 13C-NMR spectroscopy and HR mass methods (see in Supporting Information Figs. S1–S13). We also estimated the thermal properties of the ligand-based iridium complexes using TGA and DSC thermograms (see in Supporting Information Figs. S14–S17). Both complexes (1 and 2) showed stoutness up to 340 °C and experienced no phase change up to 250 °C. Overall, the synthesized iridium complexes exhibited sufficient thermal stability.

Synthetic scheme of cyclometalating ligand and iridium complexes.
A cyclic voltammetry (CV) experiment was performed to study the energy level of the newly developed complexes, as shown in Fig. S18. By comparing them with the oxidation potential of the ferrocene, we could determine the highest occupied molecular orbital (HOMO) levels of complexes 1 and 2. The electrochemical property of these Ir(III) complexes was estimated by CV experiments using tetrabutylammonium perchlorate as an electrolyte in dimethylformaide solution. The HOMO energy level was determined by comparing the oxidation potential of ferrocene, a standard material, and the synthesized materials. Both compounds 1 and 2 showed clear oxidative fluctuation at 0.78, and 0.76 V, respectively. According to the equation in Fig. S18, the HOMO energy levels were confirmed to be − 5.23, and − 5.21 eV, respectively40. The band gap energy (Eg) of both compounds was same value at 2.08 eV, which was obtained from the onset value of the absorption spectra in the solution of each compounds. The LUMO energy levels were calculated by subtracting the band gap energy from the HOMO energy.
Photophysical properties
Figure 3 show the UV–Visible absorption and photoluminescence spectra of our two complexes 1 and 2 in dichloromethane solution and vacuum-deposited thin-film with the host material at room-temperature (298 K). Spin allowed π–π* transitions were shown to be responsible for the high absorption band below 450 nm. Relatively weak absorption bands in the range 500–700 nm in both spectra were assigned to mixed 1MLCT and 3MLCT (singlet and triplet metal-to-ligand charge transfer) with an inter-ligand charge transfer (1LLCT). The photoluminescence (PL) emission peaks of complexes 1 and 2 in dichloromethane solution were observed at 760 nm with FWHM (full-width at half maximum) of 79 nm and at 755 nm with FHWM of 86 nm, respectively. Complex 1 was slightly 5 nm red shifted compared with complex 2, which was due to the ancillary ligand effect. The result can be compared with the well-known already reported two iridium complexes Ir(piq)2acac (λmax = 622 nm) and Ir(piq)2tmd (λmax = 628 nm) based on the phenyl-isoquinoline ligand, and this significant red-shift was due to the electron-rich and significant extension of the conjugative structure of the CPDT unit41,42. These Ir(III) complexes show the absolute PL quantum yield (PLQY) and transient decay of complexes 1 (3.2%, 386 ns) and 2 (3%, 323 ns), respectively (see in Fig. S19 for the observed lifetime). Based on the equation below, the radiative and non-radiative constant were calculated.

UV–Visible absorbance and PL in solution (black line), thin-film (red line) at room temperature (298 K): (a) Ir(mCPDTiq)2tmd and (b) Ir(mCPDTiq)2acac.
All numerical data and PLQY of previous literature are summarized in Table 1 and Table S1, respectively. Although PLQY is slightly low (Table S1), it is noteworthy that the radiative constant of the complexes was significantly lower or similar to those reported in other previous literature24,26,27.
To determine how the organic molecules are oriented when deposited as a thin film, angle dependent p-polarized photoluminescence spectrum with the given emitters deposited on the substrate was measured, as shown in Fig. 4. Birefringence of each complex was considered for the theoretical fitting. The horizontal dipole orientation (Θh) of complexes 1 and 2 were 0.80 and 0.81, which were highly horizontally oriented compared to the other organic emitters and well matched to the fitted curve, respectively. Complexes 1 and 2 were preferred to be oriented horizontally because they exhibited a heteroleptic structure. The structure of the auxiliary ligand affects the dipole moment of the emitters to be arranged horizontally, thereby enhancing the device characteristics43,44.

