
Abstract
Shared resources can instigate pathogen spread due to large congregations of individuals in both natural and human modified resources. Of current concern is the addition of pollinator habitat in conservation efforts as it attracts bees of various species, potentially instigating interspecific sharing of pathogens. Common pathogens have been documented across a wide variety of pollinators with shared floral resources instigating their spread in some, but not all, cases. To evaluate the impact of augmented pollinator habitat on pathogen prevalence, we extracted RNA from samples of eight bee species across three families and screened these samples for nine pathogens using RT-qPCR. We found that some habitat characteristics influenced pathogen detection; however, we found no evidence that pathogen detection in one bee species was correlated with pathogen detection in another. In fact, pathogen detection was rare in wild bees. While gut parasites were detected in 6 out of the 8 species included in this study, viruses were only detected in honey bees. Further, virus detection in honey bees was low with a maximum 21% of samples testing positive for BQCV, for example. These findings suggest factors other than the habitat itself may be more critical in the dissemination of pathogens among bee species. However, we found high relative prevalence and copy number of gut parasites in some bee species which may be of concern, such as Bombus pensylvanicus. Long-term monitoring of pathogens in different bee species at augmented pollinator habitat is needed to evaluate if these patterns will change over time.
Similar content being viewed by others

Introduction
Shared resources can pose health risks to organisms; this is true for naturally occurring resources such as mating grounds or watering holes1, but also for human modified resources such as supplemental wildlife feed2, hunter attractants3, and even bird feeders4. These shared resources can result in dense congregations of individuals2 potentially causing them to act as “hotspots,” leading to pathogen build up that can then spread throughout the environment1. Further, the interspecific spread within these congregations can intensify if resources are scarce or limited5. In some cases, the resource itself can harbor pathogens6, increasing pathogen spread within populations7. However, interspecific and intraspecific pathogen spread depends on the host competency of the individual and the species for each pathogen in question1. Rather than acting as a hotspot, an incompetent host in a biologically diverse community at a shared resource could act to dilute the spread of a pathogen8.
Pollinator population declines9 have been repeatedly suggested to be driven by factors including agricultural intensification, nutritional stress, habitat alteration and fragmentation, and pathogens10, all of which can interact synergistically. Habitat loss in particular has arguably received the most attention in recent years. To combat this, augmenting habitat to support pollinators is becoming an increasingly popular conservation tool, especially in agricultural settings. While such habitat has been found to support pollinator abundance and diversity11,12, it is being implemented en masse with limited scientific evidence for best practices13. Evaluating the impacts of this habitat on bee populations and bee health is critical to ensure that we are not exacerbating the exact pressures that are intended to be alleviated.
Parallels can easily be drawn between human modified pollinator habitat to support bees and the shared resource examples of watering holes, supplemental wildlife feed, and bird feeders. There is a great wealth of previous literature exploring the potential for pathogen cross-over among bee species (Tables 1 and 2), particularly because similar pressures are of concern; for example, there is evidence that high abundance of common species can intensify pathogen occurance14. Additionally, certain flower species have been found to harbor pathogens15,16; however, this could be counteracted or ameliorated with habitat characteristics17, such as increased flower community diversity18. Many studies have investigated the interspecific spread of pathogens from honey bees to wild bees19,20, specifically. However, as differing results have been documented in differing situations (Tables 1 and 2), it begs the question: will augmented pollinator habitat act to congregate individuals leading to hotspots of pathogen spread, or will these habitats attract a diverse pollinator community leading to pathogen dilution? And what role do the habitats themselves play in pathogen spread or dilution? To evaluate how pollinator habitat influences pathogen dissemination within bee communities, we evaluated the pathogen prevalence in eight bee species from three families across 2 years. To do this, we sampled newly established pollinator habitat across North Carolina as part of the North Carolina Department of Agriculture and Consumer Services’ (NCDA&CS) mandate titled “Protecting NC Pollinators.” We investigated pathogen occurrence and prevalence within Apis mellifera, within Bombus impatiens, between Apis mellifera and Bombus impatiens, and within six other bee species that have rarely if ever been quantified in this context.
Materials and methods
Sample collection
Samples were collected at established pollinator habitat at 12 sites across North Carolina (Supplemental Tables 1 and 2) in 2017 and 2018. Collection events occurred once a month for 4 months during peak bloom at each plot, for a total of four sampling events per locations per year (hereafter referred to as Spring, Early Summer, Late Summer, and Fall), utilizing hand nets for 30 ± 10 min along haphazard transects21. All samples were collected between April and November (Supplemental Tables 1 and 2). Focus was placed on the most commonly occurring species to ensure sufficient replication. Each individual bee collected was placed into a separate 1.7 ml microcentrifuge tube and transported back to the lab on dry ice where they were then stored at − 80 °C until further processing. At each station during each sampling event, the flower cover and flower diversity within the plot was documented and categorized into low, medium, or high. Flower cover was categorized based on what percentage of the sampling plot was in bloom at the time the samples were collected, with 0–30% bloom corresponding to low, 31–50% to medium, and 51% or more to high. Flower diversity was categorized based on how many different plant species were in bloom at the time the samples were collected, with low corresponding to 80–100% of the plot in bloom with one flower species, medium to 60–79%, and high to 59% or less (see Levenson and Tarpy22 for more details).
