
Abstract
The diverse clinical manifestation of essential tremor (ET) has led to the question whether the different phenotypes may affect the clinical outcome and progression. This study aimed to estimate the clinical characteristics and natural history of ET and ET-plus. A total of 221 patients with ET were included, 117 (52.9%) reclassified as ET and 104 (47.1%) as ET-plus. Patients with ET-plus were significantly older in age at onset (P < 0.001); had a higher frequency of cranial tremors (P < 0.001), neurological comorbidities (P < 0.001) and psychiatric comorbidities (P = 0.025); more tremor progression (P < 0.001); and poorer response to medical treatment (P < 0.001) compared to ET patients. Regression analysis revealed that late-onset tremor (OR 11.02, 95% CI 2.79–43.53), neurological comorbidities (OR 3.38, 95% CI 1.56–7.31), psychiatric comorbidities (OR 4.29, 95% CI 1.48–12.44), cranial tremors (OR 2.10, 95% CI 1.02–4.30), and poor response to medical treatment (OR 3.67, 95% CI 1.87–7.19) were associated with ET-plus diagnosis. ET and ET-plus differ in the age of onset, tremor distribution, comorbidities, treatment response rate, and progression. Identifying the ET phenotypes may increase the clinical value in therapeutic strategies and clinical research in the future.
Similar content being viewed by others

Introduction
Essential tremor (ET) is a common neurological disorder affecting about 0.3–0.9% of the general population worldwide, and the prevalence increases with advancing age1,2. Classically, ET has a benign progression. However, it can be progressive and produce functional disability in some cases. The diagnosis of ET is mainly based on clinical examination, with a clinical characteristic of symmetrical postural and kinetic tremor of the upper limbs with or without head and voice tremors3. In the past, ET was considered a monosymptomatic idiopathic neurological disorder. However, emerging evidence indicates that ET is phenotypically heterogeneous with variable motor and non-motor symptoms (NMS).
According to the 2018 consensus statement on tremor disorders by the International Parkinson and Movement Disorder Society (IPMDS), ET has been redefined as an isolated action tremor syndrome of bilateral upper limb action tremor for at least 3 years, with or without a tremor in other locations, and without other neurological signs4. Whereas ET patients with additional subtle neurological signs or “soft neurological signs”, such as tremor at rest, impaired tandem gait, questionable dystonic posturing, memory impairment, or other signs that do not suffice to make an additional diagnosis, are considered as ET-plus4. These changes aim to provide a better method of differentiating clinical phenotypes of ET and might aid future research studies.
Here we reclassified the diagnosis of ET in Thai patients according to the new classification, emphasizing differences in demographics, clinical manifestations, and progression between ET and ET-plus. This study aimed to estimate the proportion, clinical characteristics, and natural history of ET and ET-plus.
Materials and methods
A retrospective medical record review for all Thai patients diagnosed with ET (International Classification of Diseases, 10th Revision, Clinical Modification, ICD-10-CM code: G 25.0) at the out-patient department, Thammasat University Hospital, was performed from January 2019 to December 2021. The diagnoses of ET and ET-plus were reclassified according to the consensus statement on the classification of tremors, IPMDS diagnostic criteria4. Patients were excluded if they had: (a) inadequate tremor information in their medical records; (b) tremor duration under 3 years; (c) isolated focal voice or head tremors; (d) tremor related to physiological tremor, orthostatic tremor, task- and position-specific tremors, drug-induced tremor, psychogenic tremor; or (e) tremors that related to other neurological diseases such as Parkinson’s disease (PD) or dystonia. Late-onset and senile ET groups were defined as age at tremor onset ≥ 46 years and > 65 years, respectively5,6. The reclassification was performed by a movement disorder specialist based on medical records. The protocol for this study was approved by the Human Research Ethics Committee of Thammasat University (MTU-EC-IM-0-151/64). Informed consent was given a formal waive because of the retrospective design by the institutional ethics committee. All the methods were carried out in accordance with the relevant guidelines and regulations.