Angular dependence of PL intensity of (a) Ir(mCPDTiq)2tmd, (b) Ir(mCPDTiq)2acac. Fitted line (experimental data), horizontal and isotropic line (simulation) of dipole orientation.
Electroluminescence (EL) performance (device performances)
The short radiative lifetimes, efficient NIR emissions and highly horizontal oriented Ir(III) complexes prompted us to look into their EL characteristics further. The device structure of the OLEDs was as follows: indium tin oxide (ITO)/molybdenum trioxide (MoO3, 10 nm)/N,N′-Di(1-naphthyl)-N,N′-diphenyl-(1,1′-biphenyl)-4,4′-diamine (NPB, 40 nm)/Bebq2: NIR dopant (x wt%, 30 nm)/bathophenanthroline (Bphen, 20 nm)/Liq (1 nm)/Al (100 nm). ITO was patterned on a glass substrate. In the device structure, MoO3, NPB, Bebq2, Bphen, Liq, and Al were used for the hole injection layer (HIL), hole transport layer (HTL), host material, electron transport layer (ETL), electron injection layer (EIL), and cathode, respectively. In particular, Bebq2 is well known to act as an effective host during the doping process with the iridium complex and to facilitate energy transfer45,46.
The device optimization procedure was carried out with different doping concentrations (x = 5, 10, and 20 wt%) of the NIR dopant (complexes 1, 2). Moreover, all the devices were encapsulated with Al2O3 (30 nm) using the atomic layer deposition (ALD) method to measure the lifetime. The energy level and molecular structure of the materials and schematic device structure are shown in Fig. 5a. The materials of each layer were selected in consideration of the energy level, and the structure was designed to facilitate charge balance and lower driving voltage.

Electrical and optical properties of the NIR OLEDs. (a) Energy level alignment diagram and molecular structure (b) EL spectra by concentration (c) J–V–R (d) EQE of the NIR OLEDs depending on the doping concentrations.
The normalized EL spectrum of the NIR OLEDs are shown in Fig. 5b as a function of the complexes with the doping concentration. The EL spectra of all the devices are identical to PL, suggesting that exciton was well confined inside the EML. However, the phenomenon in which a peak appears in a visible region around 500 nm was observed according to the doping concentration. The peak in the visible area decreases when more dopant is injected at a high concentration. This phenomenon could be explained by the fact that the host material’s (Bebq2) PL peak (see Fig. S20) emerges at low doping concentrations. The higher the concentration, the easier it is for energy to transfer from the host to the dopant, thus the peak in the visible light area decreases47 (see Fig. 5b, Fig. S21).
The current density–voltage–radiance (J–V–R), light distribution characteristics and EQE are shown in Fig. 5c,d. All devices ran neatly without leakage current and had a Lambertian emission distribution (Fig. S22). All numerical data are described in Table 2. The maximum RC was 723 and 606 mW/sr/m2/V, EQEmax was 0.48 and 0.37%, and Von was extremely low at 2.2 V for complexes 1 and 2 at a doping concentration of 10 wt%, respectively. In addition, the stability and reliability after over 120 h based on median Lethal Time (LT50) were proved for complexes 1 and 2 at 10 wt%, respectively (Fig. S23).
Both complexes showed the highest performance at 10 wt%, indicating that they run at significantly lower voltages compared to the previously reported NIR OLEDs, which means that a balanced charge was supplied and transported to the device as an optimal structure, resulting in direct trapping of the carrier.
The NIR OLED developed through in study showed the best performance for complex 1 at a doping concentration of 10 wt%. The numerical values (Rmax, Vmax, and RC) of previous studies are presented in Table S2. Among them, the RC was calculated by dividing the maximum radiance by the maximum voltage. Compared to the other reported Ir(III)-based NIR OLED in the literature, there are some deficiencies in the highest radiance and EQE due to low the PLQY, but there is the advantage of a high RC. This is presumed to be caused by the fact that the current density of the NIR OLED device developed in this study is about 20 times high. Because the EQE is affected by the PLQY, it may be low when the PLQY is low. However, through optimization of the overall structure of the device, it was possible to implement a device with a high RC by achieving a low driving voltage and a high radiance at a relatively low voltage due to good current injection.
