
Abstract
Recent findings of microplastics in marine food such as fish, crabs and shrimps necessitate the need to develop biodegradable packaging materials. This study reports on the development of a biodegradable packing material from cellulose nanofibril-polyvinyl alcohol nanocomposite embedded with silver nanoparticles. Microcrystalline cellulose was isolated from sugarcane bagasse via the kraft process followed by conversion of cellulose I to cellulose II using NaOH/urea/water solution. The nanofibrils were then isolated using (2,2,6,6-Tetramethylpiperidin-1-yl) oxyl (TEMPO) and used as a reinforcing element in polyvinyl alcohol composite prepared through solvent casting. The tensile strength, water solubility, optical properties, water vapor permeability and wettability of the prepared films were then evaluated. The antimicrobial potency of the films was evaluated using the disc diffusion antimicrobial assay against selected microorganisms.
Similar content being viewed by others


Introduction
Escalated enquiries and disturbing reports have been stockpiling over the years concerning how unhealthy plastics are to the consumer more so with reference to food packaging1,2. Marine pollution by plastic debris3,4 and the infiltration of microplastics through the food web to cause hormonal imbalances, asthma, infertility, diabetes, cardiovascular disease and cancer are just but to sum up the magnitude of this problem5,6,7,8. A great urge in the scientific community has thereof been nurtured towards inaugurating bio-polymeric materials as sustainable substitutes to these toxic plastics9. This type of new packaging involves the use of natural sources or derivatives from organic monomers as raw materials10. Biodegradable polymers are classified depending on the raw materials used for their manufacture and the manufacturing processes and methods used to make them11,12,13. They could be obtained from biological sources such as animals, plants, agricultural remains and fossils or synthesized chemically from biopolymer monomeric units like sugars, amino acids etc14,15. Petroleum baseds biodegradable polymers such as polyvinyl alcohol (PVA), polyglycolic acid (PGA) and polylactic acid (PLA) are touted as vaible alternatives for use as packaging materials15,16,17.
Biopolymers, though highly biodegradable and a seemingly great substitute for petroleum based polymers, suffer several major drawbacks. Production of high value composites is hampered by their high processing costs and dismal performance though this is largely overcommed through polymer blending with nanoparticles or other biobased polymers18,19. This approach has been used to produce nanocomposites with enhanced physicochemmical properties such as environmental degradation, mechanical stability, UV shielding, and antimicrobial properties that leverages on the strength of each of the constituent materials20,21,22,23. Polymer blending has become one of the modern techniques for the development of new polymeric materials as it allows the manufacture of products with a larger scope of properties24. The biopolymer provides a compatible environment for the nanoparticles to utilise their high aspect ratio and surface area for maximum reinforcement25,26. PVA composites for various applications have greatly been explored with materials such as graphene, chitosan, cellulose etc27,28,29,30. This is because PVA has an opulence of surface hydroxyl groups making it highly hydrophilic, is non-toxic, has good biodegradability, provides a good framework for nano-composition and has great mechanical properties31. Cellulose alone, being insoluble in water, highly crystalline and fibrous would not be the best material for flexible film formation32. Therefore, cellulose nanofibrils (CNFs) have been used in combination with PVA in several studies to boost tensile strength, antimicrobial activity, swelling properties and biodegradability of these films29,33,34,35,36. The nanofibrils have high dispersibility in polymeric matrices, as they form strong hydrogen bonds with the hydroxyl groups of the polymer to synchronise a fortifying effect to the film37. CNFs have also been applied as anchoring sites for metal ions to serve as hubs for the nano-creation of metal nanoparticles38.
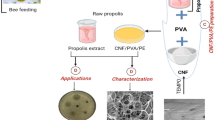
Cellulose-nano silver composite films are shown to be non-toxic, have strong antimicrobial activity and catalytic properties39,40,41. The incorporation of nanoparticles into food packaging has been beneficial because they endow various tailor-made properties in the manufacturing of polymeric films. Typically, polyolefin such as polyethylene, and biopolymers such as polylactic acid (PLA), are often fabricated in nanocomposite with engineered nanoparticles (ENPs) for functional food packaging. This is generally recognized as active packaging as the nanocomposite film can improve structural and thermal stability, endow gas/moisture barrier properties and strong antibacterial activity, ultimately extending the shelf life of the food. This study therefore, sought to prepare a composite film comprising tempo oxidized cellulose nanofibrils (TOCNF) and polyvinyl alcohol (PVA) impregnated with silver nanoparticles (AgNPs) that were generated in-situ. The films were prepared by solvent casting a solution of TOCNF, PVA and AgNPs in plastic moulds then evaporating off the liquid in an oven to give a dry nanocomposit material. The tensile strength, degree of elongation and Young’s modulus were used to evaluate the mechanical properties of the developed film while water vapor permeability, solubility, wettability and microbial inhibition were used to evaluate the films applicability as a packaging material.
Experimental
Preparation of hybrid AgNPs@PVA-CNF nanocomposite films