Pathogen screening
Eight different bee species (Apidae: Apis mellifera, Bombus impatiens, Bombus pensylvanicus, Svastra obliqua, Xylocopa virginica, Xylocopa micans; Halictidae: Halictus poeyi/ligatus; and Megachilidae: Megachile xylocopoides; Table 3) were screened for 9 different pathogens (acute bee paralysis virus [ABPV], black queen cell virus [BQCV], chronic bee paralysis virus [CBPV], deformed wing virus A [DWVa], deformed wing virus B [DWVb], Israeli acute paralysis virus [IAPV], Lake Sinai virus [LSV], Trypanosome universal primer [Trypanosome spp.], and Vairimorpha primer [as a Nosema universal primer was used during screening, results from this target will henceforth be referred to as Nosema spp. for simplicity]; as well as two reference genes (actin and apocrita 28 s [apo28s]; Supplemental Table 3). The Trypanosome universal primer was designed to amplify Chrithidia mellificae, Chrithidia bombi, and Lotmaria passim. The Nosema universal primer was designed to amplify Nosema apis and Nosema ceranae. All primer working stocks were diluted to 5 mmol. North Carolina is on the border of the range for H. poeyi and H. ligatus; because these two species are cryptic species and morphologically identical64 samples of this species complex are referred to as H. poeyi/ligatus.
Seven of the pathogens examined in this study are viruses and were selected because they are some of the most commonly occurring honey bee pathogens that have been shown to negatively affect honey bee health65. Although little is known about the true impact of most of these pathogens on native bee health and longevity14, transmission is likely given that bee species in the same area of a study have the same virus profiles44,61. The remaining pathogens are gut parasites; these pathogens were selected because they are commonly detected, known to negatively impact bee survival66 and so are economically important, interspecific transmission has been previously documented23, and infection of gut parasites has been linked to population losses in some cases67.
Sample preparation: individual bee samples
Samples of A. mellifera and B. impatiens were processed as individuals as the sample sizes of these species were the highest in our study. Samples of X. virginica and X. micans were also processed as individuals due to their large body size. When processing these individual samples, we removed each specimen from cold storage and kept it on dry ice until crushed, following an adapted protocol from Leite et al. 201268 to ensure successful pulverization and the highest quality RNA due to sample brittleness. We used two Zirconium beads (3.0 mm) for A. mellifera and B. impatiens and three Zirconium beads for X. virginica and X. micans, placing each tube into the Ivoclar Silamat S6 in order to crush the sample. Once completely pulverized, we extracted RNA using the TRIzol® Reagent69 and the Zymo Direct-zol™ RNA Miniprep Kit, following the Directzol protocol. After extraction, we assessed RNA quantity and quality using the Thermo Scientific NanoDrop ND-1000 Spectrophotometer and diluted to 200 ng of RNA per microliter. All RNA was again stored at − 80 °C until further analysis.
Sample preparation: pooled bee samples
Due to sample size and low pathogen detection (discussed below), we tested B. pensylvanicus, H. poeyi/ligatus, S. obliqua, and M. xylocopoides in pools of up to five individuals (depending on how many were collected during each sampling event) using whole bodies (summarized in Table 3). To process pooled samples, we took up to five bees per sampling event per location out of ultracold storage and immediately placed them into a sanitized ceramic mortar. Sufficient liquid nitrogen was immediately added to cover all bee material and allowed to sublimate to ensure that the samples were brittle. We then immediately and quickly pulverized samples using a pestle. Once completely crushed, we filled a new 1.7 µl tube approximately halfway with the powdered materials so as to leave enough space for the TRIzol® Reagent. As individuals of H. poeyi/ligatus are small, we combined these pooled samples into one new 1.7 tube and crushed them using the Zirconium bead protocol described above for individual samples. RNA extraction of pooled samples followed the same protocol as described above.
Sample testing
To determine the concentrations of pathogen infections in the samples, we used a two-step reverse transcriptase quantitative PCR (RT-qPCR) analysis. In step one, we synthesized cDNA with a 10-µl reaction volume using the BioBasic High Reverse Transcriptase Kit (Biobasic, Marhkam, Canada). Each well received 1 µl of extracted RNA (at a concentration of 200 ng per microliter), 2 µl of master mix, and 7 µl of lab grade water; after, we diluted the cDNA with 50 µl of water. The resulting cDNA contained a relative concentration of 3.3 ng of RNA per microliter assuming full conversion to cDNA. In step two, we performed real time PCR in triplicate on 384-well plates using Life Technologies PowerUp SYBER Green chemistry with a Quant Studio 6 Flex machine. We used 3.2 µl reaction volumes that included 1 µl of template cDNA, 1.5 µl of SYBER, 0.15 µl of primer (forward and reverse combined), and 0.55 µl of water. We included standards for absolute quantification in each plate, which involved a serial dilution of known quantities of a custom synthesized plasmid containing the targets, with one negative control containing only water also included. Even though under this protocol each plate completed 40 cycles during the PCR stage, we only included positive results that were within the range of the quantified standards. If a sample contained a positive result at a cycle number higher than the positive standards, it was not considered to be biologically relevant. Thus, the cycle number cutoff ranged from 28 to 32 cycles, depending on the target and the specific target’s standard’s results. We performed analyses using the included Quant Studio software and then normalized results to the reference gene levels using GeNorm70.