Basically, a high radiance is not very important in phototherapy using LLLT. Rather, the device must be manufactured in terms of the RC taking into consideration the driving voltage for the corresponding radiance for commercial applications. In other words, it is tremendously important to consider the power consumption of NIR OLEDs for them to be used in actual applications. The NIR OLED had an extremely low Von and the highest RC compared to the other Ir(III)-based NIR OLEDs shown in Fig. 6.

Radiance capacity depending on Von (Plotted by referring to NIR OLEDs based on Ir(III) complex).
Conclusion
In summary, heteroleptic Ir(III) complexes are designed and synthesized with a high horizontal dipole ratio of dipole orientation (complex 1: 80%, complex 2: 81%) and a short radiative lifetime (complex 1: 390 ns, complex 2: 320 ns). Because of its high electron density and long π conjugation length, this structure can close the energy gap and emit the NIR wavelength (760 nm). Notably, the NIR OLED with our newly designed Ir(III) complex has a significantly lower Von, overwhelmingly high RC, and better reliability than previous Ir(III) complex-based NIR OLEDs. Hence, our study serves to present the future direction of NIR OLEDs for practical applications in the industry.
Methods
General information and materials
All starting reactants were purchased from Sigma Aldrich and Alfa Aesar. The palladium catalyst (Tetrakis(triphenylphosphine)palladium(0)) was purchased from Umicore. Iridium hydrate was supplied by Furuya Metal Korea. All commercially purchased reagents were used without any further purification, and a series of intermediates were synthesized according to the reported method48. Moreover, (4,4-dimethyl-4H-cyclopenta[2,1-b:3,4-b']dithiophen-2-yl)trimethylstannane (4) was prepared according to the literature49.
Measurement
All spectra (1H NMR and 13C NMR) were measured using a 300 MHz (Bruker) and a 500 MHz (DRX) spectrometer. Mass spectra (FAB and EI data) were acquired with a high-resolution mass spectrometer using a JMS-700 (Jeol). To analyze the thermal stability of prepared iridium complexes, thermogravimetric analysis was conducted using TA 2050 TGA instruments. All differential scanning calorimetry (DSC) data were measured using a TA Instruments 2100 DSC and heated from room temperature to 800 °C (10 °C/min.) Ultraviolet–visible absorption spectra were acquired on a LAMBDA-900 spectrophotometer (PerkinElmer). Cyclic voltammetry (CV) was measured to study the electrochemical properties of the prepared materials. A three-electrode system was used with a carbon glass electrode, an Ag/AgCl electrode and platinum wire as the working electrode, reference electrode and counter electrode, respectively. For detailed experimental conditions, a cyclic voltammogram was recorded using tetrabutylammonium perchlorate as an electrolyte in a 0.1 M dimethylformamide solution under nitrogen gas at a scan rate of 50 mV/s.
Synthesis of 4H-cyclopenta[2,1-b:3,4-b’] dithiophen-4-one (1)
This reaction was done following the literature method48. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.07 (d, J = 4.8 Hz, 2 H), 7.02 (d, J = 4.8 Hz, 2 H).
Synthesis of 4H-cyclopenta[2,1-b:3,4-b’] dithiophene (2)
This reaction was done following the literature method48. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.21 (d, J = 5.1 Hz, 2 H), 7.12 (d, J = 5.1 Hz, 2 H), 3.5 (s, 2 H).
Synthesis of 4,4-dimethyl-4H-cyclopenta[2,1-b:3,4-b’] dithiophene (3)
This reaction was done following the literature method48. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.19 (d, J = 4.8 Hz, 2 H), 7.02 (d, J = 4.8 Hz, 2 H), 1.48 (s, 6 H).
Synthesis of (4,4-dimethyl-4H-cyclopenta[2,1-b:3,4-b’] dithiophen-2-yl) trimethylstannane (4)
This reaction was done following the literature method49. 1H NMR (300 MHz, CD2Cl2): δ (ppm) = 6.98 (d, J = 4.8 Hz, 1 H), 6.90 (s, 1 H), 6.84 (d, J = 4.8 Hz, 1 H), 1.27 (s, 6 H), 0.31—0.12 (m, 9 H).