Raw bagasse was washed with distilled water to remove impurities and dried overnight in an oven set at 80 °C42,43. Cellulose was then obtained from the dried biomass through treating the dried biomass with 10% NaOH (1:10 w/v) at 80 °C for 3 h, washing and drying to constant weight. The alkali-treated biomass was subsequently treated twice with 30% H2O2 (1:6 w/v) with the addition of 50 ml 10% NaOH as a catalyst and left to react for 6 h for bleaching to occur. 1 g of cellulose was then dispersed in 10 ml NaOH/urea/water solution (10:10:80 by volume) then frozen at − 10 °C for 24 h44. After 24 h, the resultant cellulose solution thawed to obtain a transparent solution which was then coagulated with acetic acid (5 ml dope:1 ml acid), washed with distilled water and oven-dried to obtain cellulose II41,45. To obtain CNFs, the 20 g of cellulose were dispersed in 200 ml of water in which 6.4 mmol of TEMPO and 97 mmol of NaBr had previously been dissolved in it. The oxidation was initiated by dropwise addition of 50 ml of 14 vol.% sodium hypochlorite (NaOCl) solution for 10 min at room temperature and the reaction was allowed to progress for 3 h while maintaining the pH at 10 by periodic addition of 0.5 M NaOH solution. After 3 h, the reaction was quenched by addition of ethanol, washed through centrifugation and dried in an oven set at 80 °C41,46,47,48. Hybrid AgNPs@PVA-CNFs were prepared by dispersing 0.2 g of CNF and 1 g of Polyvinyl alcohol (PVA) in 50 ml of 100 mM AgNO3 solution. The reaction vessel was then wrapped with aluminium foil and heated at 80 °C for 4 h to allow for the hydrothermal reduction of silver ion to silver nanoparticles in PVA-CNF41,49,50. Pure PVA films without AgNPs@CNFs were also prepared as a control.
FT-IR functional group analysis
The functional groups present in the AgNPs@PVA-CNF nanocomposites were determined using a Shimadzu FT-IR spectrophotometer (Shimadzu, Japan). The KBr pellets of the samples were prepared by grinding 10 mg of samples with 250 mg KBr (FT-IR grade). The 13 mm KBr pellets were prepared in a standard device under a pressure of 75 kN cm−2 for 3 min. The spectral resolution was set at 4 cm−1 and the scanning range from 400 to 4000 cm−141,51,52,53.
Optical properties of the composite films
To study the optical characteristics of the composite films, a Shimadzu UV–Vis 1800 spectrophotometer (Shimadzu, Japan) was used to evaluate the amount of light transmitted by the films. The transparency of the films and their UV-shielding abilities were determined by calculating their percent transmittance of light at 280 and 660 nm, respectively54.
Thickness of the film
The thickness of the films was measured using a micrometre screw gauge to the nearest 0.001 mm. Measurements were made in at least seven random locations of each preconditioned film and the values were reported as mean ± standard deviation (SD). The mean values were used to determine the tensile strength of the dried films55,56,57,58.
Tensile strength
The tensile strength was measured using an in-house fabricated tensile strength device constructed from a laboratory clamp and stand and metal suspensions. The rectangular-cut (10 × 50 mm) film specimens were sandwiched on both ends between two small metal brackets which were then clamped to a slotted mass hanger where successive 5 g increments of weights were added. The extension lengths were recorded after each addition and the total weight at film breakage was recorded. The tensile strength was calculated by dividing the total load at the breakpoint (in newtons) by the original cross-sectional area of the segment of the specimen that broke (in square meters) according to the following equation58,59
where W = total load, A = cross sectional area of the film.
The percent elasticity (% E) was calculated using the following equation:
where LB and LI are length at break and initial length of the films respectively. The elastic modulus (E) was determined from the slope of the stress–strain curve60.
Water vapor permeability, wettability and degree of solubility
To determine the films’ solubility and swelling capacity, film pieces (20 × 20 mm) were dried at 105 °C to constant weight followed by immersion in 50 mL of distilled water and shaken gently for 24 h at 25 °C. The solutions were poured onto a filter paper (Whatman #1) to recover the undissolved films which were then weighed, and the wettability calculated using Eq. (3):
The film solubility was determined as the percentage dry matter of the remaining film after immersion in water. Film pieces (20 × 20 mm) were dried at 105 °C to constant weight followed by immersion in 50 mL of distilled water and shaken gently for 24 h at 25 °C. The solutions were poured onto a filter paper (Whatman #1) to recover the undissolved films. The samples were then rinsed with distilled water and dried at 105 °C to constant weight and the solubility was calculated according to Eq. (4) 55,57.
To determine the water vapor transmission rate, 40 mL of distilled water was poured into a beaker which was then covered with the film and air tightened by adhesive tape to prevent the escape of vapour as the beaker was heated. The beaker was weighed (Wi) and placed in an oven at 50 °C for 24 h and then re-weighed again (Wf). The WVTR (g/m2h) was then calculated using the Eq. (5):
where Wi and Wf are the initial and final weight of the beaker (g) respectively and A is the cross-sectional area of the beaker (m2)60,61.
Disk diffusion antimicrobial tests
The antimicrobial activity of the prepared films was evaluated using five different pathogenic microbes as test strains: Escherichia coli (Ec), Bacillus subtilis (Bs), Staphylococcus aureus (Sa), Pseudomonas aeruginosa (Pa) and Candida albicans (Ca) by an inhibition zone assay method41,62. To prepare the agar diffusion assay 20 mL of 38 g/L solution of Mueller Hinton Agar was autoclaved at 121 °C for 15 min and cooled to 45 °C. Using a sterile pipette, 100 µL of the bacterial broth culture of the test organism was inoculated with 10 mL aliquots of nutrient broth, spread evenly with a sterile spreader onto sterile petri dishes to get a bacterial lawn and incubated at 37 °C for 24 h. The test circular films with a diameter of 0.5 cm were cut and placed in triplicate on the surface of the agar plates. After 24 h incubation at 37 °C, the inhibition zone diameters were measured using a vaneer caliper to the nearest millimetre in comparison to inhibition diameters of vancomayacin and reported as mean ± standard deviations. Pure PVA film was used as the control41,63.
Results and discussion
Effect of reaction parameters on AgNPs synthesis
In situ synthesis of AgNPs within cellulose nanofibrils matrix was followed through measurement of its plasmon resonance peak as shown in Figs. 1,2.

Schematic illustration showing the substitution of Ag+ ions onto PVA-TOCNFs, and the subsequent formation of AgNPs@PVA-TOCNFs.