In 2018, we collected a subset of the flowers on which the bees were foraging and conducted pathogen screening in order to determine if they contained similarly detectable levels of the pathogens. Five flower heads per sampling event were removed, placed in individual bags, and transported back on dry ice as was done with the bee samples. We screened these flowers for pathogens as detailed above; however, no pathogens were detected (data not included) and as such we did not analyze these data.
Statistical analysis
Since there is an overdispersion of zeros in our dataset, we used a Zero Inflated Negative Binomial model (ZINB)71 with a logit link. Detection levels of pathogens were analyzed in two ways; copy number (standardized to the reference gene), and relative intensity (categorized into non-detect (ND) if zero, and low, medium, or high based on the bottom two, third, and fourth quartiles of the natural log transformed copy numbers of each pathogen, respectively; Supplemental Table 4). Relative intensity was calculated within each target, not across targets. To explore the potential pathogen dissemination between A. mellifera and B. impatiens, we included season, flower cover, and flower diversity as independent variables and pathogen copy number as dependent variables in a ZINB model. We then used an ANOVA in base R to compare A. mellifera presence and relative intensity to B. impatiens presence and relative intensity.
When constructing our ZINB models, year was not found to significantly impact pathogen detection in A. mellifera (all p > 0,34), except for BQCV detections (p < 0.05). As such, year was only included as a random effect in models when analyzing BQCV detection in A. mellifera. Additionally, sampling location was not found to significantly impact pathogen detection in A. mellifera (all p > 0.16), except for Nosema spp. detections (p < 0.0001). However, in order to maintain statistical power, it was not included in any of our models. Both year (p < 0.0001) and station (p < 0.005) were found to significantly impact Trypanosome spp. detections in B. impatiens, however through an AIC based approach for best model selection these variables were not included in our final model.
Due to low sample size and low pathogen presence, we were not able to conduct further analyses on the pathogen results from the remaining six bee species; however, these findings are summarized descriptively below. All analyses were conducted in RStudio (version 3.6.2) using base R72, the pscl73 package, and the boot74 packages.
Results
We originally collected and screened 616 bee samples; however, we removed 114 samples from analysis as the amplification levels of one or both of the reference genes were at an unacceptably low level. As such, we included a total of 502 samples in our analysis—411 individually processed samples and 91 pooled samples (Fig. 1 and Table 3).

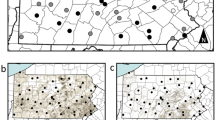
Visual comparison of viral matrices. Each block shows samples from a particular location with A. mellifera (Am) [on the left-hand matrix of each block] and B. impatiens (Bi) [on the right-hand matrix]. Samples from 2017 are displayed at the top of each block and 2018 on the bottom. Each row represents an individual sample, and each column represents a different target listed in alphabetical order (A = ABPV; B = BQCV; C = CBPV; Da = DWVa; Db = DWVb; I = IAPV; L = LSV; T = Try. spp.; and N = Nos. spp.). Relative intensity is represented with a color gradient from low (bright yellow) to high (bright red). This figure was created using Adobe Illustrator, Microsoft Excel, and Microsoft Word. Thank you to Kirsten Benson for creating the base map.
A visual representation of the results for Apis mellifera and Bombus impatiens are displayed in Fig. 1. Each block displays the samples screened at each sampling location. Within each block, results are displayed for both A. mellifera (the left-hand matrix of each block) and B. impatiens (the right-hand matrix of each block) as well as for each year (2017 results are shown on the top half of each block and 2018 results on the bottom). Each row within a viral matrix represents an individual sample tested. Each column represents the target that sample was screened against listed in alphabetical order (A = ABPV; B = BQCV; C = CBPV; Da = DWVa; Db = DWVb; I = IAPV; L = LSV; T = Try. spp.; and N = Nos. spp.). Thus, each cell displays whether a target was detected in a specific sample. While we calculated pathogen presence discretely with quartiles (Supplemental Table 4), we display those results here on a continuous gradient where no detection of a target is represented in white, low detection of a target (corresponding to Q1 and Q2) is represented in yellow, medium detection of a target (Q3) is represented in shades of orange, and high detection of a target (Q4) is represented in red. As each target may be detected at different efficiencies during testing, these quartiles are calculated within each target, not across targets.