Synthesis of 1-(4,4-dimethyl-4H-cyclopenta[2,1-b:3,4-b']dithiophen-2-yl)isoquinoline (5)
(4,4-dimethyl-4H-cyclopenta[2,1-b:3,4-b']dithiophen-2-yl)trimethylstannane (10.0 g, 48.5 mmol), 1-choloroisoquinoline (7.9 g, 48.5 mmol) were dissolved in toluene (500 mL) and purged with nitrogen gas for 30 min. Then a palladium catalyst (Pd(PPh3)4, 1.1 g, 1.0 mmol) was added to the reaction mixture and stirred at 105 °C for 12 h. After cooling to room temperature, the reactant mixture was quenched by water and it was extracted with methylene chloride three times. Then the organic solution portion was dried using anhydrous magnesium sulfate and evaporated. After solvent was removed, the product was purified by column chromatography using hexane and ethyl acetate (Yield: 7.4 g, 82%). 1H NMR (300 MHz, CD2Cl2): δ (ppm) = 8.54 (d, J = 8.4 Hz, 1 H), 8.40 (d, J = 5.7 Hz, 1 H), 7.80 (d, J = 7.5 Hz, 1 H), 7.65–7.54 (m, 3 H), 7.47 (d, J = 5.4 Hz, 1 H), 7.19 (d, J = 4.8 Hz, 1 H), 6.99 (d, J = 4.8 Hz, 1 H), 1.46 (s, 6 H). 13C NMR (300 MHz, CD2Cl2): δ (ppm) = 161.42, 161.10, 153.41, 143.63, 142.07, 138.06, 137.24, 135.31, 129.99, 127.64, 127.29, 126.42, 126.35, 125.54, 122.84, 120.95, 119.23, 52.71, 45.38, 24.94 HRMS-EI + (m/z): [M] + calcd for C20H15NS2, 333.06; found, 333.0648.
Synthesis of Ir(III) dimer [(mCPDTiq)2Ir(μ-Cl)]2 (6)
A cyclometalating ligand, 1-(4,4-dimethyl-4H-cyclopenta[2,1-b:3,4-b']dithiophen-2-yl)isoquinoline (5) (7.5 g, 22.5 mmol) and iridium(III) chloride hydrate (3.1 g, 10.2 mmol) were dissolved in ethyl glycol and distilled water. The reaction mixture was stirred overnight under nitrogen gas. Then, the precipitated solids were filtered and washed with water and a small amount of diethyl ether. These crude products were thoroughly dried and then used without a further purifying procedure.
Synthesis of Ir(mCPDTiq)2 (tmd) (7)
A prepared μ-chloro-bridged dimer [(mCPDTiq)2Ir(μ-Cl)]2 (5 g, 2.8 mmol), 2,2,6,6-tetramethyl-3,5-heptanedione (2.1 g, 11.2 mmol) and sodium carbonate (2.9 g, 28.0 mmol) were dissolved in ethyl glycol. The three-neck flask was refluxed for 12 h at 120 °C, and then the cooled reaction mixture was poured into iced water and the precipitated solid was filtered and the filtered solids were extracted with dichloromethane and water. The organic portion was dried using anhydrous magnesium sulfate and evaporate under reduced pressure. After removal of the organic solvent, the black-purple product was purified by column chromatography using hexane and ethyl acetate (Yield: 1.2 g, 42%). 1H NMR (300 MHz, CD2Cl2, d2): δ (ppm) = 9.01–8.98 (m, 2 H), 7.97 (d, J = 6.6 Hz, 2 H), 7.87–7.83 (m, 2 H), 7.75–7.71 (m, 4 H), 7.16–7.14 (d, J = 4.8 Hz, 2 H), 7.12 (d, J = 5.1 Hz, 1 H), 5.51 (s, 1 H), 1.58 (s, 12 H), 0.79 (s, 18 H). 13C NMR (500 MHz, CD2Cl2): δ (ppm) = 194.34, 167.44, 165.19, 164.68, 149.95, 141.86, 141.67, 138.16, 136.92, 133.62, 130.81, 127.19, 126.98, 126.57, 126.43, 124.15, 120.68, 114.50, 93.57, 91.89, 91.57, 89.98, 86.87, 85.78, 85.41, 54.14, 53.9, 53.78, 53.63, 53.42, 53.26, 53.21,53.06, 52.70, 45.78, 40.90, 27.57, 23.99, 22.91. HRMS-FAB + (m/z): [M + H] + calcd for C51H47IrN2O2S4, 1040.21; found, 1040.2174.