Changes in the surface plasmon reasonance peak (SPR) during the formation of AgNPs@CNFs.
As shown in Fig. 1, when the CNFs were introduced to the AgNO3 solution, the Ag+ ions were paired by ion exchange to the negatively charged carboxylates on the CNF surface. These silver ions were subsequently reduced to silver nanoparticles in a temperature-dependent reaction that was also monitored using UV–Vis spectroscopy (Fig. 2)64. During the reduction of silver ions with CNFs, the colour of the solution turned from colourless to dark reddish-brown as the reaction progressed with a subsequent formation of an absorption peak at 412 nm. This peak has been associated with the plasmon resonance peak of AgNPs in solution and is the first qualitative indicator of the presence of the nanoparticles in solution65. At the beginning, the broader peak was due to fewer AgNPs formed after the conversion of a few Ag+ to Ag0 but as the reaction progressed, the intensity and height of the SPR peak increased over time since more Ag0 are being synthesized65. Figure 2 shows the final colour of the AgNPs suspension which turned from colourless to a weakly yellow colour, then to dark reddish-brown after a while. This is a clear indication of the formation of AgNPs. This brown colour change is very important during film casting since it acts as a shield for UV radiation66. Colour uniformity of the film is an indication of an even distribution of the AgNPs. TOCNFs can absorb di/trivalent metal cations due to the introduction of carboxyl groups at C6 position of TOCNF immobilise the AgNPs49,67. In comparison, the blank (pure AgNO3 solution) showed no spectral change in the λmax or peak intensity over time. This is the SPR peak66, slightly shifting position as more AgNPs are agglomerated and which is the most favourable spectroscopic indicator for the formation of silver nanoparticles68. Figure 3 illustrates the effect of temperature variation and first-order plot of the formation of AgNPs 5 h of reaction time.

Effect of Temperature on the formation of AgNPs (A). Inset First order plot (B) of formation of AgNPs.
There was no significant change in the SPR peaks for temperatures 23 °C and 40 °C. A slight and very faint peak was seen after a while for the 60 °C solution. In comparison to the other tests, AgNPs prepared at 80 °C agglomerated significantly fast, just as also recorded by68. This shows that the preparation temperature for AgNPs plays an important role in their formation. Reaction time is also an important parameter. AgNPs began to form with an increasing concentration within the first 20 min and proceeded on steadily until the last minute. The increase in absorbance can be explained by the increase in the amount of the absorbing species (AgNPs)69,70 and from the data obtained it was found that the rate of AgNPs formation followed first-order kinetics given the high R-value obtained as shown in Fig. 3b. While some might argue that the presence of AgNPs in food packaging material poses a greater risk due to leaching of the nanoparticles, AgNPs antimicrobial packaging material are a promising form of active food packing71. To this end, the addition of AgNPs in polymeric matrices can influence the film permeability which subsequently influences product quality as it prevents the growth of food microorganisms such as E. coli and S. aureus71.
Functional groups present in films
Infrared spectroscopy was used to examine the nano-filling effects of AgNPs onto the PVA film and the results are depicted in Fig. 4.

FTIR spectra of the PVA-based films (a) Ag-PT, (b) PVA-TOCNF, (c) Pure PVA.
The wide absorption peaks around 3394 cm−1 represent the stretching vibrations of hydrogen-bonded (− OH) groups of PVA, TOCNF, PVA-CNF-AgNPs60. This suggests good hydrophilicity of the films as the films can form hydrogen bonds with water72. From IR specta of PVA vibrational bands located at 2937, 1592, 1394, and 1068 cm−1 were associated with CH2 bending, OH vibrations from adsorbed water, CH2 stretching, and C–O–C vibrational frequencies73. On the other hand, TOCNF and nanocomposite spectra had vibrational bands at 1592, 1413, 1075 and 915 cm−1 associated with C = O, symmetric CH2 bending, C–O stretching and CH2 rocking. It should be noted that while PVA had a vibrational band at 1597 region, this band could not be linked to C = O band since pure PVA does not have carbonyl functional groups as compared to TOCNF and films which do have this group74. In the presence of metallic species, it has been reported that the vibrational band of C = O present in COOH shifts due to formation of carboxylate group containing metal ions. A weak band at 2943 cm−1 characteristic for C-H stretching75 is common to all films. Additional characteristic absorption bands at 1400 and 1075 cm−1 are also of C–H and O–H bending vibrations, respectively41,65.
Tensile strength and transmittance of composite films
Figures 5 and 6 show the mechanical properties of the two prepared films.

Comparison of the mechanical properties of the pure PVA and AgNPs@PVA-CNF composite films.

Stress and strain curves of PVA and AgNPs@CNF-PVA composite films.
From the results, the tensile strength was increased from 1666 to 4001.7 Pa when CNF and AgNPs were incorporated in PVA matrix. This shows that blending AgNPs-TOCNF into the PVA matrix produces stronger interactions between the nanofiller and polymer matrix which limits the matrix motion49. While this is case, percent elongation on the other hand decreased when the filler materials were added to the matrix which is an indication of increased flexibility of the films. On the other hand, the optical transmittance of the composite films was lower as compared to pure PVA films. Pure PVA films had a transmittance of 66.5% while AgNPs@CNF-PVA had a percent transmittance of 9.51 at 600 nm. Light-induced reaction led to the destruction of chlorophyll which leads to bleaching of certain vegetables, destruction of riboflavin in milk, oxidation of vitamin C, oxidation of carotenoid pigments and discoloration of fresh meat76. Hence the use of packing material that prevents exposure to light prevents food spoilage resulting from light-induced chemical spoilage. Thus, the low transmittance of AgNPs@CNF-PVA prevents exposure of food to light and in the long run reduces food spoilage because of light-induced chemical reactions76.
Water vapour permeability (WVP), wettability (W) and solubility (S)
Figure 7 shows WVP, WS, and wettability values of the prepared films.