Apis mellifera was the only bee species in which we detected any of the viruses in our study. The most commonly detected pathogen in A. mellifera was BQCV (40 individuals), followed by DWVa (32), LSV (27), Trypanosome spp. (26), Nosema spp. (22), ABPV (1), IAPV (1), and finally CBPV with no detections (Table 3). Further, many individuals were found to be simultaneously infected with multiple pathogens, with two individuals infected with four pathogens (Fig. 3). We found that LSV had the highest copy number overall, but that BQCV (29.0% of positive detections) and Trypanosome spp. (52.9% of positive detections) more often fell into the high category of relative intensity. Due to low, or no, positive detections, we were unable to analyze ABPV, CBPV, DWVb, and IAPV results for A. mellifera. From the pathogens we were able to analyze, we found that BQCV copy number was significantly highest in the spring (logθ = − 1.34; DF = 11; p < 0.0001, SE ± 1.10), and was lowest at medium flower diversity (p < 0.005, SE ± 2.04). LSV did not significantly change across the sampling season or flower diversity (all p values > 0.18), but we detected the highest copy numbers in low flower cover (logθ = − 1.07; DF = 17; p < 0.0001, SE ± 1.51). Conversely, we detected the highest Trypanosome spp. copy number at high flower cover (logθ = − 0.25; DF = 17; p < 0.0005, SE ± 1.20; Fig. 2) and when flower diversity was low (p < 0.0001, SE ± 1.27). Additionally, copy number of Trypanosome spp. was highest in late summer (p < 0.01, SE ± 1.71; Fig. 3). To analyze the Nosema spp. results, flower diversity was removed from the model. We found that copy number detection level of Nosema spp. was highest in fall (logθ = − 0.44; DF = 13; p < 0.001, SE ± 1.63) and spring (p < 0.0001, SE ± 1.33) and was not significantly impacted by flower cover (p = 0.64). DWVa was not significantly influenced by any of the variables in our model (all p values > 0.06).

Copy number per 1 µl of template (approximately 3.3 ng of RNA) of Trypanosome spp. for A. mellifera, B. impatiens, and B. pensylvanicus across the different levels of flower cover.

Copy number per 1 µl of template (approximately 3.3 ng of RNA) of Trypanosome spp. for A. mellifera, B. impatiens, and B. pensylvanicus across the sampling season.
We only analyzed Trypanosome spp. copy number within B. impatiens as no viruses were detected in any of our B. impatiens samples and only one individual was detected with Nosema spp. Copy numbers within B. impatiens (68 individuals) were higher than copy numbers in A. mellifera. We found that Trypanosome spp. copy number was significantly lowest in the fall (logθ = − 0.85; DF = 17; p < 0.005, SE ± 0.91; Fig. 3) and significantly highest with medium flower diversity (p < 0.05, SE ± 0.88) and low (p < 0.01, SE ± 0.70; Fig. 2) flower diversity. Trypanosome spp. copy number was not significantly influenced by flower cover (all p values > 0.24).
When exploring potential pathogen dissemination between A. mellifera and B. impatiens we focused on Trypanosome spp. detections, as this was only pathogen detected in both species with high sample numbers. We did not find any evidence that pathogen detection of one species was correlated with the pathogen detection of the other. Presence of a positive Trypanosome spp. detection in A. mellifera had no correlation with any relative intensity category in B. impatiens (all p values > 0.15). Similarly, the relative intensity of Trypanosome spp. in A. mellifera had no correlation to the presence or relative intensity of Trypanosome spp. in B. impatiens (all p values > 0.18).
While we did not find any positive detections of viruses in the other bee species tested in this study, we did find gut pathogens. Trypanosome spp. were detected in B. pensylvanicus (6 pools), H. poeyi/ligatus (6), S. obliqua (1), and X. micans (1 individual; Table 3 and Figs. 2 and 3). Nosema spp. was also detected in B. pensylvanicus (5 pools) and S. obliqua (1). Within these gut pathogen results, B. pensylvanicus had the highest copy number detection level for Nosema spp., by an entire order of magnitude, followed by S. obliqua and then A. mellifera. Bombus pensylvanicus had the highest copy number detection level of Trypanosome spp. again followed by S. obliqua,and then B. impatiens.
Discussion
Apis mellifera was the only pollinator species in which we detected any viruses. However, we detected gut pathogens across most of the bee species tested. Some pathogen copy numbers—such as BQCV and Nosema spp. in A. mellifera, and Trypanosome spp. in both A. mellifera and B. impatiens—significantly changed across the sampling season, a finding that is similar to previous literature17. While other pathogen copy numbers—such as LSV in A. mellifera and Trypanosome spp. in both A. mellifera and B. impatiens—were significantly influenced by flower cover; however, this occurred in opposite directions where LSV was highest at low flower cover and Trypanosome spp. were highest at high flower cover. Similar to previous literature75, Trypanosome spp. detection levels were highest in low flower diversity. While Trypanosome spp. detection patterns were similar in A. mellifera and B. impatiens, we found no evidence of correlations between these two species. These results suggest that the habitat is not acting as a pathogen hotspot but rather some other mechanism may be more critical in pathogen dissemination within bee communities. One explanation could be that even though shared floral resources have been documented as a source of spread for some pathogens40, the occurrence may actually be rather rare16 and its success depends on the bee15 and flower species in question16. It has also been suggested that non-host bees can reduce infection levels through the dilution effect51. It is possible that as time progresses and bees continue to utilize these habitats, the pathogens pressures will intensify intraspecifically. Further long-term testing will be necessary to evaluate this possibility.