Synthesis of Ir(mCPDTiq)2 (acac) (8)
(Yield: 34%) 1H NMR (300 MHz, CD2Cl2, d2): δ (ppm) = 9.02–8.98 (m, 2 H), 8.09 (d, J = 7.8 Hz, 2 H), 7.91–7.88 (m, 2 H), 7.78–7.75 (m, 4 H), 7.20–7.18 (d, J = 6.6 Hz, 2 H), 7.17–7.16 (d, J = 4.8 Hz, 2 H), 6.76–6.74 (d, J = 4.8 Hz, 2 H), 5.37 (s, 1 H), 1.77 (s, 6 H), 0.80 (s, 6 H), 0.45 (s, 6H). 13C NMR (300 MHz, CDCl3): δ (ppm) = 184.68, 167.21, 164.75, 142.96, 141.75, 138.36, 137.02, 133.68, 130.90, 127.21, 127.05, 126.55, 124.24, 120.61, 114.71, 100.59, 45.87, 28.43, 24.03, 23.05. HRMS-FAB + (m/z): [M + H] + calcd for C45H35IrN2O2S4, 956.1211; found, 956.1216.
UV–vis absorbance and PL measurements
Thin-films (50 nm) were vacuum deposited on a glass substrate under a high vacuum (2 \(\times\) 10−6 torr). The thickness of the complexes was measured using a quartz thickness-monitor. Then, UV–vis absorption and PL spectra were measured using a Fluorolog-3 (HORIBA). The excitation wavelength was set to 430 nm in both complexes. Moreover, the PLQY of the Ir(III) complexes was measured using a Fluorolog-3 (HORIBA).
Measurement of the angle dependent p-polarized photoluminescent spectrum (ADPL)
The emission layer was deposited on a bare 50 nm-thick glass substrate. Then, glass encapsulation was done in a nitrogen (N2)-filled glove box to avoid degradation from the air. A full angle dependent p-polarized PL spectrum was obtained with the goniometer based motorized intensity measurement system (Phelos, Fluxim). Simulation and fitting with the experimental data were made with a customized MATLAB code describing the coherent dipole radiation theory.
Device fabrication and measurement
All organic and inorganic materials were purchased from Sigma-Aldrich and iTASCO. The devices were fabricated on a patterned ITO glass substrate with a sheet resistance of 15 \(\Omega\) per square. MoO3, NPB, Bphen, Liq, and Al were deposited at a rate of 0.5, 1, 1, 0.1, and 2 Å/s, respectively. The host (Bebq2) and guest (complexes 1 and 2) were co-deposited at various doping concentrations under a high vacuum of 2 \(\times\) 10−6 torr. All the devices were encapsulated within Al2O3 using the atomic layer deposition (ALD) process under 70℃. The current density–voltage-radiance (J–V–R) and angle-resolved EL intensity characteristics of the fabricated devices were obtained with a source-measure unit (Keithley 2400) using a calibrated photodiode (FDS 100, Thorlab) and a fiber optic spectrometer (BW_UVNb, StellarNet) held on a motorized goniometer (PRM1/MZ8, Thorlabs) in a nitrogen (N2)-filled glove box. The EQE (ηEQE) and PE (ηPE) of the devices were estimated from the measured full angular characteristics without Lambertian simplification50. The operating lifetime was monitored using a McScience Polaronix M6000 OLED Lifetime Tester.
Data availability
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
References
Song, J., Lee, H., Jeong, E. G., Choi, K. C. & Yoo, S. Organic light-emitting diodes: Pushing toward the limits and beyond. Adv. Mater. 32, 1–17 (2020).
