
Abstract
Progesterone receptors (PRs) are implicated in various cancers since their presence/absence can determine clinical outcomes. The overstimulation of progesterone can facilitate oncogenesis and thus, its modulation through PR inhibition is urgently needed. To address this issue, a novel stacked ensemble learning approach (termed StackPR) is presented for fast, accurate, and large-scale identification of PR antagonists using only SMILES notation without the need for 3D structural information. We employed six popular machine learning (ML) algorithms (i.e., logistic regression, partial least squares, k-nearest neighbor, support vector machine, extremely randomized trees, and random forest) coupled with twelve conventional molecular descriptors to create 72 baseline models. Then, a genetic algorithm in conjunction with the self-assessment-report approach was utilized to determine m out of the 72 baseline models as means of developing the final meta-predictor using the stacking strategy and tenfold cross-validation test. Experimental results on the independent test dataset show that StackPR achieved impressive predictive performance with an accuracy of 0.966 and Matthew’s coefficient correlation of 0.925. In addition, analysis based on the SHapley Additive exPlanation algorithm and molecular docking indicates that aliphatic hydrocarbons and nitrogen-containing substructures were the most important features for having PR antagonist activity. Finally, we implemented an online webserver using StackPR, which is freely accessible at http://pmlabstack.pythonanywhere.com/StackPR. StackPR is anticipated to be a powerful computational tool for the large-scale identification of unknown PR antagonist candidates for follow-up experimental validation.
Similar content being viewed by others

Introduction
Progesterone receptor (PR) has emerged as a potential therapeutic target due to its increasingly recognized role in cancer development and progression. PR is implicated in various cancers such as breast and gynecological cancers (i.e., endometrial and ovarian cancers). However, their most studied implications lie in breast cancer. Cancer is still a major public health concern worldwide, with breast cancer ranking as the most commonly occurring in women. As of 2020, an estimated 2.3 million women were diagnosed with breast cancer resulting in 685,000 deaths globally1. In addition, ovarian and endometrial cancers ranked sixth and eighth out of the top ten cancers in women with incidence rates of 313,959 and 417,367, respectively2. Furthermore, according to the presence or absence of Estrogen receptor (ER) and PR, as well as human epidermal growth factor receptor 2 (Her2), breast cancer can be defined into 3 major groups. These include ER/PR-positive/Her2-negative tumors occurring in approximately 70% of patients, Her2 positive tumors in 15–20% of patients, and triple-negative tumors that lack all receptors in 15% of patients3.
PR belongs to the steroid nuclear receptor family which consists of other members such as ER, androgen receptor (AR), glucocorticoid receptor (GR), and mineralocorticoid receptor (MR). The role of PR in breast cancer through ER modulation has been investigated and as such, the expression of PR can estimate breast cancer prognosis4,5. Progesterone, the female sex hormone and endogenous ligand for PR, is involved in the development of the mammary glands, in the maintenance of pregnancy, and in the female menstrual cycle6,7. In the last decade, extensive research using murine models has recognized the role of progesterone in the development of breast cancer8. Additionally, these studies indicate that progesterone along with estrogen is a significant hormone involved in mammary gland development9. Triggered by estrogen, progesterone stimulates the growth of murine mammary stem cells (MSC) and inherently promotes the regeneration of the mammary gland10. Subsequently, breast cancer risk is amplified due to the increase in MSC numbers in response to progesterone11,12. This facilitates oncogenesis through the accumulation of replication mutations13,14. Therefore, modulation of progesterone through PR inhibition is vital for decreasing MSC and thus, slowing accumulation.
As stated above, breast cancer cells need estrogen and progesterone to grow hence, creating ER/PR positive or negative tumors. Determining the hormone response status of a tumor is the gold standard method and is crucial for patients to suitably benefit from endocrine therapy. Endocrine therapy used for hormone-positive breast cancer patients constitutes an easy-to-approach option for patients as it is generally well tolerated. However, A cohort study conducted on patients with breast cancer indicated that those with ER-positive/PR-negative tumors had a lower survival rate than those with ER-positive/PR-positive tumors15,16. This study suggests that, despite being ER-positive, the loss of PR is associated with the development of resistance in such tumors17. Thus, the research and development of PR-targeted compounds offer a great opportunity.
The lack of specificity regarding PR-targeting drugs exhibits a significant barrier in the clinical translation of PR-targeted therapies. Until now, the use of steroidal PR agonists has been mainly in the areas of oral contraception and postmenopausal hormone therapy18,19,20. However, the in vitro cell growth inhibitory effects afforded by PR antagonists particularly in ovarian, breast, prostate, and bone cancer cells, are now garnering attention as a potential anti-cancer regimen21,22,23,24,25. Furthermore, despite high in vitro antagonist activity against PR, recent clinical trials with mifepristone, a selective progesterone receptor modulator (SPRM), have largely been unsuccessful for ovarian cancer23,26. Despite achieving substantial activity to counter breast cancer in clinical trials, SPRMs have not been well endured by most participants due to their great affinity and responsiveness to GR24. Recently, interest in SPRMs for the treatment of breast cancer has been revived due to the discovery of potent progesterone antagonists having minimal antiglucocorticoid activity, such as telapristone acetate (TPA) and ulipristal acetate (UPA). TPA and UPA are currently in phase II and phase III clinical trials for treating endometriosis and breast cancer, and abnormal uterine bleeding, respectively19,27,28,29. Vilaprisan is another SPRM that underwent efficacy and safety assessment in phase III clinical trials of the oral treatment for uterine fibroids30. However, reports of hepato-cytotoxicity by SPRMs in other clinical trials halted the aforementioned one31. Similarly, concerns regarding hepato-cytotoxicity have limited the advancement of onipristone, a full progesterone antagonist, despite promising activity against breast and gynecological cancers. The clinical trial with onipristone is currently being re-evaluated with a lower drug dosage32. In this regard, there appears to be particular promise in developing newer PR antagonists.
The conventional process of novel drug discovery represents a costly, labor-, and time-intensive venture. These days, the use of computer-aided drug discovery methods has increasingly played essential and fundamental roles as part of the drug discovery process to alleviate the burden of labor and expense. As such, over the past decade, several computational approaches (i.e., molecular docking, quantitative structure–activity relationship (QSAR), and deep-learning) have been utilized for the exploration of PR modulators pertaining to 3D QSAR and docking studies of steroidal33 and non-steroidal34,35,36,37 analogs and, more recently, deep learning-based QSAR38. However, all these approaches might be limited in the quick and accurate identification of new PR antagonists from large-scale uncharacterized compounds. On the other hand, machine learning (ML) methods can utilize only SMILES information without the need for 3D structural information, highlighting their great efficiency in the large-scale identification of compounds.
Thus, in this study, we developed StackPR, a novel and stacked computational model for accurate and large-scale identification of compounds against PR by using only SMILES notation without the use of 3D structural information. Firstly, we comprehensively investigated the impact of different molecular descriptors and ML algorithms in the identification of PR antagonists by utilizing six popular ML algorithms (i.e., logistic regression (LR), partial least squares (PLS), k-nearest neighbor (KNN), support vector machine (SVM), extremely randomized trees (ET) and random forest (RF)) coupled with twelve conventional molecular descriptors. Secondly, a total of 72 different classifiers were developed and considered as baseline models. Then, a genetic algorithm cooperating with the self-assessment-report approach (GA-SAR) algorithm was employed to determine m out of the 72 baseline models for constructing the optimal meta-predictor using the stacking strategy. Thirdly, the SHapley Additive exPlanations (SHAP) algorithm was used to indicate the most important features for StackPR. Finally, molecular docking was conducted to determine the top compounds having a high binding affinity towards PR. The top-scoring compound was then further analyzed for its binding interactions and substructures.