General demographic data, including sex, current age, age of tremor onset, family history of tremor syndromes, past medical illness, clinical characteristics, and clinical course, were collected. The tremor progression and treatment responsiveness were rated based on the clinical course documented in medical records at 6 months–1 year of follow-up. We redefined tremor progression as static or definite progression and the pharmacological responsiveness as a poor or satisfactory response. The continuous variables were summarized as the mean ± standard deviation (SD) or median, and categorical variables were summarized as numbers and percentages. Comparisons of quantitative data were carried out using the independent t-test or Mann–Whitney test, and qualitative data were compared using the chi-squared test or Fisher’s exact test. The strength of association between nominal variables was determined using the Lambda value. Binary logistic regression analysis was applied to identify significant factors associated with ET-plus. All tests were two-sided, and a P value of less than 0.05 was considered statistically significant.
Results
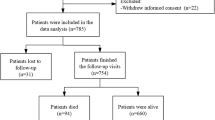
A total of 355 medical records of patients with a diagnosis of ET were reviewed. Of these, 52 (14.6%) records were excluded due to inadequate information on tremors. The remaining 303 (85.4%) were reclassified using the 2018 consensus statement. Eighty-one patients (26.7%) did not meet the diagnostic criteria due to a duration of tremor under 3 years (50), isolated focal tremor (16), and tremor-related to other neurological disorders (15). The remaining 221 (62.3%) patients met the diagnostic criteria of ET syndromes. These included 117 (33.0%) reclassified as ET and 104 (29.3%) as ET-plus (Fig. 1).

Flowchart of the reclassification of 355 patients diagnosed with ET.
In patients who fulfilled the clinical diagnosis of ET syndromes, 167 (75.6%) were diagnosed and followed up by neurologists, and 54 (24.4%) patients were followed up by general practitioners. In contrast, 70.1% of patients who do not meet the diagnostic criteria were diagnosed by general practitioners. There was a strong association between the fulfilled clinical diagnosis and the specialty of the physician who made the diagnosis (lambda value = 0.37).
Regarding topography of tremor manifestations, 209 (94.6%) patients reported relatively symmetrical bilateral hand involvement, 51 (23.1%) had head tremor, 19 (8.6%) had voice tremor, 11 (5.0%) had jaw tremor, 10 (4.5%) had leg tremor, and 10 (4.5%) had both head and voice tremor. In addition, female gender and late-onset ET were significantly associated with cranial tremors compared to male patients (38.2% vs 18.3%, P < 0.001) and early-onset ET (33.2% vs 18.2%, P = 0.029). In terms of comorbidities, 22 (10.0%) of the patients had a history of stroke, four (1.8%) had epilepsy, 13 (5.9%) had cognitive impairment, and 18 (8.1%) had polyneuropathy. Most ET patients reported no to minimal tremor progression, with only 21 (9.5%) reporting definite progression of their tremor. The response to pharmacological treatments, either beta-blockers or antiepileptics, was approximately 45.7%.
Comparisons between patients with ET and ET-plus
In patients who met the diagnostic criteria of ET syndromes, 117 (52.9%) were reclassified as ET and 104 (47.1%) as ET-plus. The mean age of the patients was 67.79 ± 19.03 years, with an age range of 18–96 years. The demographic and clinical characteristics of the patients are shown in Table 1. Patients with ET-plus were significantly older in age (77.06 ± 11.41 vs. 59.55 ± 20.63 years, P < 0.001) and age of tremor onset (70.33 ± 11.42 vs. 53.30 ± 20.61 years, P < 0.001), and had a higher frequency of cranial tremors (41.3% vs. 19.7%, P < 0.001), neurological comorbidities (44.2% vs. 12.0%, P < 0.001), and psychiatric comorbidities (21.2% vs. 10.3%, P = 0.025) compared to ET patients. ET-plus patients also showed poorer response to pharmacological treatment (76.9% vs. 34.2%, P < 0.001) and more frequent progression of their tremor (20.2% vs. 0.0%, P < 0.001) compared to ET patients. Regression analysis identified the following factors as significantly associated with ET-plus: late-onset tremor (OR 11.02, 95% CI 2.79–43.53), neurological comorbidities (OR 3.38, 95% CI 1.56–7.31), psychiatric comorbidities (OR 4.29, 95% CI 1.48–12.44), cranial tremors (OR 2.10, 95% CI 1.02–4.30), and poor response to medical treatment (OR 3.67, 95% CI 1.87–7.19) (Table 2). There was no difference in sex, family history of tremor, disease duration, or rate of lower limb tremor between groups.