Park, Y. et al. Cell proliferation effect of deep: Penetrating microcavity tandem NIR OLEDs with therapeutic trend analysis. Sci. Rep. 12, 1–11 (2022).
Jeon, Y. et al. A wearable photobiomodulation patch using a flexible red-wavelength OLED and its in vitro differential cell proliferation effects. Adv. Mater. Technol. 3, 1–10 (2018).
Jeon, Y. et al. Sandwich-structure transferable free-form OLEDs for wearable and disposable skin wound photomedicine. Light Sci. Appl. 8, 1–16 (2019).
Choi, S., Jeon, Y., Kwon, J. H., Ihm, C. & Kim, S. Y. Wearable photomedicine for neonatal jaundice treatment using blue organic light-emitting diodes (OLEDs): Toward textile-based wearable phototherapeutics. Adv. Sci. https://doi.org/10.1002/advs.202204622 (2022).
Kim, H. et al. A flexible and wavelength-designable polymer light-emitting diode employing sandwich-encapsulation for wearable skin rejuvenation photomedicine. Adv. Mater. Interfaces 8, 1–11 (2021).
Jeon, Y. et al. Parallel-stacked flexible organic light-emitting diodes for wearable photodynamic therapeutics and color-tunable optoelectronics. ACS Nano 14, 15688–15699 (2020).
Chung, H. et al. The nuts and bolts of low-level laser (Light) therapy. Ann. Biomed. Eng. 40, 516–533 (2012).
Kwak, K., Thanthirige, V. D., Pyo, K., Lee, D. & Ramakrishna, G. Energy gap law for exciton dynamics in gold cluster molecules. J. Phys. Chem. Lett. 8, 4898–4905 (2017).
Zampetti, A., Minotto, A. & Cacialli, F. Near-infrared (NIR) organic light-emitting diodes (OLEDs): Challenges and opportunities. Adv. Funct. Mater. 29, 1905825 (2019).
Fang, F. et al. Near-infrared thermally activated delayed fluorescence nanoparticle: A metal-free photosensitizer for two-photon-activated photodynamic therapy at the cell and small animal levels. Small 18, 1–10 (2022).
Xue, J. et al. Highly efficient thermally activated delayed fluorescence via J-aggregates with strong intermolecular charge transfer. Adv. Mater. 31, 1–9 (2019).
Ye, H. et al. Near-infrared electroluminescence and low threshold amplified spontaneous emission above 800 nm from a thermally activated delayed fluorescent emitter. Chem. Mater. 30, 6702–6710 (2018).
Yuan, Y. et al. Over 10% EQE near-infrared electroluminescence based on a thermally activated delayed fluorescence emitter. Adv. Funct. Mater. 27, 1–5 (2017).
Kim, D. H. et al. High-efficiency electroluminescence and amplified spontaneous emission from a thermally activated delayed fluorescent near-infrared emitter. Nat. Photonics 12, 98–104 (2018).
Tuong Ly, K. et al. Near-infrared organic light-emitting diodes with very high external quantum efficiency and radiance. Nat. Photonics 11, 63–68 (2017).
Cao, L., Li, J., Zhu, Z. Q., Huang, L. & Li, J. Stable and efficient near-infrared organic light-emitting diodes employing a platinum(II) porphyrin complex. ACS Appl. Mater. Interfaces 13, 60261–60268 (2021).
Graham, K. R. et al. Extended conjugation platinum(II) porphyrins for use in near-infrared emitting organic light emitting diodes. Chem. Mater. 23, 5305–5312 (2011).
Wang, S. F. et al. Highly efficient near-infrared electroluminescence up to 800 nm using platinum(II) phosphors. Adv. Funct. Mater. 2002173, 1–9 (2020).
Sommer, J. R. et al. Efficient near-infrared polymer and organic light-emitting diodes based on electrophosphorescence from (tetraphenyltetranaphtho[2,3]porphyrin)platinum(II). ACS Appl. Mater. Interfaces 1, 274–278 (2009).
Lee, T. C. et al. Rational design of charge-neutral, near-infraredemitting osmium(ll) complexes and oled fabrication. Adv. Funct. Mater. 19, 2639–2647 (2009).