Moisture content, water vapor permeability, and water contact angle of agar and agar/cellulose composite film.
WVP is an important property in food packaging applications. Packaging material is the barrier between the exterior surrounding atmosphere and the packaged material inside74. The film is required to reduce to the minimum the moisture transfer from the outer surrounding, which would tamper with the freshness of the packaged food. In this study, pure PVA film had the very highest value: 467.49 g/smPa, whereas AgNPs-TOCNFs/PVA blended film had a very low value: 34.27 g/smPa. PVA is very hydrophilic making the film very unstable in aqueous solutions. This means more water vapour easily passes through the pure PVA film60. The TOCNFs in the composite film acts as an impermeable nanofiller to the PVA making the film more tortuous. It also complexes the molecular structure of the PVA providing the -COOH groups to bind to the − OH groups of the PVA thus reducing their hydrophilicity77. Extra moisture in the food allows for the development of microorganisms which in turn will render the food unsuitable for consumption after a very short span. With a very low WVP value, a film can extend food shelf-life up to considerable periods. As shown in Fig. 7, PVA is very soluble in water giving water solubility values of up to 60%. Upon blending the film with AgNPs-TOCNFs the film achieves more stability in water as the value considerably goes down to 26%. Several extrinsic factors such as temperature, humidity, oxygen, and water vapour have been reported as contributing factors to food spoilage during storage as bacterial growth is preferably favoured in moist conditions76, making the food harmful for human consumption71.
X-ray diffractograms
Figure 8 depicts X-ray diffractograms of CNF, PVA and AgNPs@PVA-CNF.

X-ray diffractograms of (a) CNF, (b) PVA and (c) AgNPs@PVA-CNF composite films.
Figure 8 shows the XRD patterns and corresponding crystallinity index (CrI) of CNF, PVA and AgNPs@PVA-CNF. Cellulose is composed of both crystalline and amorphous domains which tend to influence the XRD diffractogram observed41,43,78. The diffractogram of cellulose isolated from bagasse composed of a peaks at at 2 θ angles of 16°, 22°, 34°, respectively, and were attributed to the diffraction planes of (101), (002) and (040) crystalline plane cellulose 41,43,78. The diffractograms of PVA on the other, had one sharp peak with a shoulder at 2 θ = 19° and another small peak at 2 θ = 40° which upon incorporation of CNF within its marix led to overlapping of CNF and PVA peak that resulted in a broad peak centred at 2 θ = 20°. Sharp crystalline reflections, with a strong maximum at d = 4.68 Å (2 θ = 19.4°) and a shoulder at d = 4.43 Å (2θ = 20°), typical of the crystalline atactic PVA and they correspond to 101̄ and 101 reflections, respectively79. With respect to AgNPs@PVA-CNF, sharph distinct peaks at 2θ = 38, 44, 64, 77, and 82° associated with 111, 200, 220, 311 diffraction planes of silver nanoparticles respectively.
TGA thermograms of nanocomposite films
The thermal profile of PVA before and after incorporation of CNF and AgNPs were evaluated and the results are depicted in Figs. 9,10.

TGA thermograms of PVA and AgNPs@PVA-CNF composite films.

TGA thermograms of PVA and AgNPs@PVA-CNF composite films.
As can be seen from Figs. 9,10, the degradation temperature of the neat PVA films was 365 °C. On the other hand, the degradation temperature shifted to 170 °C when AgNPs@CNF was incorporated within its matrix. This shift in the degradation temperature was attributed to presence of CNF and AgNPs within the composite matrix. It should be noted that CNF has been shown to have two degradation cycles with the first one occurring between 50 and 100 °C attributed to adsorbed water while the second degradation cycle occurring between 220 and 380 °C attributed to breakdown of glucose chains41,80. In this study, the composite films had three degradation points at 45, 170 and 336 °C attributed to adsorbed water, breakdown of glucose units in cellulose and characteristic degradation of PVA via dehydration (or elimination of water from the PVA molecules), which resulted in the formation of polyene inter-mediate41,80. However, while it has been reported that strong hydrogen bonding interaction between the TOCNs and PVA matrix increase the thermal stability of the resultant composite films, this was not the case in this study. Here, presence of AgNPs within the matrix seemed to have played a role in the reduction of the thermal stability as the degradation temperature of the resultant film were lower than those observed in the neat films. It has been observed that that the disruption of hydrogen bond interactions due to the association of AgNPs with the COOH group introduced at C6 leads to the decrease in thermal stability of AgNPs@CNF41,80.
SEM micrographs
Figure 11 depicts SEM micrographs of CNF and AgNPs@CNF composites.

SEM micrographs of (a) CNF and (b) AgNPs@CNF.
From Fig. 11, it can be observed that cellulose was composed of rigid smooth fibers entangled amongst themselves without presence of AgNPs. In situ reduction of AgNPs could be observed in fibers afterwards as the fiber surface was decorated with small spherical AgNPs within the fibril network. It should be noted that celluloe nanofibrils agglomerate upon drying hence the absence of isolated fibrils with smaller sizes. However, the sizes of CNF from bagasse have been reported in a different study which also reported on the presence of AgNPs41. From Fig. 12, presence of AgNPs was confirmed from the SEM micrographs of nanocomposite consisting of AgNPs, CNF and PVA in which the sizes of AgNPs embedded within the composite network was found to be between 60 and 240 nm.

Size distribution of AgNPs embedded within PVA-CNF composite. Inset SEM micrographs of AgNPs@PVA-CNF.
Antimicrobial activity of composite films
The presence of the inhibition zone was used to assess the antimicrobial effect of the film towards the different strains of bacteria and the results are depicted in Figs. 13,14.

Inhibition of nanocomposite against C. albicans (Ca), B. subtili (Bs), and P. aeruginosa (Pa), E. coli (Ec).