Gut parasites are currently considered a serious threat to several bee species, especially bumble bees67; of particular concern in North America is the American Bumble Bee (B. pensylvanicus). In our study, B. pensylvanicus had the greatest positive detections of gut parasites out of all the bee species tested, supporting the hypothesis that gut parasites pose a threat to their populations. At the time of writing this paper—but after the period when samples were collected and analyzed—the United States Fish and Wildlife Service (FWS) has announced a 90-day findings petition for B. pensylvanicus populations in order to inform decisions surrounding its population status, and status reviews are underway in state FWS offices. Currently in North Carolina, B. pensylvanicus is listed at “W3: Rare but Questionable Documentation”76 and “Vulnerable/Apparently Secure”76, meaning more documentation is needed on this species before making any regulatory decision. Information from this study will be important in making future conservation decisions surrounding this and other species, and data from this study has already been shared with the NCFWS to do so. As gut pathogens are considered a threat to this species’ population77, monitoring should be continued in future work. However, one consideration is noting the species of gut pathogens being detected. While all samples in this study were screened for N. apis and ceranae, preliminary results showed that some samples tested positive for the Nosema spp. primer but did not test positive for N. ceranae; however, as the results were inconclusive, the data are not included here. This suggests native bee populations are facing their own gut parasites, especially Nosema species78, that are not as commonly tested for. This has already been documented in previous literature32, especially in bumble bees with regards to the health consequences of Nosema bombi infections79. It is important to note, though, that detecting a pathogen neither equates to infection nor demonstrates specific health impacts of the pathogen28. For example, it has been suggested that the presence of N. ceranae in B. terrestris may be due to ingested spores passing through the gut rather than true infection27. Future research should prioritize evaluating the true infectivity and health impacts of these pathogens on a variety of bee species, taking into consideration the use of species specific pathogen primers.
Many studies have previously found the presence of what are traditionally called ‘honey bee’ viruses in various native bee species, something this study does not confirm. Given that several other recently published papers have also documented fewer detections than previous research34,57,63, the unexpected results require speculation as to why. Unlike most other studies, we collected honey bee samples as individual foragers rather than groups from nest entrances or even inside managed beehives. This could have resulted in lower infection levels in our samples (e.g., heavily infected bees may not live long enough or be sufficiently healthy to forage) resulting in reduced pathogen detection and spread. Another factor that plays a key role is viral sequence variation. Previous research has documented different variants circulating in different regions of the world18. The presence of different viral variants could reduce interspecific spread between bee species and also impact detectability of such variants. Alternatively, floral diversity has been documented as an important factor for pathogen sharing and infection levels18,75. Thus, plant diversity could potentially be used as a tool to intentionally limit pathogen sharing between honey bees and native bees at these augmented habitats. This is something that should be investigated further in future research and taken into consideration when establishing new pollinator habitat.
Another factor to consider when comparing the results from this study to previously published work is the techniques and methods used to screen for pathogens. Nearly every aspect of sample processing could influence the detection of pathogens including the brand of chemical used, primer efficiencies, the amount of starting template, specific techniques used, and even machine functioning. In a study on mouse behavior it was found that lab environment alone caused significant differences in results, even when standardizing protocols80,81. As noted in Tables 1 and 2 of this paper, previous research evaluating co-occurrence of pathogens among honey bees and other species has varied in technique used, starting template amount, and sample processing—all of which could bias results. When coupled with other considerations such as spurious PCR amplification (as is known to occur at 30 cycles and above82) and the influence of pathogen infection itself on detection ability within a sample83, comparing across studies should be done with extreme caution so as to avoid potentially misleading comparisons.
As planted habitat for pollinators will likely continue to be used as a tool in pollinator conservation, we should take care to establish this habitat with plant species that provide floral resources while limiting pathogen transmission. We should also prioritize conducting long-term monitoring of the bees within these habitats to ensure it continues to protect pollinator populations and their health over time.
References
Paull, S. H. et al. From superspreaders to disease hotspots: Linking transmission across hosts and space. Front. Ecol. Environ. 10, 75–82 (2012).
Sorensen, A., Van Beest, F. M. & Brook, R. K. Impacts of wildlife baiting and supplemental feeding on infectious disease transmission risk: A synthesis of knowledge. Prev. Vet. Med. 113, 356–363 (2014).