Materials and methods
Dataset construction
The dataset used in this study was collected from the ChEMBL database (Target ID: CHEMBL208; version 25)39, which consisted of an initial set of 5,240 compounds with activity for PR. After the data curation process, a final data set of 1,168 compounds were obtained using IC50 (half-maximal inhibitory concentration) as the bioactivity unit for further investigation. IC50 denotes the amount of drug required for the inhibition of a biological process by half, thus stipulating the potency measure of an antagonist drug in pharmacological research40. Furthermore, Beck et al.41 reports that IC50 can be used effectively in building robust and reliable models to support SAR (structure–activity relationship) studies in drug discovery. Bioactivity thresholds of IC50 ≤ 1 μM and ≥ 10 μM were applied to the final data set of 1,168 to separate compounds into active and inactive groups, respectively. Compounds with biological activity IC50 values ranging between 1 and 10 μM (i.e., intermediate compounds), which consisted of 445 compounds, were not selected for this study. As a result, a high-quality dataset was obtained which consisted of 463 active (positive samples) and 260 (negative samples) inactive compounds. Of these compounds, 370 active and 208 inactive compounds are considered as the training dataset (called TRN515), and the remaining compounds (93 active and 52 inactive compounds) are used as the independent test dataset (called IND145).
Feature engineering
Molecular fingerprints stipulate data regarding the substructures that are inherently present in a molecule and thus, are important in QSAR studies. Using the built-in function of the PADEL-descriptor software, salt was removed and tautomers were standardized during structure pre-processing. Herein, the SMILES notation was used as the input for descriptor calculations. SMILES strings are a useful representation of molecules while gaining the advantages in terms of their storage and handling42. We utilized twelve molecular fingerprints, to generate structural features of the investigated compounds. The twelve fingerprints consisted of AP2D, Circle, CKD, CKDExt, CKDGraph, Estate, FP4, FP4C, Hybrid, KR, MACCS, and PubChem. Details of each fingerprint descriptor can be found in Table 1. Herein, all molecular descriptors can be extracted using the Python environment43.
StackPR framework
In this section, we describe our stacked ensemble learning framework (named StackPR) designed for the high-throughput identification of PR antagonists. Unlike other conventional ensemble strategies, the stacking strategy integrates the strengths of different predictive models without human intervention to generate the final meta-predictor44,45,46,47. To date, numerous previous studies have indicated that the final meta-predictor can potentially attain a more stable predictive performance48,49,50. The overall workflow for the development of StackPR contains three major steps (i.e., baseline model construction, new feature vector generation, and meta-predictor development) as provided in the paragraphs hereafter (Fig. 1).

System flowchart of the proposed StackPR. The overall workflow for the development of StackPR contains three major steps: dataset construction, baseline model construction and new feature vector generation and meta-predictor development.
In the first step, we used twelve different molecular descriptors (AP2D, Circle, CKD, CKDExt, CKDGraph, Estate, FP4, FP4C, Hybrid, KR, MACCS, and PubChem) in combination with six ML algorithms (LR, PLS, KNN, SVM, ET, and RF) to generate a collection of baseline models. Specially, all the molecular descriptors were normalized in the range of 0–1, when training the six ML classifiers. As a result, a total of 72 baseline models were created. Herein, we utilized ET, LR, RF, and SVM classifiers with their optimal parameters, where the search range is recorded in Table 2. In the meanwhile, KNN and PLS classifiers were generated by using their default parameters. All the baseline models were built and optimized using the Scikit-learn v0.22.0 package51 with a tenfold cross-validation test. In addition, we evaluated and analyzed the effect of molecular descriptors and ML classifiers in PR antagonist identification by performing a tenfold cross-validation test to determine the best-performing baseline model in terms of Matthew’s coefficient correlation (MCC). In addition, we evaluated and analyzed the effect of molecular descriptors and ML classifiers in PR antagonist identification by performing a tenfold cross-validation test to determine the best-performing baseline model in terms of Matthew’s coefficient correlation (MCC).
In the second step, all the 72 baseline models were utilized for constructing a new feature vector. Each baseline model can provide the information on the predicted confidence of being PR antagonists. Herein, the predicted confidence is treated as a probabilistic feature (PF), where PF is in the range of 0–1. For a given compound C, its feature vector is created by concatenating all PFs generated by all the 72 baseline models, which can be defined by:
where \({PF}_{BM\left(i\right)}\) is the ith PF derived from the ith baseline model (\(BM(i)\)) of compound C. Thus, \(\mathrm{nFeat}(C)\) is the 72-D feature vector and considered as a new feature vector.
In the last step, a meta-predictor was built by using the 72-D feature vector coupled with the RF algorithm (called mRF model). To improve the discriminative ability of the new feature vector, in this study, we also utilized the GA-SAR algorithm52 to determine m out of the 72 PFs, where the number of m was set within the range of 5–20 with an interval of 1. Specifically, the chromosome of the GA-SAR algorithm contains two main genes, binary (GA-gene) and parametric (GA-chrom). The binary (GA-gene) and parametric (GA-chrom) are the dominant genes of the chromosome of the GA-SAR algorithm, which are used for feature and parameter optimizations, respectively. For the mRF model, the chromosome consists of n = 72 binary genes (bgi) for selecting m important PFs and 3-bit genes for optimizing the parameters of mRF (n_estimators \(\in\) {20, 50, 100, 200, 500}). If bgi = 1, the ith feature is selected to construct the mRF model; otherwise, the ith feature is excluded from the optimal feature set.
Performance evaluation
The performance evaluation results of StackPR and its baseline models were examined in terms of five well-known performance metrics, including MCC, F-value, sensitivity (Sn) and specificity (Sp), and accuracy (ACC)53,54,55 as described follows:
where TP and TN indicate the number of true positives and true negatives, respectively. Moreover, FP and FN represent the number of false positives and false negatives, respectively56,57,58,59,60,61.
Molecular docking
Molecular docking was performed as a virtual screening to identify potential inhibitors for human progesterone receptors. In this study, 1168 bioactive compounds with known bioactivity described using IC50, were collected from the ChEMBL database39 and preprocessed as input ligands for investigation. The molecular structure of the ligands was generated and optimized to achieve low-energy conformers using the OpenBabel software62. The co-crystal structure of the human progesterone receptor in complex with asoprisnil (PDB ID: 2OVH) was retrieved from the Protein Data Bank63 to be used as a receptor molecule for virtual screening. The protein structure was preprocessed by removing water molecules and adding Gasteiger charges and missing hydrogen atoms. The protein structure was additionally cleaned up by repairing bonds and removing non-polar hydrogens and lone pair atoms using MGLTools64. Consequently, grid boxes with the dimensions of 40 × 40 × 60 Angstrom were applied to the center of ligands inside the binding cavity of the protein receptor. Parameters for molecular docking were defined by default with a seed number of 1000 using Autodock Vina65,66. The method was validated using ligand re-binding approaches and the calculated RMSD of the atomic position between co-crystallized ligand and re-binding ligand was observed to be 0.322 Angstroms which is acceptable for further investigations. The binding energy (Kcal/mol) was calculated during virtual screening using the built-in scoring function of Autodock Vina and the compounds exhibiting the lowest binding energy were chosen for further investigation. The docked poses and binding interactions were visualized using the PyMOL Molecular Graphics System, version 2.2.3 (Schrödinger LLC, 2010).