Yuan, Y. et al. Boosting efficiency of near-infrared organic light-emitting diodes with Os(II)-based pyrazinyl azolate emitters. Adv. Funct. Mater. 30, 1–8 (2020).
Liao, J. L. et al. Near infrared-emitting tris-bidentate Os(ii) phosphors: Control of excited state characteristics and fabrication of OLEDs. J. Mater. Chem. C 3, 4910–4920 (2015).
Park, Y. et al. Thienothiophenyl-isoquinoline iridium complex-based deep red to near-infrared organic light-emitting diodes with low driving voltage and high radiant emittance for practical biomedical applications. Adv. Photonics Res. 2, 2100121 (2021).
Wang, X. Q. et al. Over 800 nm emission via harvesting of triplet excitons in exciplex organic light-emitting diodes. J. Phys. Chem. Lett. 12, 6034–6040 (2021).
Xue, J. et al. Homoleptic facial Ir(III) complexes via facile synthesis for high-efficiency and low-roll-off near-infrared organic light-emitting diodes over 750 nm. Chem. Mater. 29, 4775–4782 (2017).
You, C. et al. σ-π and p-π conjugation induced NIR-emitting iridium(iii) complexes anchored by flexible side chains in a rigid dibenzo[a, c]phenazine moiety and their application in highly efficient solution-processable NIR-emitting devices. J. Mater. Chem. C 8, 7079–7088 (2020).
Xin, L., Xue, J., Lei, G. & Qiao, J. Efficient near-infrared-emitting cationic iridium complexes based on highly conjugated cyclometalated benzo[g]phthalazine derivatives. RSC Adv. 5, 42354–42361 (2015).
Tao, R. et al. High-efficiency near-infrared organic light-emitting devices based on an iridium complex with negligible efficiency roll-off. J. Mater. Chem. C 1, 6446–6454 (2013).
Zhang, Y., Li, Q., Cai, M., Xue, J. & Qiao, J. An 850 nm pure near-infrared emitting iridium complex for solution-processed organic light-emitting diodes. J. Mater. Chem. C 8, 8484–8492 (2020).
Cui, Y. et al. The critical role of anode work function in non-fullerene organic solar cells unveiled by counterion-size-controlled self-doping conjugated polymers. Chem. Mater. 30, 1078–1084 (2018).
Kumar, C. V. et al. Solution processed organic solar cells based on A-D-D′-D-A small molecule with benzo[1,2-b:4,5-b′]dithiophene donor (D′) unit, cyclopentadithiophene donor (D) and ethylrhodanine acceptor unit having 6% light to energy conversion efficiency. J. Mater. Chem. A 3, 4892–4902 (2015).
Lo, C. K. et al. Every atom counts: Elucidating the fundamental impact of structural change in conjugated polymers for organic photovoltaics. Chem. Mater. 30, 2995–3009 (2018).
Cai, Y. et al. Achieving organic solar cells with efficiency over 14% based on a non-fullerene acceptor incorporating a cyclopentathiophene unit fused backbone. J. Mater. Chem. A 8, 5194–5199 (2020).
Kim, H. et al. A tuned alternating D-A copolymer hole-transport layer enables colloidal quantum dot solar cells with superior fill factor and efficiency. Adv. Mater. 32, 1–7 (2020).
Cheng, P., Li, G., Zhan, X. & Yang, Y. Next-generation organic photovoltaics based on non-fullerene acceptors /639/301/299/946 /639/624/399 review-article. Nat. Photonics 12, 131–142 (2018).
Yi, Y. Q. Q. et al. Small molecule acceptors with a nonfused architecture for high-performance organic photovoltaics. Chem. Mater. 31, 904–911 (2019).
Kim, K. H. et al. Controlling emitting dipole orientation with methyl substituents on main ligand of iridium complexes for highly efficient phosphorescent organic light-emitting diodes. Adv. Opt. Mater. 3, 1191–1196 (2015).
Kim, K. H. & Kim, J. J. Origin and control of orientation of phosphorescent and TADF dyes for high-efficiency OLEDs. Adv. Mater. 30, 1–19 (2018).