In the ET-plus group, 40 (38.5%) of the 104 patients had multiple soft neurological signs; 47 (45.2%) had mild memory impairment, 43 (41.3%) had rest tremor, 37 (35.6%) had tandem gait impairment, 16 (15.4%) had questionable dystonia, and three (2.9%) had mild dysmetria in the upper limbs. Patients with ET-plus also had a higher frequency of epilepsy, cognitive impairment, depression, and hallucination than patients with ET.
Discussion
We describe the clinical phenotype and characteristics of 221 patients with ET syndromes. These include 117 (52.9%) reclassified as ET and 104 (47.1%) as ET-plus. Therefore, only one-third of cases initially diagnosed as ET persisted in being diagnosed as ET under the new classification. This observation supported the existence of phenotypic differences in ET. In addition, more than a quarter of patients initially diagnosed with ET did not meet the new diagnostic criteria for ET. These results suggest that, without the defined diagnostic criteria, the concept of ET varies substantially among clinicians. Acknowledgment of the new classification and diagnosis criteria is crucial for improving the value of this syndrome in clinical diagnosis and research.
To the best of our knowledge, this is the first study to assess the clinical phenotypes, demographics, and characteristics of ET syndromes in Thai patients. Our data showed a concordant prevalence of ET that increased with advancing age1,2,7. We found a slightly higher prevalence in females than males, as was previously reported7. However, most recent epidemiological studies have found a predominance of affection for the male gender1,2. This inverse proportion might be an impact of a higher survived female elderly in the Thai population and conducting a hospital-based prevalence survey. Male patients are less likely to attend a hospital with mild symptoms. We observed a bimodal age distribution for the age of onset with a small early peak in the first and second decades of life and a later rise in the sixth and seventh decades, as described in previous studies6,8 (Fig. 2). Late-onset and senile ET were found in over 80% and 55% of the cases, respectively. This distribution might be an effect due to recall bias and the nature of our hospital, a tertiary referral center. A positive family history of ET was found in 50–55% of patients in our study, lower than the previous reports of 60–75%6,8,9. This difference might be affected by the amount of missing data from medical records in our study. In terms of the topography of tremor manifestations, our study showed a distribution of tremors concordant with earlier reports5,8,9,10. Head tremor was observed in approximately one-quarter of the patients and predominantly in female patients, while voice tremor was the third most commonly involved body part in ET5,8,9.

Histogram of the age-at-onset distribution of ET and ET-plus.
In this study, we found a slightly higher prevalence of ET compared to ET-plus. Since the diagnosis of ET-plus in our study is mainly based on the presence of soft neurological signs recorded in the medical history, the prevalence of ET-plus in our study might be lower than the true prevalence. However, we could identify several factors associated with ET-plus: older age at onset, the presence of cranial tremors, poor response to medical treatment, and history of psychiatric and neurological comorbidities. These factors agreed with previous studies11,12,13.
ET-plus patients were significantly older in age and age at tremor onset compared to ET patients, resulting in a substantially higher prevalence of comorbidities and degenerative neuropsychiatric disorders. In addition, our ET-plus patients also had a higher prevalence of cranial tremors and a poorer response to medical treatments. These findings were consistent with previous reports that showed that cranial tremors were associated with more severe hand tremors and are less responsive to beta-blockers9,14.
An association between ET and dystonia is well recognized but not frequently observed in a clinical setting8,15. Only trained neurologists and movement disorder specialists might clinically notice the differences. In a previous cohort database, about half of the ET patients evaluated at the movement disorders clinic had a variety of patterns of associated dystonia, including cervical dystonia, writer’s cramp, spasmodic dysphonia, and cranial dystonia8. According to the new consensus statement on tremor disorders, the clinical interpretation of questionable dystonia has been left to investigator4. So, the diagnosis of ET-plus is even more challenging with a high rate of discordance15,16. The proportion of questionable dystonia in ET-plus patients from the previous studies varied between 0 and 91%, depending on the research methodology and assessment tools11,12,13,16,17,18. A higher prevalence rate of cranial tremors and poorer responsiveness to medical treatment in ET-plus patients might result from questionable or subtle signs of undetected cranial-cervical dystonia. Regarding tremor progression, all ET patients had no-to-minor tremor progression. In contrast, approximately 20% of ET-plus patients reported worsening tremor with time.