Results and discussion
Analysis of applicability domain
Applicability domain (AD) analysis is key for reliable model predictions which can be utilized for all models45,64,65. Several approaches for AD analysis have been proposed however, herein the t-distributed stochastic neighbor embedding (t-SNE)66,67 was used for visual investigation of the feature distribution pertaining to the twelve molecular fingerprints. Supplementary Fig. S1A–L elucidates the 2D feature distribution space where the TRN515 and IND145 datasets are represented by red and green dots, respectively. In order for the compounds to fall within the AD of the model, compounds from the IND145 dataset must be present in the defined boundary of the TRN515 dataset. Conversely, compounds are considered outside the AD of the model if they fall beyond the established boundary stated above. From Supplementary Fig. S1, the chemical spaces for all twelve molecular fingerprints were observed to be overlapping for both the TRN515 and IND145 datasets. Thus, it can be inferred that the AD is well defined for these twelve molecular fingerprint-based models developed herein.
Performance evaluation of different molecular descriptors and ML algorithms
Here, we assessed the performance of several ML classifiers trained using utilizing twelve molecular descriptors and six ML algorithms. All the 72 classifiers (6 MLs × 12 descriptors) were assessed by using tenfold cross-validation and independent tests. The performance evaluation results of the 72 classifiers are provided in Fig. 2 and Supplementary Tables S1–S4. Note that the classifier with the highest MCC was deemed to be the best-performing classifier in this study. Supplementary Table S3 shows that the top-five molecular descriptors having the highest cross-validation performance were Circle, KR, CKD, CKDExt, and Hybrid with corresponding average ± standard deviation and MCC of 0.834 ± 0.018, 0.802 ± 0.023, 0.797 ± 0.024, 0.796 ± 0.014, and 0.784 ± 0.013, respectively. In the case of performance results of the six ML methods, Supplementary Table S4 shows that ET and RF achieve superior performance in terms of MCC with a range of 0.768–0.770. It could be noticed that the top-five classifiers having the highest cross-validation performance consisted of ET-Circle, RF-Circle, SVM-Circle, LR-Circle, and LR-CKD with corresponding MCC of 0.850, 0.846, 0.843, 0.842 and 0.841, respectively (Fig. 2 and Supplementary Table S1). Interestingly, four out of the top-five classifiers were developed using the Circle descriptor. This indicates that the Circle descriptor was beneficial for the identification of PR antagonists. For the performance on the IND145 dataset, the top-five classifiers yielded MCC with a range of 0.849–0.881. Although the best-performing classifier (ET-Circle) could perform well in the identification of PR antagonists, ensemble models that can automatically integrate the individual strengths of the above-mentioned classifiers are admissible45,49,50,67.

Performance evaluations of top 30 baseline models. (A, B) Cross-validation ACC and MCC of top 30 baseline models. (C, D) Independent test ACC and MCC of top 30 baseline models.
Construction of StackPR
Although the ET-Circle model has yielded good performances, its overall prediction performance is still unsatisfactory for real therapeutic applications. Thus, we herein employed an ensemble approach that could take advantage of several ML-based classifiers to construct a stable stacked model. Specifically, we built two mRF models coupled with two types of new feature vectors, including the 72-D and m-D feature vectors (referred herein as the optimal model and control, respectively). The 72-D feature vector was obtained by all the 72 PFs, while the m-D feature vector was obtained from the GA-SAR algorithm. Table 3 provides the overall performance of the 72-D and m-D feature vectors. After performing the GA-SAR algorithm, we obtained the optimal feature set having m = 10 selected PFs derived from ten baseline models of KNN-AP2D, LR-CKDExt, SVM-CKDExt, ET-CKDExt, KNN-MACCS, RF-PubChem, SVM-KR, LR-FP4, RF-Circle, and PLS-Hybrid. As shown in Table 3, the 10-D feature vector achieves the overall best performance compared with the 72-D feature vector on both the TRN515 and IND145 datasets in approximately all performance metrics, except for AUC. Remarkably, ACC, Sn, and MCC of the 10-D feature vector were 3.45, 4.30, and 7.31% higher than the 72-D feature vector on the independent test dataset. Altogether, the mRF model in conjunction with the 10-D feature vector (called the optimal feature vector) is deemed to be the final model herein and referred to as StackPR for the convenience of discussion.
Stacking improves the prediction performance
In this section, we aim to reveal the effectiveness of the stacking strategy by comparing the performance of StackPR with the top five baseline models as judged by the cross-validation MCC. These baseline models included ET-Circle, RF-Circle, SVM-Circle, LR-Circle, and LR-CKD. The detailed results of their performance evaluations are provided in Fig. 3, Table 4, and Supplementary Table S5. Table 4 shows that StackPR’s ACC, Sn, MCC, and F-value were better than the top five baseline models on both the TRN515 and IND145 datasets. Interestingly, ACC, Sn, and MCC of StackPR were 0.950, 0.970, 0.893, and 0.958, which were 1.90, 2.70, 4.31, and 1.52%, respectively, higher than the best-performing baseline model ET-Circle in the TRN515 dataset. StackPR also attained a better performance as compared with ET-Circle on the IND145 dataset. For the performance on the independent test dataset, StackPR’s ACC, Sn, and MCC were 0.966, 0.978, 0.925, and 0.973, respectively. To be specific, these three-performance metrics were higher than that of the ET-Circle model by 2.07, 3.23, 4.34, and 1.67%, respectively. However, StackPR provided a slightly lower AUC as compared to the ET-Circle model (0.978 versus 0.983), StackPR could identify more true compounds against PR (TP) in terms of both the TRN515 (299 versus 290) and IND145 (91 versus 88) datasets (Table 4 and Supplementary Table S5). These results indicated that the stacking approach used in StackPR effectively integrated the advantages of the baseline models, contributing to the improvement in performance.

Performance comparison of StackPR with the top five baseline models on the Main-TRN (A, B) and Main-IND (C, D) datasets. Prediction results of StackPR with the top five baseline models in terms of MCC, Sn, Sp and MCC (A, C). ROC curves and AUC values of StackPR with the top five baseline models (B, D).