Gortázar, C., Acevedo, P., Ruíz-Fons, F. & Vicente, J. Disease risks and overabundance of game species. Eur. J. Wildl. Res. 52, 81–87 (2006).
Brittingham, M. C. & Temple, S. A. Avian disease and winter bird feeding. Passeng. Pigeon 50, (1998).
Franz, M., Kramer-Schadt, S., Greenwood, A. D. & Courtiol, A. Sickness-induced lethargy can increase host contact rates and pathogen spread in water-limited landscapes. Funct. Ecol. 32, 2194–2204 (2018).
Galbraith, J. A., Stanley, M. C., Jones, D. N. & Beggs, J. R. Experimental feeding regime influences urban bird disease dynamics. J. Avian Biol. 48, 700–713 (2017).
Moyers, S. C., Adelman, J. S., Farine, D. R., Thomason, C. A. & Hawley, D. M. Feeder density enhances house finch disease transmission in experimental epidemics. Philos. Trans. R. Soc. B Biol. Sci. 373(1745), 20170090 (2018).
Keesing, F., Holt, R. D. & Ostfeld, R. S. Effects of species diversity on disease risk. Ecol. Lett. 9, 485–498 (2006).
Mathiasson, M. E. & Rehan, S. M. Status changes in the wild bees of north-eastern North America over 125 years revealed through museum specimens. Insect Conserv. Divers. 12, 278–288 (2019).
Vanbergen, A. J. & Initiative, I. P. Threats to an ecosystem service: pressures on pollinators. Front. Ecol. Env. 11, 251–259 (2013).
Buhk, C. et al. Flower strip networks offer promising long term effects on pollinator species richness in intensively cultivated agricultural areas. BMC Ecol. 18(1), 1–13 (2018).
Morandin, L. A. & Kremen, C. Hedgerow restoration promotes pollinator populations and exports native bees to adjacent fields. Ecol. Appl. 23, 829–839 (2013).
Williams, N. M. et al. Native wildflower plantings support wild bee abundance and diversity in agricultural landscapes across the United States. Ecol. Appl. 25, 2119–2131 (2015).
Graystock, P. et al. Dominant bee species and floral abundance drive parasite temporal dynamics in plant-pollinator communities. Nat. Ecol. Evol. 4, 1358–1367 (2020).
Adler, L. S. et al. Disease where you dine: Plant species and floral traits associated with pathogen transmission in bumble bees. Ecology 99, 2535–2545 (2018).
Alger, S. A., Burnham, P. A. & Brody, A. K. Flowers as viral hot spots: Honey bees (Apis mellifera) unevenly deposit viruses across plant species. PLoS ONE 14(9), e0221800 (2019).
McNeil, D. J. et al. Bumble bees in landscapes with abundant floral resources have lower pathogen loads. Sci. Rep. 10, 1–12 (2020).
Daughenbaugh, K. F. et al. Metatranscriptome analysis of sympatric bee species identifies bee virus variants and a new virus, andrena-associated bee virus-1. Viruses 13, 291 (2021).
Alger, S. A., Alexander Burnham, P., Boncristiani, H. F. & Brody, A. K. RNA virus spillover from managed honeybees (Apis mellifera) to wild bumblebees (Bombus spp.). PLoS ONE 14, e0217822 (2019).
Ravoet, J. et al. Widespread occurrence of honey bee pathogens in solitary bees. J. Invertebr. Pathol. 122, 55–58 (2014).
Hayes, S. E., Tuiwawa, M., Stevens, M. I. & Schwarz, M. P. A recipe for weed disaster in islands: A super-generalist native pollinator aided by a ‘Parlourmaid’ plant welcome new arrivals in Fiji. Biol. Invasions 21, 1643–1655 (2019).
Levenson, H. & Tarpy, D. R. Pollinator community response to planted pollinator habitat in agroecosystems over time. Authorea https://doi.org/10.22541/au.164191433.37143936/v1 (2022).
Graystock, P., Yates, K., Darvill, B., Goulson, D. & Hughes, W. O. H. Emerging dangers: Deadly effects of an emergent parasite in a new pollinator host. J. Invertebr. Pathol. 114, 114–119 (2013).
Genersch, E., Yue, C., Fries, I. & De Miranda, J. R. Detection of Deformed wing virus, a honey bee viral pathogen, in bumble bees (Bombus terrestris and Bombus pascuorum) with wing deformities. J. Invertebr. Pathol. 91, 61–63 (2006).
Müller, U., McMahon, D. P. & Rolff, J. Exposure of the wild bee Osmia bicornis to the honey bee pathogen Nosema ceranae. Agric. For. Entomol. 21, 363–371 (2019).
Strobl, V., Yañez, O., Straub, L., Albrecht, M. & Neumann, P. Trypanosomatid parasites infecting managed honeybees and wild solitary bees. Int. J. Parasitol. 49, 605–613 (2019).
Gisder, S. et al. Rapid gastrointestinal passage may protect Bombus terrestris from becoming a true host for Nosema ceranae. Appl. Environ. Microbiol. 86(12), e00629-20 (2020).