In terms of non-motor features, ET-plus patients had a higher frequency of cognitive and psychiatric symptoms compared to ET patients. Many reports of neuropsychological performance in ET patients have shown a pattern of attentional and executive dysfunction resembling that observed in subcortical dementia, reflecting the disruption in the cerebello-thalamo-frontal network19. Alternately, they may be a feature of concomitant neurodegenerative diseases associated with ET-plus, such as PD or Alzheimer’s disease19. Regarding psychiatric disorders, depression and hallucination were more common in our ET-plus patients. These mental disturbances may be secondary to more severe tremor symptoms in ET-plus or differences in pathologic brain lesions. While restless leg syndrome (RLS) has been reported associated with ET20, our study found only one documented case in the ET-plus group. This result might reflect an unawareness of RLS symptoms in Thai ET patients. Screening of RLS and other NMS with an NMS questionnaire might benefit in early detection and management of NMS in ET patients. A recent study has shown that many components of ET-plus, including motor and non-motor features, correlated with age and tremor duration21. This finding indicated that these components could develop in a more advanced stage of ET or even represent a transitional stage between ET and other degenerative diseases. Therefore, ET-plus may represent a disease stage rather than a distinct disease subtype21. Interestingly, our patients' disease duration between ET and ET-plus was not significantly different, and all ET patients in our study reported a static in their tremor progression. These findings might suggest that these two phenotypes could represent a difference in a trait condition and rate of progression, or it could be just a pitfall from a recall bias in the retrospective study.
When applying the ET-plus criteria, we faced challenges in a group of patients with signs of both ET and parkinsonism, representing a spectrum of an “ET-PD syndrome”22. Most of these patients were excluded from the study because they had a PD diagnosis that might relate to their tremors. The dual-disease pitfall and uncertain definition of soft neurological signs are problematic issues in this classification for clinical implications and epidemiological studies of ET23,24. Whether this is a coincidence or a true association, it is possible that longstanding ET patients might develop PD later in life. In a previous comprehensive cohort database of ET patients, approximately 20% of patients with a history of ET later developed parkinsonism8. On the other hand, 7.8% of PD patients reported a history of pre-existing ET25. To date, the relationship between ET and PD remains controversial22. However, there is growing evidence that these two disorders are pathogenically related26. A recent study showed a higher prevalence of rapid eye movement (REM) sleep behavior disorder, a prodromal marker of α-synucleinopathies in ET-plus patients, than in the general population11. We also observed resting hand and jaw tremors commonly seen in PD in approximately 40% and 9% of ET-plus patients. Moreover, 20% of ET-plus patients required additional treatment with levodopa. These findings suggested that ET-plus might be a prediagnostic phase of PD or a “placeholder” stage before developing other degenerative disorders, reflecting an area of current scientific uncertainty 11,27,28.
Pathological and neuroimaging studies found that ET patients had cerebellar atrophy and an increased number of damaged Purkinje cell (PC) axons10,22. Furthermore, recent studies have shown disruption of climbing fiber (CF)-PC connections in ET cerebellar cortex, including an abnormal extension of CFs into the outer portion of the cerebellar cortex, increased synaptic contacts of CFs on spiny branchlets of PCs, an increased number of CF-PC lateral crossing fibers29,30,31. These abnormalities are clinical–pathological heterogeneity and associated with tremor severity, sex, and cranial distribution29,30,31. Patients with midline tremors and tandem gait disorders had more cerebellar atrophy and more Purkinje cell axonal swellings with torpedo formation in the cerebellar vermis32. Although, a recent postmortem study has not demonstrated pathological differences in cerebellar cortex between ET and ET-plus cases33. With additional multiple soft signs, ET-plus patients may have a broader range of pathological lesions in the cerebello-thalamo-cortical circuit. More future research is needed to identify relations between clinical and pathological evidence of ET and ET-plus.
We acknowledge that our present study has some limitations. First, our study was based on data from retrospective chart reviews in one tertiary center, and the number of participants was limited. Many individuals with ET are asymptomatic and are not referred to our center. A prospective, multi-center registry with a larger sample size should be considered in the future. Second, the concept of ET-plus was relatively new and not widely acknowledged by general practitioners and neurologists. Subtle neurological deficits and soft neurological signs such as questionable dystonia and impaired tandem gait may have gone undetected. A video-based assessment with a uniform protocol would provide more detail on neurological signs and improve diagnostic accuracy. Lastly, the patients and clinicians subjectively evaluated the progression and treatment response without using assessment tools. An objective assessment with validated tools and a long-term evaluation is needed for future research protocols.