Analysis of new feature vector
In this section, we investigated the performance of the optimal feature vector by testing and comparing its performance with the twelve conventional molecular descriptors. Supplementary Tables S6–S7 provide the predictive performance of the optimal feature vector against the twelve conventional molecular descriptors. For the sake of fairness, the RF classifier was employed to train different models coupled with the twelve molecular descriptors and build respective prediction models. For the convenience of the comparison purpose, we conducted the performance comparison of the optimal feature vector with only the top five molecular descriptors having the highest cross-validation MCC, including Circle, KR, MACCS, FP4, and CKDExt. We noticed that the top five beneficial molecular descriptors achieved the overall best performance as compared with the top five beneficial molecular descriptors on both the TRN515 and IND145 datasets as judged by ACC, Sn, and Sp (Supplementary Tables S6–S7). Remarkably, for the performance on the IND145 dataset, the optimal feature vector’s ACC, Sn, and MCC were 2.07–6.90, 2.15–4.3,0, and 4.34–15.18%, respectively, higher than that of the top five molecular descriptors. In addition, the 2D feature space of the optimal feature vector and the top five molecular descriptors were depicted by using the t-distributed stochastic neighbor embedding (t-SNE)68,69, where the red and green dots represent positive (active compounds) and negative (inactive compounds) samples, respectively (Fig. 4). Overall, we observe that the distributions of the five top-five beneficial molecular descriptors did not show a clear separation between the two classes (Fig. 4A–E). On the other hand, the optimal feature vector was able to provide a clear separation between the two classes (Fig. 4F). Altogether, these results indicate that our proposed 10-D feature vector derived from the combination of several molecular descriptors and ML algorithms had a more discriminative ability to capture the key information of active and inactive compounds against PR, contributing to the performance improvement.

t-distributed stochastic neighbor embedding (t-SNE) distribution of positive and negative samples on the training dataset.
Mechanistic interpretation of StackPR
To better understand the impact governing each feature of the StackPR model, an analysis of the feature importance was conducted. Herein, we utilized the SHAP framework70 to calculate the value of each feature while also shedding light on the output of the model. SHAP is a game-theoretic approach used to explain the output of any ML classifier and represents a crucial role in many bioinformatics applications. The positive and negative SHAP values indicate that the prediction model better favors active or inactive compounds, respectively. Figure 5 shows that the top five informative PFs based on SHAP values were derived from RF-Circle, SVM-KR, LR-CKDExt, ET-CKDExt, and SVM-CKDExt. Interesting, RF-Circle is found in both the top-performing classifier and the top-ranked informative PFs based on SHAP values. As can be seen in Fig. 6, the top-twenty most important informative features as deduced by SHAP values were PubChemFP499, PubChemFP335, PubChemF383, PubChemFP542 PubChemFP566, PubChemFP652, PubChemFP382, PubChemFP338, PubChemFP260, PubChemFP569, PubChemFP392, PubChemFP712, PubChemFP420, PubChemFP717, PubChemFP780, PubChemFP840, PubChemFP590, PubChemFP145, PubChemFP430, and PubChemFP467. In addition, as stated above, high SHAP value (positive scale) with high feature value (represented by red) which are most likely to have an impact on the substructure of the compounds were seen in twelve out of top-twenty informative features (Fig. 6 and Table 5) representing, five aliphatic hydrocarbons (i.e., PubChemFP335, PubChemFP712, PubChemFP717, PubChemFP780, PubChemFP430), five nitrogen-containing (i.e., PubChemFP338, PubChemFP569, PubChemFP392, PubChemFP145, PubChemFP467), one alcohol (i.e., PubChemFP840) and one carbonyl group (i.e., PubChemFP420; which is common to several classes of organic compounds such as aldehyde and ketone). Therefore, it can be inferred that aliphatic hydrocarbons and nitrogen-containing compounds represent substructures that have a high impact on PR antagonism. Further exploration into the PubChem substructure descriptions (Table 5), offers observations that bioisoteric transformations occur when the CH group of compounds containing heteroaromatic rings are substituted with a N atom (i.e., PubChemFP145). This substation exerts antagonistic effects by mirroring the binding of natural ligands71. Furthermore, the above-mentioned features belong mainly to N-methylmethanamine, ethylenediamine, isopropylamine etc. which are all precursors of many significant PR antagonists such as mifepristone, lonaprisan and vilaprisan. However, several steroidal SPRMs with a dimethylamino substituent have shown an increase in liver enzyme upon continued use72. Nevertheless, novel analogues of mifepristone with increased PR selectivity over GR has been identified73. As for the aliphatic substructures observed in Table 5, Nishiyama et al.74, identified that alkyl substitutions at 4-Alkyl analogs of phenanthridin-6(5H)-one skeleton show potent PR antagonistic activity. Additionally, the 1-propynyl substituent at the C-17 position of mifepristone accounts for its high PR binding affinity75. Similarly, Richardson et al.76, discovered that alkyl substituents on the N1 of the indole skeleton played an important role in the binding affinity of compounds to PR whereby changing the substituent from methyl to ethyl and then from ethyl to isopropyl afforded 7- and fivefold improvements in potency, respectively. Moreover, the indole with the isopropyl substituent showed higher PR selectivity over GR and AR in both functional and binding assays76. Taken together, the important PubChem features as determined by SHAP are effective as potential PR antagonist substructures.

Feature importance from StackPR as ranked by SHAP values based on the training datasets. Such SHAP values represent the directionality of features where positive and negatives SHAP values influences the predictions toward positive and negative samples, respectively.

Feature importance from the RF-PubChem model as ranked by SHAP values based on the training datasets. Such SHAP values represent the directionality of features where positive and negatives SHAP values influences the predictions toward positive and negative samples, respectively.
Case study
In this section, we used molecular docking to visualize the binding interactions of compounds to PR in comparison to known modulators by using the AutoDock Vina software66. Out of all the compounds, the top-ten compounds with the highest docking scores indicating the highest binding affinity were selected for evaluation. As can be seen in Supplementary Fig. S2, the docking score of the top-ten compounds were − 12.67, − 12.33, − 11.82, − 11.67, − 11.48, − 11.11, − 11.10, − 11.06, − 11.03, − 11.01 kcal/mol with corresponding IC50 values of 0.33, 0.2, 0.025, 3.64, 0.27, 34, 4.6, 0.63, 0.63 and 19 nM, respectively. Of note, the IC50 values are all in the nM range highlighting that all the top-ten compounds showed strong binding affinity to PR as determined through in vitro experiments.
A further in-depth analysis of these top-ten compounds (Supplementary Fig. S2) reveals the top scaffolds pertaining to a typical steroid skeleton consisting of fused rings with three six-member rings and one five-member ring77 (nine out of ten compounds), nitrogen-containing compounds (nine out of ten compounds), and aliphatic compounds (all ten compounds). The compounds with the aliphatic substructures have a structural similarity not only to progesterone but also to already known PR antagonists such as asoprisnil, mifepristone and onapristone78. In addition, various derivatives of mifepristone such as aglepristone, lilopristone, onapristone, and telapristone have been synthesized and some are currently in clinical trials. Nine out of the top-ten compounds, resembles asoprisnil and mifepristone, which are well-known PR antagonists. Thus, the binding of PR to these compounds with high affinity is valid.