Inhibition zone assay of pure AgNPs@CNF-PVA films and vancomycin on the growth of C. albicans (Ca), E. coli (Ec), P. aeruginosa (Pa), B. subtili (Bs) and P. mirabilis (Pm).
From Figs. 13,14, pure PVA film did not show any antimicrobial activity, since there was no inhibition zone recorded. In comparison, the AgNPs@CNF-PVA composite film formed strong inhibition zones around the disk as shown in Fig. 9, thus it can be considered to have antimicrobial abilities. By comparing these results with that of known standards as depicted in Fig. 10, Vancomycin had inhibitoty effects against all the bacteria used in this study apart from C. albicans and B. subtili (Bs). The AgNPs@CNF-PVA composite film ensures some degree of inhibition across all the tested strains, unlike the standards. The antimicrobial efficacy was evaluated through the analysis of known antibacterial standards which showed limited toxicity towards certain bacteria and demonstrated enhanced antimicrobial effects towards others as shown in Fig. 14. Silver exhibits strong bactericidal properties for a wide spectrum of bacteria, viruses and fungi71,81,82. AgNPs can attach themselves onto the bacterial membrane forming clusters that consistently tamper with the integrity of the cell wall to cause membrane damage, leading to bacterial death83,84. Metallic silver in itself is inert but it can react with moisture from the atmosphere to give silver ions85 which are highly toxic to most bacteria86,87. With these antibacterial test results, the hybrid AgNPs@CNF-PVA composite film has the potential for antibacterial packaging material.
Conclusion
Hybrid antimicrobial AgNPs@CNF-PVA composite films were prepared and the applicability to function as a food packaging material was determined through measurement of their water vapour permeability (WVP), wettability (W) and solubility (S). The films were found to have excellent mechanical properties given their high tensile strength, Young’s modulus and their percent elongation. The composite films were found to have good antimicrobial properties as they were able to inhibit the growth of both gram-positive, gram-negative bacteria and a fungus used in this study. This inhibition was attributed to the presence of silver nanoparticles which have been reported to have antimicrobial properties against various microorganisms as in the case of this study. As such, with further modifications and blending with other polymers, this composite material can be an excellent replacement for petroleum-based packaging material which have been reported to be environmental contaminants.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Bott, J., Störmer, A. & Franz, R. Migration of nanoparticles from plastic packaging materials containing carbon black into foodstuffs. Food Addit. Contam. 31, 1769–1782. https://doi.org/10.1080/19440049.2014.952786 (2014).
Rudel, R. A. et al. Food packaging and bisphenol A and bis(2-ethyhexyl) phthalate exposure: Findings from a dietary intervention. Environ. Health Perspect. 119, 914–920. https://doi.org/10.1289/ehp.1003170 (2011).
Eriksen, M. et al. Plastic pollution in the world’s oceans: More than 5 trillion plastic pieces weighing over 250,000 Tons Afloat at sea. PLoS ONE 9, 1–15. https://doi.org/10.1371/journal.pone.0111913 (2014).
Wilcox, C., Van Sebille, E., Hardesty, B. D. & Estes, J. A. Threat of plastic pollution to seabirds is global, pervasive and increasing. Proc. Natl. Acad. Sci. U. S. A. 112, 11899–11904. https://doi.org/10.1073/pnas.1502108112 (2015).
Smith, M., Love, D. C., Rochman, C. M. & Neff, R. A. Microplastics in seafood and the implications for human health. Curr. Environ. Heal Rep. 5, 375–386. https://doi.org/10.1007/s40572-018-0206-z (2018).
Melzer, D. et al. Association of urinary bisphenol A concentration with heart disease: Evidence from NHANES 2003/06. PLoS ONE https://doi.org/10.1371/journal.pone.0008673 (2010).
Singh, S. & Li, S. S. L. Phthalates: Toxicogenomics and inferred human diseases. Genomics 97, 148–157. https://doi.org/10.1016/j.ygeno.2010.11.008 (2011).
Manikkam, M., Tracey, R., Guerrero-Bosagna, C. & Skinner, M. K. Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PLoS ONE https://doi.org/10.1371/journal.pone.0055387 (2013).
Siracusa, V., Rocculi, P., Romani, S. & Rosa, M. D. Biodegradable polymers for food packaging: A review. Trends Food Sci. Technol. 19, 634–643. https://doi.org/10.1016/j.tifs.2008.07.003 (2008).
Cha, D. S. & Chinnan, M. S. Biopolymer-based antimicrobial packaging: A review. Crit. Rev. Food Sci. Nutr. 44, 223–237. https://doi.org/10.1080/10408690490464276 (2004).
Arora, A. & Padua, G. W. Review: Nanocomposites in food packaging. J. Food Sci. 75, 43–49. https://doi.org/10.1111/j.1750-3841.2009.01456.x (2010).
Rhim, J. W., Park, H. M. & Ha, C. S. Bio-nanocomposites for food packaging applications. Prog. Polym. Sci. 38, 1629–1652. https://doi.org/10.1016/j.progpolymsci.2013.05.008 (2013).
Mensitieri, G. et al. Processing and shelf life issues of selected food packaging materials and structures from renewable resources. Trends Food Sci. Technol. 22, 72–80. https://doi.org/10.1016/j.tifs.2010.10.001 (2011).
Ojha, S. et al. Nanotechnology in Crop Protection (Elsevier, 2018).
Younes, B. Classification, characterization and the production processes of biopolymers used in the textiles industry. J. Text. Inst. 108, 674–682. https://doi.org/10.1080/00405000.2016.1180731 (2017).