In summary, ET is a heterogeneous syndrome with variable clinical signs and symptoms. ET and ET-plus patients differ in the age of onset, tremor distribution, comorbidities, treatment response, and progression rate. Identifying the ET phenotypes may increase the clinical value of this syndrome in identifying pathogenetic hypotheses, therapeutic strategies, and clinical research in the future.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Louis, E. D. & Ferreira, J. J. How common is the most common adult movement disorder? Update on the worldwide prevalence of essential tremor. Mov. Disord. 25, 534–541. https://doi.org/10.1002/mds.22838 (2010).
Song, P. et al. The global prevalence of essential tremor, with emphasis on age and sex: A meta-analysis. J. Glob. Health 11, 04028. https://doi.org/10.7189/jogh.11.04028 (2021).
Deuschl, G., Bain, P. & Brin, M. Consensus statement of the Movement Disorder Society on Tremor. Ad Hoc Sci.Committee. Mov. Disord. 13(Suppl 3), 2–23. https://doi.org/10.1002/mds.870131303 (1998).
Bhatia, K. P. et al. Consensus Statement on the classification of tremors from the task force on tremor of the International Parkinson and Movement Disorder Society. Mov. Disord. 33, 75–87. https://doi.org/10.1002/mds.27121 (2018).
Deuschl, G. & Elble, R. Essential tremor–neurodegenerative or nondegenerative disease towards a working definition of ET. Mov. Disord. 24, 2033–2041. https://doi.org/10.1002/mds.22755 (2009).
Hopfner, F. et al. Early- and late-onset essential tremor patients represent clinically distinct subgroups. Mov. Disord. 31, 1560–1566. https://doi.org/10.1002/mds.26708 (2016).
Tallon-Barranco, A. et al. Clinical features of essential tremor seen in neurology practice: A study of 357 patients. Parkinson. Relat. Disord. 3, 187–190. https://doi.org/10.1016/s1353-8020(97)00031-x (1997).
Lou, J. S. & Jankovic, J. Essential tremor: Clinical correlates in 350 patients. Neurology 41, 234–238. https://doi.org/10.1212/wnl.41.2_part_1.234 (1991).
Chen, W. et al. Topography of essential tremor. Parkinson. Relat. Disord. 40, 58–63. https://doi.org/10.1016/j.parkreldis.2017.04.012 (2017).
Rajput, A., Robinson, C. A. & Rajput, A. H. Essential tremor course and disability: A clinicopathologic study of 20 cases. Neurology 62, 932–936. https://doi.org/10.1212/01.wnl.0000115145.18830.1a (2004).
Huang, H. et al. Clinical characteristics of patients with essential tremor or essential tremor plus. Acta Neurol. Scand. 141, 335–341. https://doi.org/10.1111/ane.13209 (2020).
Prasad, S. & Pal, P. K. Reclassifying essential tremor: Implications for the future of past research. Mov. Disord. 34, 437. https://doi.org/10.1002/mds.27615 (2019).
Rajalingam, R., Breen, D. P., Lang, A. E. & Fasano, A. Essential tremor plus is more common than essential tremor: Insights from the reclassification of a cohort of patients with lower limb tremor. Parkinson. Relat. Disord. 56, 109–110. https://doi.org/10.1016/j.parkreldis.2018.06.029 (2018).
Louis, E. D., Ford, B. & Frucht, S. Factors associated with increased risk of head tremor in essential tremor: A community-based study in northern Manhattan. Mov. Disord. 18, 432–436. https://doi.org/10.1002/mds.10395 (2003).
Pandey, S., Bhattad, S. & Hallett, M. The problem of questionable dystonia in the diagnosis of “Essential Tremor-Plus”. Tremor Other Hyperkinet. Mov. (N Y) 10, 27. https://doi.org/10.5334/tohm.539 (2020).
Rajan, R., Pandey, S., Anandapadmanabhan, R. & Srivastava, A. K. Interrater and intrarater agreement on the 2018 consensus statement on classification of tremors. Mov. Disord. 33, 1966–1967. https://doi.org/10.1002/mds.27513 (2018).
Pandey, S. & Bhattad, S. Questionable dystonia in essential tremor plus: A video-based assessment of 19 patients. Mov. Disord. Clin. Pract. 6, 722–723. https://doi.org/10.1002/mdc3.12838 (2019).