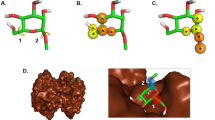
The docked pose of the compound with the highest docking score (i.e., CHEMBL259879; − 12.67 kcal/mol) was further investigated for its binding interactions to PR (PDB ID: 2OVH) and is portrayed in Fig. 7A and C. In addition, the binding interactions of PR (PDBID: 2OVH) with its co-crystallized structure, a SPRM (i.e., asoprisnil) in the docked pose were also elucidated using PyMOL (Fig. 7A,B). Asoprisnil exhibits antagonistic activities in endometrium, ovary, and breast tissues79. As observed from Fig. 7B, the docking pose between PR and asoprisnil, reveal vital interactions in the active site that consists of hydrogen bonds with NE2 and NH2 of residues Q725 and R766 at a distance of 2.91 Å and 2.74 Å, respectively, which are depicted with the blue dash lines. Additionally, residues L721, W755 and L797, were observed to form hydrophobic interactions at distances of 3.53 Å, 3.48 Å and 3.96 Å, respectively (gray dash lines). In addition, a water bridge (light purple dash lines) was shown connecting NH1 of R766 and O14 of asoprisnil. Furthermore, a perpendicular pi-stacking (depicted as a green dash line) with the phenyl ring of residue F778 was also observed at a distance of 5.32 Å. On the other hand, the docked pose of CHEMBL259879 bound to PR (Fig. 7C) reveals the interacting binding pocket residues to be comprised of Q725, and R766 forming hydrogen bonds (shown as blue dash lines) at distances of 3.66 Å, and 3.11 Å, respectively. Moreover, residues L715, L718, L721, L726, L763, W755, F778, L797, and Y890 showed binding through hydrophobic interactions that are represented with gray dash lines. In addition, two pi-pi interactions were observed, one being a staggered stacking (i.e., parallel pi-stacking, shown as a light green dash line) with W755 and the other being a pi-teeing (i.e., perpendicular pi-stacking, shown as a dark green dash line) with F778 at distances of 4.10 Å and 5.12 Å, respectively. Both these types of pi-pi interactions are known to be electrostatically attractive80. These results indicate that the top compound as revealed by molecular docking (i.e., CHEMBL259879) forms interactions in the binding pocket of PR that are comprised of more residues than asoprisnil. These outcomes are in accordance with SPRM binding analysis highlighting the relevance of hydrophobicity in the interactions of the ligand and catalytic residues, L718, and Q72535. Therefore, these interactions could be useful in determining the top substructures needed for a high-affinity PR antagonist. Additionally, competitive binding to PR was determined from the ability of compound CHEMBL259879 to exhibit antagonistic activity in a CHO (chinese hamster ovary) cell line-based assay in PubChem’s bioassay record with an IC50 of 0.33 nM81,82. Overall, these results indicate that CHEMBL259879 could be a good candidate for PR antagonism.

Superimposed docked pose of PR with co-crystallized ligand (PDB ID: 2OVH) (i.e., asoprisnil, A) and the top scoring compound as measured by AutoDock Vina (i.e., CHEMBL259879, A). Close-up views of the binding cavity of PR-asoprisnil (B) and PR-CHEMBL259879 (C). Hydrogen bond and hydrophobic residues are shown in red and green colored text boxes, respectively.
Taking it a step further, we superimposed the ligands asoprisnil and CHEMBL259879 in the PR binding pocket to elucidate their molecular similarities and differences. As can be seen in Fig. 8, CHEMBL259879 overlays well over asoprisnil with differing scaffold substitutions at C11 and C17. A cyclopentone at C11 and a benzene with a pyridine substitution at C17 was revealed for CHEMBL259879 while asoprisnil has a benzaldehyde oxime at C11 and a dimethoxymethyl at C17. Furthermore, a key role in the binding of ligands to PR and/or GR receptor is observed to be modifications made near the C17 propinyl group (Fig. 8)83,84. In addition, substitutions pertaining to ethyl and pentynyl at the hydrophobic C17 produces higher PR antagonism as compared to that of asoprisnil or mifepristone85. Minor changes in the C17 such as a phenyl group with small, stabilizing electron-withdrawing substituents, (i.e., F, Cl, Br, and CF3), was shown to prominently increase the potency of compounds while hightening selectivity over GR86. As can be seen in Supplementary Fig. S2, two of the top-ten compounds (i.e., compounds (3) and (10)) contained those with the above-mentioned properties.

Overlaid structures of PR-Asoprisnil and PR-CHEMBL259879 where the carbons are colored orange and yellow, respectively. The C-11 and C-17 substituents are highlighted to show their effect on antagonist activity and PR selectivity, respectively.
The substitution of 4-(dimethylamino) phenyl group at the C11 position (Fig. 8) is known to define the degree of antagonistic activity, as per the literature83,84. In addition, small substituents such as methyl revealed powerful agonistic properties against PR in comparison to phenyl derivatives which displayed varying degrees of antagonistic activity83,84. Interestingly, we can observe that nine of the top-ten compounds (i.e., compounds (1), (2), (3), (4), (5), (7), (8), (9) and (10)) contain substitutions of either one or two phenyl derivatives at the C11 position. Furthermore, research suggests that, nitrogen heterocycle substitution at C11 (i.e., compounds (1), (4), (5), (9) and (10)) especially in compounds devoid of a center of electronegativity in this region show the highest antagonistic activity82,87. Altogether, the top-ten compounds as determined through molecular docking, have the potential to be good candidate compounds as they have been validated for binding affinity in vitro with IC50 in the nM range. Furthermore, these compounds exhibit molecular substitutions which have been reported as favorable for high PR antagonism while simultaneously increasing selectivity over GR.
StackPR web server
To guarantee that the StackPR predictor can perform the high-throughput identification of PR antagonists in a cost-effective manner, we have developed a user-friendly webserver (named StackPR) which is available at http://pmlabstack.pythonanywhere.com/StackPR. To obtain the prediction results, users are recommended to input the SMILES notation in the textbox. In addition, a step-by-step guideline on the usage of the StackPR webserver is provided at http://pmlabstack.pythonanywhere.com/about_StackPR.
Conclusions
Breast cancer is the most detected cancer among women while gynecological cancers such as ovarian and endometrial cancers rank sixth and eighth, respectively. PR, a steroid nuclear receptor has emerged as a potential therapeutic target owing to its implications for the prognosis and development of breast cancer. Despite extensive research and high in vitro antagonistic activity of many compounds to PR, none have passed phase III clinical trials as yet. Thus, the discovery of newer PR antagonists is greatly needed. Here, we present StackPR, a novel stacked ML-based approach for the high-throughput identification of PR antagonists. We integrated six popular ML algorithms (i.e., ET, KNN, LR, PLS, RF, and SVM) coupled with twelve conventional molecular descriptors (i.e., AP2D, Circle, CKD, CKDExt, CKDGraph, Estate, FP4, FP4C, Hybrid, KR, MACCS, and PubChem) to develop the final meta-predictor using a stacking strategy. Experimental results showed that StackPR achieved impressive predictive performance with ACC, MCC, and AUC of 0.966, 0.925and 0.978, respectively on the independent test dataset. The high MCC value indicates that the proposed StackPR can effectively complement experimental studies to reduce the number of both false-positive and false-negative cases. Furthermore, analysis results based on the SHAP algorithm and molecular docking indicate that aliphatic hydrocarbons and nitrogen-containing substructures as well as substitutions at the C-11 and C-17 carbons of the steroid skeleton were the most important features for having PR antagonist activity. Moreover, non-steroidal derivatives also offer great promise particularly if potent substituents are involved. Remarkably, the docking pose of the top-scoring compound when further analyzed for binding interactions, revealed that hydrogen bonds and hydrophobic interactions were in accordance with reported PR antagonist activities. We anticipate that StackPR will be a powerful and useful computational tool for the large-scale identification of unknown PR antagonist candidates for follow-up experimental validation.
Data availability
All the data used in this study are available at http://pmlabstack.pythonanywhere.com/StackPR.
References
World Health Organization. Breast Cancer. https://www.who.int/news-room/fact-sheets/detail/breast-cancer#:~:text=In%202020%2C%20there%20were%202.3,the%20world's%20most%20prevalent%20cancer. Accessed 9 April 2022.