Lopez-Rubio, A., Gavara, R. & Lagaron, J. M. Bioactive packaging: Turning foods into healthier foods through biomaterials. Trends Food Sci. Technol. 17, 567–575. https://doi.org/10.1016/j.tifs.2006.04.012 (2006).
Arrieta, M. P., Samper, M. D., Aldas, M. & López, J. On the use of PLA-PHB blends for sustainable food packaging applications. Materials (Basel) 10, 1–26. https://doi.org/10.3390/ma10091008 (2017).
Souza, V. G. L. & Fernando, A. L. Nanoparticles in food packaging: Biodegradability and potential migration to food—A review. Food Packag. Shelf Life 8, 63–70. https://doi.org/10.1016/j.fpsl.2016.04.001 (2016).
Tang, X. Z., Kumar, P., Alavi, S. & Sandeep, K. P. Recent advances in biopolymers and biopolymer-based nanocomposites for food packaging materials. Crit. Rev. Food Sci. Nutr. 52, 426–442. https://doi.org/10.1080/10408398.2010.500508 (2012).
Tahir, N., Bhatti, H. N., Iqbal, M. & Noreen, S. Biopolymers composites with peanut hull waste biomass and application for crystal violet adsorption. Int. J. Biol. Macromol. 94, 210–220. https://doi.org/10.1016/j.ijbiomac.2016.10.013 (2017).
Abdullayev, E. & Lvov, Y. Halloysite clay nanotubes as a ceramic “skeleton” for functional biopolymer composites with sustained drug release. J. Mater. Chem. B 1, 2894–2903. https://doi.org/10.1039/c3tb20059k (2013).
Rashid, A., Bhatti, H. N., Iqbal, M. & Noreen, S. Fungal biomass composite with bentonite efficiency for nickel and zinc adsorption: A mechanistic study. Ecol. Eng. 91, 459–471. https://doi.org/10.1016/j.ecoleng.2016.03.014 (2016).
Fortunati, E. et al. Carbon nanotubes and silver nanoparticles for multifunctional conductive biopolymer composites. Carbon 49, 2370–2379. https://doi.org/10.1016/j.carbon.2011.02.004 (2011).
Zhan, F. et al. Facile solvothermal preparation of Fe 3 O 4 -Ag nanocomposite with excellent catalytic performance. RSC Adv. 9, 878–883. https://doi.org/10.1039/c8ra08516a (2019).
Rangabhashiyam, S. & Selvaraju, N. Evaluation of the biosorption potential of a novel Caryota urens inflorescence waste biomass for the removal of hexavalent chromium from aqueous solutions. J. Taiwan Inst. Chem. Eng. 47, 59–70. https://doi.org/10.1016/j.jtice.2014.09.034 (2015).
Yang, Z., Peng, H., Wang, W. & Liu, T. Crystallization behavior of poly(ε-caprolactone)/layered double hydroxide nanocomposites. J. Appl. Polym. Sci. 116, 2658–2667. https://doi.org/10.1002/app (2010).
Zhao, R. et al. Water-insoluble sericin/β-cyclodextrin/PVA composite electrospun nanofibers as effective adsorbents towards methylene blue. Colloids Surfaces B 136, 375–382. https://doi.org/10.1016/j.colsurfb.2015.09.038 (2015).
Zubair, N. A., Rahman, N. A., Lim, H. N. & Sulaiman, Y. Production of conductive PEDOT-coated PVA-GO composite nanofibers. Nanoscale Res. Lett. https://doi.org/10.1186/s11671-017-1888-0 (2017).
Pang, Z. et al. A room temperature ammonia gas sensor based on cellulose/TiO2/PANI composite nanofibers. Colloids Surfaces A 494, 248–255. https://doi.org/10.1016/j.colsurfa.2016.01.024 (2016).
Sargazi, G., Afzali, D., Mostafavi, A. & Ebrahimipour, S. Y. Synthesis of CS/PVA biodegradable composite nanofibers as a microporous material with well controllable procedure through electrospinning. J. Polym. Environ. 26, 1804–1817. https://doi.org/10.1007/s10924-017-1080-8 (2018).
Cai, R. et al. Synthesis and antimicrobial activity of mesoporous hydroxylapatite/zinc oxide nanofibers. Mater. Des. 87, 17–24. https://doi.org/10.1016/j.matdes.2015.08.004 (2015).
Yang, X., Reid, M. S., Olsén, P. & Berglund, L. A. Eco-friendly cellulose nanofibrils designed by nature: Effects from preserving native state. ACS Nano 14, 724–735. https://doi.org/10.1021/ACSNANO.9B07659/SUPPL_FILE/NN9B07659_SI_002.MP4 (2020).
Lu, H. et al. Electrospun water-stable zein/ethyl cellulose composite nanofiber and its drug release properties. Mater. Sci. Eng. C 74, 86–93. https://doi.org/10.1016/j.msec.2017.02.004 (2017).
Cai, J. et al. Electrospun composite nanofiber mats of Cellulose@Organically modified montmorillonite for heavy metal ion removal: Design, characterization, evaluation of absorption performance. Compos. Part A 92, 10–16. https://doi.org/10.1016/j.compositesa.2016.10.034 (2017).
Liao, N. et al. Electrospun bioactive poly (e{open}-caprolactone)-cellulose acetate-dextran antibacterial composite mats for wound dressing applications. Colloids Surfaces A 469, 194–201. https://doi.org/10.1016/j.colsurfa.2015.01.022 (2015).
Aboamera, N. M. et al. An effective removal of organic dyes using surface functionalized cellulose acetate/graphene oxide composite nanofibers. Cellulose 25, 4155–4166. https://doi.org/10.1007/s10570-018-1870-8 (2018).
Fahma, F., Hori, N., Iwata, T. & Takemura, A. Preparation and characterization of polychloroprene nanocomposites with cellulose nanofibers from oil palm empty fruit bunches as a nanofiller. J. Appl. Polym. Sci. 131, 1–6. https://doi.org/10.1002/app.40159 (2014).
Yang, G. et al. Hydrothermal synthesis of bacterial cellulose/AgNPs composite: A “green” route for antibacterial application. Carbohydr. Polym. 87, 2482–2487. https://doi.org/10.1016/j.carbpol.2011.11.017 (2012).
Shankar, S., Wang, L. F. & Rhim, J. W. Preparations and characterization of alginate/silver composite films: Effect of types of silver particles. Carbohydr. Polym. 146, 208–216. https://doi.org/10.1016/j.carbpol.2016.03.026 (2016).