Cardona, C. M., Li, W., Kaifer, A. E., Stockdale, D. & Bazan, G. C. Electrochemical considerations for determining absolute frontier orbital energy levels of conjugated polymers for solar cell applications. Adv. Mater. 23, 2367–2371 (2011).
Su, Y. J. et al. Highly efficient red electrophosphorescent devices based on iridium isoquinoline complexes: Remarkable external quantum efficiency over a wide range of current. Adv. Mater. 15, 884–888 (2003).
Tian, N. et al. Structure-property relationship of red- and green-emitting iridium(III) complexes with respect to their temperature and oxygen sensitivity. Eur. J. Inorg. Chem. 2010, 4875–4885 (2010).
Kim, K. H., Ahn, E. S., Huh, J. S., Kim, Y. H. & Kim, J. J. Design of heteroleptic Ir complexes with horizontal emitting dipoles for highly efficient organic light-emitting diodes with an external quantum efficiency of 38%. Chem. Mater. 28, 7505–7510 (2016).
Kim, K. H., Moon, C. K., Lee, J. H., Kim, S. Y. & Kim, J. J. Highly efficient organic light-emitting diodes with phosphorescent emitters having high quantum yield and horizontal orientation of transition dipole moments. Adv. Mater. 26, 3844–3847 (2014).
Fukagawa, H. et al. Highly efficient and stable red phosphorescent organic light-emitting diodes using platinum complexes. Adv. Mater. 24, 5099–5103 (2012).
Kim, D. H. et al. Highly efficient red phosphorescent dopants in organic light-emitting devices. Adv. Mater. 23, 2721–2726 (2011).
Rajakannu, P. et al. Naphthalene benzimidazole based neutral Ir(III) emitters for deep red organic light-emitting diodes. Inorg. Chem. 59, 12461–12470 (2020).
Drozdov, F. V. et al. Novel cyclopentadithiophene-based D-A copolymers for organic photovoltaic cell applications. Macromol. Chem. Phys. 214, 2144–2156 (2013).
Wang, M. et al. Linear conjugated polymer backbones improve alignment in nanogroove-assisted organic field-effect transistors. J. Am. Chem. Soc. 139, 17624–17631 (2017).
Lee, Y. H. et al. Blue TADF emitters based on B-heterotriangulene acceptors for highly efficient OLEDs with reduced efficiency roll-off. ACS Appl. Mater. Interfaces 13, 45778–45788 (2021).
Acknowledgements
This work was supported by the Engineering Research Center of Excellence (ERC) Program supported by the National Research Foundation (NRF), Korean Ministry of Science & ICT (Grant No. NRF-2017R1A5A1014708). This research was supported by National R&D Program through the National Research Foundation of Korea (NRF) funded by Ministry of Science and ICT (2021M3C1C3097646). This work was supported by the Industrial Strategic Technology Development Program (20011059) funded by MOTIE, Korea. This work was supported by the Technology development Program (2022-00141495) funded by the Ministry of SMEs and Startups (MSS, Korea). This work was supported by the Technology Innovation Program (20018379, Development of high-reliability light-emitting fiber-based woven wearable displays) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea). This work was also supported by the Technology Innovation Program (20018954, Development of low-power pulse oximetry sensors for continuous health monitoring) funded by the Ministry of Trade, Industry & Energy (MOTIE).
Author information
Authors and Affiliations
Contributions
K.-C.C., Y.-H.K., and S.Y. supervised the manuscript. Y.P. and W.L. optimized and analyzed the characteristics of materials and OLED structure. G.L. designed and synthesized two types of NIR materials. All authors discussed the results of the experiment and participated in the preparation of the manuscript. Y.P., G.L., and W.L. contributed equally as main authors. K.-C.C., Y.-H.K., and S.Y. contributed equally as the corresponding author.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, Y., Lee, G.S., Lee, W. et al. Heteroleptic Ir(III)-based near-infrared organic light-emitting diodes with high radiance capacity. Sci Rep 13, 1369 (2023). https://doi.org/10.1038/s41598-023-27487-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-27487-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