GLOBOCAN. Estimated number of incident cases worldwide, females, all ages. International Agency for Research on Cancer 2022 (2020). https://gco.iarc.fr/today/online-analysis-multi-bars?v=2020&mode=cancer&mode_population=countries&population=900&populations=900&key=total&sex=2&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&nb_items=10&group_cancer=1&include_nmsc=0&include_nmsc_other=1&type_multiple=%257B%2522inc%2522%253Atrue%252C%2522mort%2522%253Afalse%252C%2522prev%2522%253Afalse%257D&orientation=horizontal&type_sort=0&type_nb_items=%257B%2522top%2522%253Atrue%252C%2522bottom%2522%253Afalse%257D.
Onitilo, A. A., Engel, J. M., Greenlee, R. T. & Mukesh, B. N. Breast cancer subtypes based on ER/PR and Her2 expression: Comparison of clinicopathologic features and survival. Clin. Med. Res. 7(1–2), 4–13. https://doi.org/10.3121/cmr.2009.825 (2009).
Li, Z., Wei, H., Li, S., Wu, P. & Mao, X. The role of progesterone receptors in breast cancer. Drug Des. Dev. Ther. 16, 305–314. https://doi.org/10.2147/DDDT.S336643 (2022).
Mohammed, H. et al. Progesterone receptor modulates ERalpha action in breast cancer. Nature 523(7560), 313–317. https://doi.org/10.1038/nature14583 (2015).
Brisken, C. et al. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc. Natl. Acad. Sci. USA 95(9), 5076–5081. https://doi.org/10.1073/pnas.95.9.5076 (1998).
Graham, J. D. & Clarke, C. L. Physiological action of progesterone in target tissues. Endocr. Rev. 18(4), 502–519. https://doi.org/10.1210/edrv.18.4.0308 (1997).
Lydon, J. P. et al. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 9(18), 2266–2278. https://doi.org/10.1101/gad.9.18.2266 (1995).
Brisken, C. Progesterone signalling in breast cancer: A neglected hormone coming into the limelight. Nat. Rev. Cancer 13(6), 385–396. https://doi.org/10.1038/nrc3518 (2013).
Ranjan, M. et al. Progesterone receptor antagonists reverse stem cell expansion and the paracrine effectors of progesterone action in the mouse mammary gland. Breast Cancer Res. 23(1), 78. https://doi.org/10.1186/s13058-021-01455-2 (2021).
Asselin-Labat, M. L. et al. Control of mammary stem cell function by steroid hormone signalling. Nature 465(7299), 798–802. https://doi.org/10.1038/nature09027 (2010).
Joshi, P. A. et al. Progesterone induces adult mammary stem cell expansion. Nature 465(7299), 803–807. https://doi.org/10.1038/nature09091 (2010).
Tomasetti, C. & Vogelstein, B. Cancer etiology: Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347(6217), 78–81. https://doi.org/10.1126/science.1260825 (2015).
Tomasetti, C., Li, L. & Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355(6331), 1330–1334. https://doi.org/10.1126/science.aaf9011 (2017).
Zheng, Z. Y., Bay, B. H., Aw, S. E. & Lin, V. C. A novel antiestrogenic mechanism in progesterone receptor-transfected breast cancer cells. J. Biol. Chem. 280(17), 17480–17487. https://doi.org/10.1074/jbc.M501261200 (2005).
Tomlinson, I. P., Nicolai, H., Solomon, E. & Bodmer, W. F. The frequency and mechanism of loss of heterozygosity on chromosome 11q in breast cancer. J. Pathol. 180(1), 38–43. https://doi.org/10.1002/(SICI)1096-9896(199609)180:1%3c38::AID-PATH638%3e3.0.CO;2-C (1996).
Cui, X., Schiff, R., Arpino, G., Osborne, C. K. & Lee, A. V. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J. Clin. Oncol. 23(30), 7721–7735. https://doi.org/10.1200/JCO.2005.09.004 (2005).
Singhal, H. et al. Progesterone receptor isoforms, agonists and antagonists differentially reprogram estrogen signaling. Oncotarget 9(4), 4282–4300. https://doi.org/10.18632/oncotarget.21378 (2018).
Lee, O. et al. Selective progesterone receptor modulators in early-stage breast cancer: A randomized, placebo-controlled phase II window-of-opportunity trial using telapristone acetate. Clin. Cancer Res. 26(1), 25–34. https://doi.org/10.1158/1078-0432.CCR-19-0443 (2020).
Afhuppe, W. et al. Global gene expression profiling of progesterone receptor modulators in T47D cells provides a new classification system. J. Steroid. Biochem. Mol. Biol. 113(1–2), 105–115. https://doi.org/10.1016/j.jsbmb.2008.11.015 (2009).
Trabert, B., Sherman, M. E., Kannan, N. & Stanczyk, F. Z. Progesterone and breast cancer. Endocr. Rev. 41, 2. https://doi.org/10.1210/endrev/bnz001 (2020).
Zheng, N. et al. Mifepristone inhibits ovarian cancer metastasis by intervening in SDF-1/CXCR4 chemokine axis. Oncotarget 8(35), 59123–59135. https://doi.org/10.18632/oncotarget.19289 (2017).
Ponikwicka-Tyszko, D. et al. Molecular mechanisms underlying mifepristone’s agonistic action on ovarian cancer progression. EBioMedicine 47, 170–183. https://doi.org/10.1016/j.ebiom.2019.08.035 (2019).
Ritch, S. J., Brandhagen, B. N., Goyeneche, A. A. & Telleria, C. M. Advanced assessment of migration and invasion of cancer cells in response to mifepristone therapy using double fluorescence cytochemical labeling. BMC Cancer 19(1), 376. https://doi.org/10.1186/s12885-019-5587-3 (2019).
Tieszen, C. R., Goyeneche, A. A., Brandhagen, B. N., Ortbahn, C. T. & Telleria, C. M. Antiprogestin mifepristone inhibits the growth of cancer cells of reproductive and non-reproductive origin regardless of progesterone receptor expression. BMC Cancer 11, 207. https://doi.org/10.1186/1471-2407-11-207 (2011).
Rocereto, T. F. et al. A phase II evaluation of mifepristone in the treatment of recurrent or persistent epithelial ovarian, fallopian or primary peritoneal cancer: A gynecologic oncology group study. Gynecol. Oncol. 116(3), 332–334. https://doi.org/10.1016/j.ygyno.2009.10.071 (2010).
Rolla, E. Endometriosis: Advances and controversies in classification, pathogenesis, diagnosis, and treatment. F1000 Res. 8, 529. https://doi.org/10.12688/f1000research.14817.1 (2019).
Lukes, A. S. et al. Health-related quality of life with ulipristal acetate for treatment of uterine leiomyomas: A randomized controlled trial. Obstet. Gynecol. 133(5), 869–878. https://doi.org/10.1097/AOG.0000000000003211 (2019).
Liu, J. H. et al. Ulipristal acetate for treatment of uterine leiomyomas: A randomized controlled trial. Obstet. Gynecol. 132(5), 1241–1251. https://doi.org/10.1097/AOG.0000000000002942 (2018).