Shankar, S. et al. Wound healing potential of green synthesized silver nanoparticles prepared from Lansium domesticum fruit peel extract. Mater. Express 5, 159–164. https://doi.org/10.1166/mex.2015.1225 (2015).
Madivoli, E. S. et al. Cellulose-based hybrid nanoarchitectonics with silver nanoparticles: Characterization and Antimicrobial potency. J. Inorg. Organomet. Polym. Mater. 32, 854–863. https://doi.org/10.1007/s10904-021-02212-w (2022).
Di Giorgio, L., Salgado, P. R., Dufresne, A. & Mauri, A. N. Nanocelluloses from phormium (Phormium tenax) fibers. Cellulose https://doi.org/10.1007/s10570-020-03120-x (2020).
Ciolacu, D., Ciolacu, F. & Popa, V. I. Amorphous cellulose—Structure and characterization. Cellul. Chem. Technol. 45, 13–21 (2011).
Chen, X. et al. Effects of polymorphs on dissolution of cellulose in NaOH/urea aqueous solution. Carbohydr. Polym. 125, 85–91. https://doi.org/10.1016/j.carbpol.2015.02.054 (2015).
Tang, Z. et al. TEMPO-Oxidized cellulose with high degree of oxidation. Polymers (Basel) https://doi.org/10.3390/polym9090421 (2017).
Masruchin, N., Park, B. D., Causin, V. & Um, I. C. Characteristics of TEMPO-oxidized cellulose fibril-based hydrogels induced by cationic ions and their properties. Cellulose 22, 1993–2010. https://doi.org/10.1007/s10570-015-0624-0 (2015).
Iwamoto, S. et al. Comparison study of TEMPO-analogous compounds on oxidation efficiency of wood cellulose for preparation of cellulose nanofibrils. Polym. Degrad. Stab. 95(8), 1394–1398 (2010).
Saito, T. et al. Homogeneous suspensions of individualized microfibrils from TEMPO-catalyzed oxidation of native cellulose. Biomacromol 7, 1687–1691. https://doi.org/10.1021/bm060154s (2006).
Xu, X. et al. Properties of novel polyvinyl alcohol/cellulose nanocrystals/silver nanoparticles blend membranes. Carbohydr. Polym. 98, 1573–1577. https://doi.org/10.1016/j.carbpol.2013.07.065 (2013).
Fortunati, E. et al. Cellulose nanocrystals extracted from okra fibers in PVA nanocomposites. J. Appl. Polym. Sci. 128, 3220–3230. https://doi.org/10.1002/app.38524 (2013).
Ponce, C. et al. Functionalization of agave cellulose nanoparticles and its characterization by microscopy and spectroscopy techniques. Microsc. Microanal. 19, 200–201. https://doi.org/10.1017/S1431927613002997 (2013).
Madivoli, E. S. et al. Synthesis and characterization of dialdehyde cellulose nanofibers from O. sativa husks. SN Appl. Sci. 1, 1–7. https://doi.org/10.1007/s42452-019-0769-9 (2019).
Madivoli, E. S. et al. Isolation of cellulose nanofibers from Oryza sativa residues via TEMPO mediated oxidation. J. Nat. Fibers 5, 1–13. https://doi.org/10.1080/15440478.2020.1764454 (2020).
Shankar, S. & Rhim, J. W. Preparation and characterization of agar/lignin/silver nanoparticles composite films with ultraviolet light barrier and antibacterial properties. Food Hydrocoll. 71, 76–84. https://doi.org/10.1016/j.foodhyd.2017.05.002 (2017).
Zimet, P. et al. Physico-chemical and antilisterial properties of nisin-incorporated chitosan/carboxymethyl chitosan films. Carbohydr. Polym. 219, 334–343. https://doi.org/10.1016/j.carbpol.2019.05.013 (2019).
Afonso, C. R. et al. Biodegradable antioxidant chitosan films useful as an anti-aging skin mask. Int. J. Biol. Macromol. 132, 1262–1273. https://doi.org/10.1016/j.ijbiomac.2019.04.052 (2019).
Kanai, N. et al. Structural characterization of cellulose nanofibers isolated from spent coffee grounds and their composite films with poly(vinyl alcohol): A new non-wood source. Cellulose 27, 5017–5028. https://doi.org/10.1007/s10570-020-03113-w (2020).
American Society for Testing and Materials. Standard Test Method for Tensile Properties of Thin Plastic Sheeting (ASTM International, 2018).
Stevens, E. S. & Poliks, M. D. Tensile strength measurements on biopolymer films. J. Chem. Educ. 80, 810–812. https://doi.org/10.1021/ed080p810 (2003).
Chowdhury, S. et al. Poly(vinyl) alcohol crosslinked composite packaging film containing gold nanoparticles on shelf life extension of banana. Food Packag. Shelf Life 24, 100463. https://doi.org/10.1016/j.fpsl.2020.100463 (2020).
Ernest Ravindran, R. S., Subha, V. & Ilangovan, R. Silver nanoparticles blended PEG/PVA nanocomposites synthesis and characterization for food packaging. Arab. J. Chem. https://doi.org/10.1016/j.arabjc.2020.05.005 (2020).
Cai, R. et al. One-step synthesis of silver nanoparticles on polydopamine-coated sericin/polyvinyl alcohol composite films for potential antimicrobial applications. Molecules 22, 721. https://doi.org/10.3390/molecules22050721 (2017).
Madivoli, E. S. et al. Biosynthesis of iron nanoparticles using Ageratum conyzoides extracts, their antimicrobial and photocatalytic activity. SN Appl. Sci. 1, 1–9. https://doi.org/10.1007/s42452-019-0511-7 (2019).
Ito, H. et al. Cellulose nanofiber nanocomposites with aligned silver nanoparticles. Nanocomposites 4, 167–177. https://doi.org/10.1080/20550324.2018.1556912 (2018).
Pandey, S., Goswami, G. K. & Nanda, K. K. Green synthesis of biopolymer-silver nanoparticle nanocomposite: An optical sensor for ammonia detection. Int. J. Biol. Macromol. 51, 583–589. https://doi.org/10.1016/j.ijbiomac.2012.06.033 (2012).
Kanmani, P. & Rhim, J. W. Physicochemical properties of gelatin/silver nanoparticle antimicrobial composite films. Food Chem. 148, 162–169. https://doi.org/10.1016/j.foodchem.2013.10.047 (2014).