Pandey, S. & Bhattad, S. Soft signs in essential tremor plus: A prospective study. Mov. Disord. Clin. Pract. 8, 1275–1277. https://doi.org/10.1002/mdc3.13350 (2021).
Janicki, S. C., Cosentino, S. & Louis, E. D. The cognitive side of essential tremor: What are the therapeutic implications?. Ther. Adv. Neurol. Disord. 6, 353–368. https://doi.org/10.1177/1756285613489591 (2013).
Alonso-Navarro, H., Garcia-Martin, E., Agundez, J. A. G. & Jimenez-Jimenez, F. J. Association between restless legs syndrome and other movement disorders. Neurology 92, 948–964. https://doi.org/10.1212/WNL.0000000000007500 (2019).
Louis, E. D., Huey, E. D. & Cosentino, S. Features of “ET plus” correlate with age and tremor duration: “ET plus” may be a disease stage rather than a subtype of essential tremor. Parkinson. Relat. Disord. 91, 42–47. https://doi.org/10.1016/j.parkreldis.2021.08.017 (2021).
Shahed, J. & Jankovic, J. Exploring the relationship between essential tremor and Parkinson’s disease. Parkinson. Relat. Disord. 13, 67–76. https://doi.org/10.1016/j.parkreldis.2006.05.033 (2007).
Fasano, A., Lang, A. E. & Espay, A. J. What is “essential” about essential tremor?. A diagnostic placeholder. Mov. Disord. 33, 58–61. https://doi.org/10.1002/mds.27288 (2018).
Louis, E. D. “Essential Tremor Plus”: A problematic concept: Implications for clinical and epidemiological studies of essential tremor. Neuroepidemiology 54, 180–184. https://doi.org/10.1159/000502862 (2020).
Ou, R. et al. Association between positive history of essential tremor and disease progression in patients with Parkinson’s disease. Sci. Rep. 10, 21749. https://doi.org/10.1038/s41598-020-78794-1 (2020).
Fekete, R. & Jankovic, J. Revisiting the relationship between essential tremor and Parkinson’s disease. Mov. Disord. 26, 391–398. https://doi.org/10.1002/mds.23512 (2011).
Louis, E. D. Essential tremor: “Plus” or “Minus” Perhaps now is the time to adopt the term “the essential tremors”. Parkinson. Relat. Disord. 56, 111–112. https://doi.org/10.1016/j.parkreldis.2018.06.026 (2018).
Louis, E. D. et al. Essential tremor-plus: A controversial new concept. Lancet Neurol. 19, 266–270. https://doi.org/10.1016/S1474-4422(19)30398-9 (2020).
Lin, C. Y. et al. Abnormal climbing fibre-Purkinje cell synaptic connections in the essential tremor cerebellum. Brain 137, 3149–3159. https://doi.org/10.1093/brain/awu281 (2014).
Wu, Y. C. et al. Increased climbing fiber lateral crossings on purkinje cell dendrites in the cerebellar hemisphere in essential tremor. Mov. Disord. 36, 1440–1445. https://doi.org/10.1002/mds.28502 (2021).
Louis, R. J., Lin, C. Y., Faust, P. L., Koeppen, A. H. & Kuo, S. H. Climbing fiber synaptic changes correlate with clinical features in essential tremor. Neurology 84, 2284–2286. https://doi.org/10.1212/WNL.0000000000001636 (2015).
Louis, E. D. et al. Torpedoes in the cerebellar vermis in essential tremor cases vs controls. Cerebellum 10, 812–819. https://doi.org/10.1007/s12311-011-0291-0 (2011).
Gionco, J. T. et al. Essential tremor versus “ET-plus”: A detailed postmortem study of cerebellar pathology. Cerebellum 20, 904–912. https://doi.org/10.1007/s12311-021-01263-6 (2021).
Author information
Authors and Affiliations
Contributions
P.L.: conception and designed the study, data acquisition, interpretation, and analysis, manuscript preparation and revision; P.D., S.M., P.S., N.U., K.K.: data acquisition and manuscript revision. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lolekha, P., Dharmasaroja, P., Uransilp, N. et al. The differences in clinical characteristics and natural history between essential tremor and essential tremor plus. Sci Rep 12, 7669 (2022). https://doi.org/10.1038/s41598-022-11775-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-11775-8
This article is cited by
-
Cerebellar voxel-based morphometry in essential tremor
Journal of Neurology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