Tehel, A., Streicher, T., Tragust, S. & Paxton, R. J. Experimental infection of bumblebees with honeybee-associated viruses: No direct fitness costs but potential future threats to novel wild bee hosts. R. Soc. Open Sci. 7(7), 200480 (2020).
Reynaldi, F. J., Sguazza, G. H., Albicoro, F. J., Pecoraro, M. R. & Galosi, C. M. First molecular detection of co-infection of honey bee viruses in asymptomatic Bombus atratus in South America. Braz. J. Biol. 73, 797–800 (2013).
Schoonvaere, K. et al. Unbiased RNA shotgun metagenomics in social and solitary wild bees detects associations with eukaryote parasites and new viruses. PLoS ONE 11(12), e0168456 (2016).
Melathopoulos, A. et al. Viruses of managed alfalfa leafcutting bees (Megachille rotundata Fabricus) and honey bees (Apis mellifera L.) in Western Canada: Incidence, impacts, and prospects of cross-species viral transmission. J. Invertebr. Pathol. 146, 24–30 (2017).
Schoonvaere, K., Smagghe, G., Francis, F. & de Graaf, D. C. Study of the metatranscriptome of eight social and solitary wild bee species reveals novel viruses and bee parasites. Front. Microbiol. 9, 177 (2018).
Payne, A. N., Shepherd, T. F. & Rangel, J. The detection of honey bee (Apis mellifera)-associated viruses in ants. Sci. Rep. 10(1), 1–8 (2020).
Dalmon, A. et al. Possible spillover of pathogens between bee communities foraging on the same floral resource. Insects 12(2), 122 (2021).
Kojima, Y. et al. Infestation of Japanese native honey bees by tracheal mite and virus from non-native European honey Bees in Japan. Microb. Ecol. 62, 895–906 (2011).
Graystock, P. et al. The Trojan hives: Pollinator pathogens, imported and distributed in bumblebee colonies. J. Appl. Ecol. 50, 1207–1215 (2013).
Plischuk, S. et al. South American native bumblebees (Hymenoptera: Apidae) infected by Nosema ceranae (Microsporidia), an emerging pathogen of honeybees (Apis mellifera). Environ. Microbiol. Rep. 1, 131–135 (2009).
Evison, S. E. et al. Pervasiveness of parasites in pollinators. PLoS ONE 7(1), e30641 (2012).
Graystock, P., Goulson, D. & Hughes, W. O. H. The relationship between managed bees and the prevalence of parasites in bumblebees. PeerJ 2, e522 (2014).
Graystock, P., Goulson, D. & Hughes, W. O. Parasites in bloom: Flowers aid dispersal and transmission of pollinator parasites within and between bee species. Proc. R. Soc. B Biol. Sci. 282(1813), 20151371 (2015).
Tripodi, A. D., Szalanski, A. L. & Strange, J. P. Novel multiplex PCR reveals multiple trypanosomatid species infecting North American bumble bees (Hymenoptera: Apidae: Bombus). J. Invertebr. Pathol. 153, 147–155 (2018).
Singh, R. et al. RNA viruses in hymenopteran pollinators: evidence of inter-taxa virus transmission via pollen and potential impact on non-Apis hymenopteran species. PLoS ONE 5(12), e14357 (2010).
Peng, W. et al. Host range expansion of honey bee Black Queen Cell Virus in the bumble bee, Bombus huntii. Apidologie 42, 650–658 (2011).
Levitt, A. L. et al. Cross-species transmission of honey bee viruses in associated arthropods. Virus Res. 176, 232–240 (2013).
Fürst, M. A., McMahon, D. P., Osborne, J. L., Paxton, R. J. & Brown, M. J. F. Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature 506, 364–366 (2014).
Gamboa, V. et al. Bee pathogens found in Bombus atratus from Colombia: A case study. J. Invertebr. Pathol. 129, 36–39 (2015).
Radzevičiūtė, R. et al. Replication of honey bee-associated RNA viruses across multiple bee species in apple orchards of Georgia, Germany and Kyrgyzstan. J. Invertebr. Pathol. 146, 14–23 (2017).
Murray, E. A. et al. Viral transmission in honey bees and native bees, supported by a global black queen cell virus phylogeny. Environ. Microbiol. 21, 972–983 (2019).
Dobelmann, J., Felden, A. & Lester, P. J. Genetic strain diversity of multi-host RNA viruses that infect a wide range of pollinators and associates is shaped by geographic origins. Viruses 12, 13–15 (2020).
Olgun, T., Everhart, S. E., Anderson, T. & Wu-Smart, J. Comparative analysis of viruses in four bee species collected from agricultural, urban, and natural landscapes. PLoS ONE 15(6), e0234431 (2020).
Fearon, M. L. & Tibbetts, E. A. Pollinator community species richness dilutes prevalence of multiple viruses within multiple host species. Ecology 102(5), e03305 (2021).