Seitz, C. et al. Rationale and design of ASTEROID 2, a randomized, placebo- and active comparator-controlled study to assess the efficacy and safety of vilaprisan in patients with uterine fibroids. Contemp. Clin. Trials 55, 56–62. https://doi.org/10.1016/j.cct.2017.02.002 (2017).
Ciebiera, M. et al. Vilaprisan, a new selective progesterone receptor modulator in uterine fibroid pharmacotherapy-will it really be a breakthrough?. Curr. Pharm. Des. 26(3), 300–309. https://doi.org/10.2174/1381612826666200127092208 (2020).
Lewis, J. H. et al. Onapristone extended release: Safety evaluation from phase I–II studies with an emphasis on hepatotoxicity. Drug Saf. 43(10), 1045–1055. https://doi.org/10.1007/s40264-020-00964-x (2020).
So, S. S., van Helden, S. P., van Geerestein, V. J. & Karplus, M. Quantitative structure-activity relationship studies of progesterone receptor binding steroids. J. Chem. Inf. Comput. Sci. 40(3), 762–772. https://doi.org/10.1021/ci990130v (2000).
Khadijah Saghiri, I. D., Melkemi, N. & Mesli, F. QSAR study, molecular docking/dynamics simulations and ADME prediction of 2-phenyl-1H-indole derivatives as potential breast cancer inhibitors. Biointerface Res. Appl. Chem. 13(2), 154 (2022).
Soderholm, A. A., Lehtovuori, P. T. & Nyronen, T. H. Docking and three-dimensional quantitative structure-activity relationship (3D QSAR) analyses of nonsteroidal progesterone receptor ligands. J. Med. Chem. 49(14), 4261–4268. https://doi.org/10.1021/jm060234e (2006).
Jones, D. G. et al. Discovery of non-steroidal mifepristone mimetics: pyrazoline-based PR antagonists. Bioorg. Med. Chem. Lett. 15(13), 3203–3206. https://doi.org/10.1016/j.bmcl.2005.05.001 (2005).
Du, Y. et al. Aromatic beta-amino-ketone derivatives as novel selective non-steroidal progesterone receptor antagonists. Bioorg. Med. Chem. 18(12), 4255–4268. https://doi.org/10.1016/j.bmc.2010.04.092 (2010).
Matsuzaka, Y. & Uesawa, Y. DeepSnap-deep learning approach predicts progesterone receptor antagonist activity with high performance. Front. Bioeng. Biotechnol. 7, 485. https://doi.org/10.3389/fbioe.2019.00485 (2019).
Mendez, D. et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 47(D1), D930–D940. https://doi.org/10.1093/nar/gky1075 (2019).
Aykul, S. & Martinez-Hackert, E. Determination of half-maximal inhibitory concentration using biosensor-based protein interaction analysis. Anal. Biochem. 508, 97–103. https://doi.org/10.1016/j.ab.2016.06.025 (2016).
Beck, C. Y. et al. Assay operations for SAR support. in Assay Guidance Manual (Eli Lilly & Company and the National Center for Advancing Translational Sciences, 2017). https://www.ncbi.nlm.nih.gov/books/NBK91994/?report.
Carta, G., Onnis, V., Knox, A. J., Fayne, D. & Lloyd, D. G. Permuting input for more effective sampling of 3D conformer space. J. Comput. Aid. Mol. Des. 20(3), 179–190. https://doi.org/10.1007/s10822-006-9044-4 (2006).
Su, Z.-D. et al. iLoc-lncRNA: Predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics 34(24), 4196–4204 (2018).
Rao, B., Zhou, C., Zhang, G., Su, R. & Wei, L. ACPred-Fuse: Fusing multi-view information improves the prediction of anticancer peptides. Brief. Bioinform. 21(5), 1846–1855 (2020).
Qiang, X. et al. CPPred-FL: A sequence-based predictor for large-scale identification of cell-penetrating peptides by feature representation learning. Brief. Bioinform. 21(1), 11–23 (2020).
Charoenkwan, P. et al. StackDPPIV: A novel computational approach for accurate prediction of dipeptidyl peptidase IV (DPP-IV) inhibitory peptides. Methods 204, 189–198 (2021).
Charoenkwan, P. et al. StackIL6: A stacking ensemble model for improving the prediction of IL-6 inducing peptides. Brief. Bioinform. 22(6), 172 (2021).
Malik, A. A. et al. StackHCV: A web-based integrative machine-learning framework for large-scale identification of hepatitis C virus NS5B inhibitors. J. Comput. Aid. Mol. Des. 35(10), 1037–1053 (2021).
Fu, X., Cai, L., Zeng, X. & Zou, Q. StackCPPred: A stacking and pairwise energy content-based prediction of cell-penetrating peptides and their uptake efficiency. Bioinformatics 36(10), 3028–3034 (2020).
Mishra, A., Pokhrel, P. & Hoque, M. T. StackDPPred: A stacking based prediction of DNA-binding protein from sequence. Bioinformatics 35(3), 433–441 (2019).
Pedregosa, F. et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Charoenkwan, P., Schaduangrat, N., Nantasenamat, C., Piacham, T. & Shoombuatong, W. iQSP: A sequence-based tool for the prediction and analysis of quorum sensing peptides via Chou’s 5-steps rule and informative physicochemical properties. Int. J. Mol. Sci. 21(1), 75 (2020).
Azadpour, M., McKay, C. M. & Smith, R. L. Estimating confidence intervals for information transfer analysis of confusion matrices. J. Acoust. Soc. Am. 135(3), 140–146 (2014).
Charoenkwan, P., Anuwongcharoen, N., Nantasenamat, C., Hasan, M. & Shoombuatong, W. In silico approaches for the prediction and analysis of antiviral peptides: A review. Curr. Pharm. Des. 27, 2180–2188 (2021).
Charoenkwan, P., Chiangjong, W., Hasan, M. M., Nantasenamat, C. & Shoombuatong, W. Review and comparative analysis of machine learning-based predictors for predicting and analyzing anti-angiogenic peptides. Curr. Med. Chem. 29, 849–864 (2022).
Dao, F.-Y. et al. DeepYY1: A deep learning approach to identify YY1-mediated chromatin loops. Brief. Bioinform. 22(4), 356 (2021).
Yang, H. et al. A comparison and assessment of computational method for identifying recombination hotspots in Saccharomyces cerevisiae. Brief. Bioinform. 21(5), 1568–1580 (2020).
Dao, F.-Y. et al. Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics 35(12), 2075–2083 (2019).
Chen, W., Lv, H., Nie, F. & Lin, H. i6mA-Pred: Identifying DNA N6-methyladenine sites in the rice genome. Bioinformatics 35(16), 2796–2800 (2019).
Lv, H. et al. Deep-Kcr: Accurate detection of lysine crotonylation sites using deep learning method. Brief. Bioinform. 22(4), 255 (2021).
Xu, Z.-C. et al. iRNAD: A computational tool for identifying D modification sites in RNA sequence. Bioinformatics 35(23), 4922–4929 (2019).
O’Boyle, N. M. et al. Open Babel: An open chemical toolbox. J. Cheminform. 3, 33. https://doi.org/10.1186/1758-2946-3-33 (2011).
Burley, S. K. et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 49(D1), D437–D451. https://doi.org/10.1093/nar/gkaa1038 (2021).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30(16), 2785–2791. https://doi.org/10.1002/jcc.21256 (2009).
Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31(2), 455–461. https://doi.org/10.1002/jcc.21334 (2010).