Van Eerdenbrugh, B., Alonzo, D. E. & Taylor, L. S. Influence of particle size on the ultraviolet spectrum of particulate-containing solutions: Implications for in-situ concentration monitoring using UV/Vis fiber-optic probes. Pharm. Res. 28, 1643–1652. https://doi.org/10.1007/s11095-011-0399-4 (2011).
Bindhu, M. R. & Umadevi, M. Synthesis of monodispersed silver nanoparticles using Hibiscus cannabinus leaf extract and its antimicrobial activity. Spectrochim. Acta A Mol. Biomol. Spectrosc. 101, 184–190. https://doi.org/10.1016/j.saa.2012.09.031 (2013).
Prathna, T. C., Chandrasekaran, N., Raichur, A. M. & Mukherjee, A. Biomimetic synthesis of silver nanoparticles by Citrus limon (lemon) aqueous extract and theoretical prediction of particle size. Colloids Surf. B Biointerfaces 82, 152–159. https://doi.org/10.1016/j.colsurfb.2010.08.036 (2011).
Moskovits, M. & Vlčková, B. Adsorbate-induced silver nanoparticle aggregation kinetics. J. Phys. Chem. B 109, 14755–14758. https://doi.org/10.1021/jp051177o (2005).
Carbone, M., Donia, D. T., Sabbatella, G. & Antiochia, R. Silver nanoparticles in polymeric matrices for fresh food packaging. J. King Saud Univ. Sci. 28, 273–279 (2016).
Wang, Z., Qiao, X. & Sun, K. Rice straw cellulose nano fibrils reinforced poly (vinyl alcohol ) composite films. Carbohydr. Polym. 197, 442–450. https://doi.org/10.1016/j.carbpol.2018.06.025 (2018).
López-De-Dicastillo, C. et al. Development of active polyvinyl alcohol/β-cyclodextrin composites to scavenge undesirable food components. J. Agric. Food Chem. 59, 11026–11033. https://doi.org/10.1021/jf200749f (2011).
Abdulkhani, A. et al. Preparation of cellulose/polyvinyl alcohol biocomposite films using 1-n-butyl-3-methylimidazolium chloride. Int. J. Biol. Macromol. 62, 379–386. https://doi.org/10.1016/j.ijbiomac.2013.08.050 (2013).
Pavaloiu, R. D. et al. Composite films of poly(vinyl alcohol)-chitosan-bacterial cellulose for drug controlled release. Int. J. Biol. Macromol. 68, 117–124. https://doi.org/10.1016/j.ijbiomac.2014.04.040 (2014).
Huis In’t Veld JHJHI. Microbial and biochemical spoilage of foods: An overview. Int. J. Food Microbiol. 33, 1–18. https://doi.org/10.1016/0168-1605(96)01139-7 (1996).
Shankar, S. & Rhim, J. W. Preparation of nanocellulose from micro-crystalline cellulose: The effect on the performance and properties of agar-based composite films. Carbohydr. Polym. 135, 18–26. https://doi.org/10.1016/j.carbpol.2015.08.082 (2016).
Otenda, B. V. et al. Cellulose nanofibrils from sugarcane bagasse as a reinforcing element in polyvinyl alcohol composite films for food packaging. J. Nat. Fibers https://doi.org/10.1080/15440478.2020.1848712 (2020).
Ricciardi, R., Auriemma, F., De Rosa, C. & Lauprêtre, F. X-ray diffraction analysis of poly(vinyl alcohol) hydrogels, obtained by freezing and thawing techniques. Macromolecules 37, 1921–1927. https://doi.org/10.1021/MA035663Q (2004).
Tomić, N. Z. Thermal studies of compatibilized polymer blends. In Compatibilization of Polymer Blends: Micro and Nano Scale Phase Morphologies, Interphase Characterization and Properties 489–510 (Elsevier, 2019).
Ghosh, S. et al. Antimicrobial activity of highly stable silver nanoparticles embedded in agar-agar matrix as a thin film. Carbohydr. Res. 345, 2220–2227. https://doi.org/10.1016/j.carres.2010.08.001 (2010).
Martínez-Abad, A., Sánchez, G., Lagaron, J. M. & Ocio, M. J. On the different growth conditions affecting silver antimicrobial efficacy on Listeria monocytogenes and Salmonella enterica. Int. J. Food Microbiol. 158, 147–154. https://doi.org/10.1016/j.ijfoodmicro.2012.07.010 (2012).
Qing, Y. et al. Potential antibacterial mechanism of silver nanoparticles and the optimization of orthopedic implants by advanced modification technologies. Int. J. Nanomed. 13, 3311–3327 (2018).
Rai, M. K., Deshmukh, S. D., Ingle, A. P. & Gade, A. K. Silver nanoparticles: The powerful nanoweapon against multidrug-resistant bacteria. J. Appl. Microbiol. 112, 841–852. https://doi.org/10.1111/j.1365-2672.2012.05253.x (2012).
Cano, A., Cháfer, M., Chiralt, A. & González-Martínez, C. Development and characterization of active films based on starch-PVA, containing silver nanoparticles. Food Packag. Shelf Life 10, 16–24. https://doi.org/10.1016/j.fpsl.2016.07.002 (2016).
Xiu, Z. M. et al. Negligible particle-specific antibacterial activity of silver nanoparticles. Nano Lett. 12, 4271–4275. https://doi.org/10.1021/nl301934w (2012).
Xu, H. et al. Role of reactive oxygen species in the antibacterial mechanism of silver nanoparticles on Escherichia coli O157:H7. Biometals 25, 45–53. https://doi.org/10.1007/s10534-011-9482-x (2012).
Author information
Authors and Affiliations
Contributions
E.S.M prepared manuscript, conducted the research P.G.K. reviewed manuscript, conceptualization of the idea J.G. and M.E. prepared manuscript, conducted the research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Madivoli, E.S., Kareru, P.G., Gichuki, J. et al. Cellulose nanofibrils and silver nanoparticles enhances the mechanical and antimicrobial properties of polyvinyl alcohol nanocomposite film. Sci Rep 12, 19005 (2022). https://doi.org/10.1038/s41598-022-23305-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-23305-7
This article is cited by
-
Microfibrillated celluloses produced from kraft pulp of coffee parchment
Biomass Conversion and Biorefinery (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