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. Model 61(8), 3891–3898. https://doi.org/10.1021/acs.jcim.1c00203 (2021).
Xie, R. et al. DeepVF: A deep learning-based hybrid framework for identifying virulence factors using the stacking strategy. Brief. Bioinform. 22(3), 125 (2021).
Van Der Maaten, L. Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res. 15(1), 3221–3245 (2014).
Van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 11 (2008).
Lee, S. M. L. A. S.-I. A Unified Approach to Interpreting Model Predictions. Proceeding (2017).
Kumar, R. et al. The dynamic structure of the estrogen receptor. J. Amino Acids 2011, 812540. https://doi.org/10.4061/2011/812540 (2011).
Moller, C. et al. Discovery of vilaprisan (BAY 1002670): A highly potent and selective progesterone receptor modulator optimized for gynecologic therapies. ChemMedChem 13(21), 2271–2280. https://doi.org/10.1002/cmdc.201800487 (2018).
Jin, C. et al. Synthesis and identification of novel 11beta-aryl-4’,5’-dihydrospiro[estra-4,9-diene-17beta,4’-oxazole] analogs with dissociated antiprogesterone activities. Bioorg. Med. Chem. Lett. 17(21), 5754–5757. https://doi.org/10.1016/j.bmcl.2007.08.064 (2007).
Nishiyama, Y. et al. Novel nonsteroidal progesterone receptor (PR) antagonists with a phenanthridinone skeleton. ACS Med. Chem. Lett. 9(7), 641–645. https://doi.org/10.1021/acsmedchemlett.8b00058 (2018).
Spitz, I. M. Progesterone receptor antagonists. Curr. Opin. Investig. Drugs 7(10), 882–890 (2006).
Richardson, T. I. et al. Novel 3-aryl indoles as progesterone receptor antagonists for uterine fibroids. ACS Med. Chem. Lett. 2(2), 148–153. https://doi.org/10.1021/ml100220b (2011).
Greaves, R. F., Jevalikar, G., Hewitt, J. K. & Zacharin, M. R. A guide to understanding the steroid pathway: New insights and diagnostic implications. Clin. Biochem. 47(15), 5–15. https://doi.org/10.1016/j.clinbiochem.2014.07.017 (2014).
Lala, T. H. D. et al. Abstract P6-20-13: The Pure Progesterone Receptor (PR) Antagonist Onapristone Enhances the Anti-Proliferative Effects of CDK4/6 Inhibitors in Preclinical in-Vitro Breast Cancer Models. (American Association for Cancer Research, Proceeding, 2019).
Islam, M. S., Afrin, S., Jones, S. I. & Segars, J. Selective progesterone receptor modulators-mechanisms and therapeutic utility. Endocr. Rev. 41, 5. https://doi.org/10.1210/endrev/bnaa012 (2020).
MichaelLewis, C. B., Hardebeck, L. & Wireduaah, S. Modern computational approaches to understanding interactions of aromatics. In Aromatic interactions: frontiers in knowledge and application Vol. 20 (ed. Hof, D. W. J. A. F.) (Royal Society of Chemistry, 2017).
N. C. F. B. Information. PubChem Compound Summary for CID 44451278. https://pubchem.ncbi.nlm.nih.gov/compound/44451278. Accessed 15 June 2022.
Rewinkel, J. et al. 11-(pyridinylphenyl)steroids: A new class of mixed-profile progesterone agonists/antagonists. Bioorg. Med. Chem. 16(6), 2753–2763. https://doi.org/10.1016/j.bmc.2008.01.010 (2008).
Nickisch, K. et al. Synthesis and biological evaluation of partially fluorinated antiprogestins and mesoprogestins. Steroids 78(2), 255–267. https://doi.org/10.1016/j.steroids.2012.09.010 (2013).
Nickisch, K. et al. Synthesis and biological evaluation of 11’ imidazolyl antiprogestins and mesoprogestins. Steroids 92, 45–55. https://doi.org/10.1016/j.steroids.2014.08.017 (2014).
Qingxin Cui, R. B., Xu, F., Li, Q., Wang, W. & Bian, Q. New molecular entities and structure–activity relationships of drugs designed by the natural product derivatization method from 2010 to 2018 author links open overlay panel. Stud. Nat. Prod. Chem. 69, 371–415 (2021).
Kang, F. A. et al. Parallel synthesis and SAR study of novel oxa-steroids as potent and selective progesterone receptor antagonists. Bioorg. Med. Chem. Lett. 17(9), 2531–2534. https://doi.org/10.1016/j.bmcl.2007.02.013 (2007).
Winneker, R. C. et al. A new generation of progesterone receptor modulators. Steroids 73(7), 689–701. https://doi.org/10.1016/j.steroids.2008.03.005 (2008).
Steinbeck, C., Han, Y., Kuhn, S., Horlacher, O., Luttmann, E. & Willighagen, E. The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo- and Bioinformatics. J. Chem. Inf. Comput. Sci. 43(2) 493–500 (2003).
Willighagen, E. L. et al., The Chemistry Development Kit (CDK) v2. 0: atom typing, depiction, molecular formulas, and substructure searching. J. Cheminformatics 9(1), 1–19 (2017).
Hall, L. H., & Kier, L. B. Electrotopological state indices for atom types: A novel combination of electronic, topological, and valence state information. J. Chem. Inf. Comput. Sci. 35(6), 1039–1045 (1995).
Klekota, J. & Roth, F. P. Chemical substructures that enrich for biological activity. Bioinformatics, 24(21), 2518–2525 (2008).
Durant, J. L., Leland, B. A., Henry, D. R. & Nourse, J. G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci., 42(6), 1273–1280 (2002).
Kim, S. et al. PubChem Substance and Compound databases. Nucleic Acids Res, 44(D1), D1202–D1213 (2016).
LAGGNER, C. SMARTS patterns for functional group classification (2005).
Acknowledgements
This research was supported by Mahidol University to N.S. This work is also supported by College of Arts, Media and Technology, Chiang Mai University and partially supported by Chiang Mai University and Mahidol University. For the computational resources, this work was supported by Information Technology Service Center (ITSC) of Chiang Mai University.
Author information
Authors and Affiliations
Contributions
Project administration, supervision, conceptualization, analysis and investigation: W.S.; methodology, visualization, validation, software and web server development: P.C.; data collection, interpretation, and docking analysis: N.S.; writing—original draft: M.A.M., P.L., W.S., N.S. and N.A.; writing—review and editing: N.S. and W.S. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schaduangrat, N., Anuwongcharoen, N., Moni, M.A. et al. StackPR is a new computational approach for large-scale identification of progesterone receptor antagonists using the stacking strategy. Sci Rep 12, 16435 (2022). https://doi.org/10.1038/s41598-022-20143-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-20143-5
This article is cited by
-
Machine-learning-assisted high-throughput identification of potent and stable neutralizing antibodies against all four dengue virus serotypes
Scientific Reports (2024)
-
TIPred: a novel stacked ensemble approach for the accelerated discovery of tyrosinase inhibitory peptides
BMC Bioinformatics (2023)
-
DeepAR: a novel deep learning-based hybrid framework for the interpretable prediction of androgen receptor antagonists
Journal of Cheminformatics (2023)
-
StackER: a novel SMILES-based stacked approach for the accelerated and efficient discovery of ERα and ERβ antagonists
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