Sokół, R., Michalczyk, M. & Michołap, P. Preliminary studies on the occurrence of honeybee pathogens in the national bumblebee population. Ann. Parasitol. 64, 385–390 (2018).
Bravi, M. E. et al. Wild bumble bees (Hymenoptera: Apidae: Bombini) as a potential reservoir for bee pathogens in northeastern Argentina. J. Apic. Res. 58, 710–713 (2019).
Mazzei, M. et al. Detection of replicative Kashmir Bee Virus and Black Queen Cell Virus in Asian hornet Vespa velutina (Lepelieter 1836) in Italy. Sci. Rep. 9, 1–9 (2019).
Li, J. et al. Cross-species infection of deformed wing virus poses a new threat to pollinator conservation. J. Econ. Entomol. 104, 732–739 (2011).
Sachman-Ruiz, B., Narváez-Padilla, V. & Reynaud, E. Commercial Bombus impatiens as reservoirs of emerging infectious diseases in central México. Biol. Invasions 17, 2043–2053 (2015).
Jones, L. J., Ford, R. P., Schilder, R. J. & López-Uribe, M. M. Honey bee viruses are highly prevalent but at low intensities in wild pollinators of cucurbit agroecosystems. J. Invertebr. Pathol. 185, 107667 (2021).
Dolezal, A. G. et al. Honey bee viruses in wild bees: Viral prevalence, loads, and experimental inoculation. PLoS ONE 11(11), e0166190 (2016).
Mazzei, M. et al. First detection of replicative deformed wing virus (DWV) in Vespa velutina nigrithorax. Bull. Insectology 71, 211–216 (2018).
Plischuk, S. et al. Parasites and pathogens associated with native bumble bees (Hymenoptera: Apidae: Bombus spp.) from highlands in Bolivia and Peru. Stud. Neotrop. Fauna Environ. Stud. https://doi.org/10.1080/01650521.2020.1743551 (2020).
McMahon, D. P. et al. A sting in the spit: Widespread cross-infection of multiple RNA viruses across wild and managed bees. J. Anim. Ecol. 84, 615–624 (2015).
Bailes, E. J. et al. First detection of bee viruses in hoverfly (syrphid) pollinators. Biol. Lett. 14(2), 20180001 (2018).
Pritchard, Z. A. et al. Do viruses from managed honey bees (Hymenoptera: Apidae) endanger wild bees in native prairies?. Environ. Entomol. 50, 455–466 (2021).
Danforth, B. N., Mitchell, P. L. & Packer, L. Mitochondrial DNA differentiation between two cryptic Halictus (Hymenoptera: Halictidae) species. Ann. Entomol. Soc. Am. 91, 387–391 (1998).
Grozinger, C. M. & Flenniken, M. L. Bee viruses: Ecology, pathogenicity, and impacts. Annu. Rev. Entomol. 64, 205–226 (2019).
Antúnez, K. et al. Immune suppression in the honey bee (Apis mellifera) following infection by Nosema ceranae (Microsporidia). Environ. Microbiol. 11, 2284–2290 (2009).
Cameron, S. A. et al. Patterns of widespread decline in North American bumble bees. PNAS 108, 662–667 (2011).
Leite, G. M., Magan, N. & Medina, A. Comparison of different bead-beating RNA extraction strategies: An optimized method for filamentous fungi. J. Microbiol. Methods 88, 413–418 (2012).
Simms, D., Cizdziel, P. & Chomczynski, P. TRIzol: A new reagent for optimal single-step isolation of RNA. Focus (Madison) 15, 99–102 (1993).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3(7), 1–12 (2002).
Mwalili, S. M., Lesaffre, E. & Declerck, D. The zero-inflated negative binomial regression model with correction for misclassification: An example in caries research. Stat. Methods Med. Res. 17, 123–139 (2008).
R Core Team. R: Language and Environment for Statistical Computing. R Foundation for Statistical Computer (2018). Available at: https://www.r-project.org/.
Jackman, S. et al. Package ‘pscl’. (2020).
Canty, A. & Ripley, B. Package ‘boot’. (2021).
Figueroa, L. L. et al. Landscape simplification shapes pathogen prevalence in plant-pollinator networks. Ecol. Lett. 23, 1212–1222 (2020).
National Heritage Program. Species/Community Search. National Heritage Program: Natural and Cultural Resources (2021). Available at: https://ncnhp.org/data/speciescommunity-search.
Hatfield, R. et al. IUCN Assessments for North American Bombus spp. (2014).
Sersic, A. N., Masco, M. & Noy-Meir, I. Natural hybridization between species of Calceolaria with different pollination syndromes in southern Patagonia, Argentina. Plant Syst. Evol. 230, 111–124 (2001).
Otti, O. & Schmid-Hempel, P. Nosema bombi: A pollinator parasite with detrimental fitness effects. J. Invertebr. Pathol. 96, 118–124 (2007).
Crabbe, J. C., Wahlsten, D. & Dudek, B. C. Genetics of mouse behavior: Interactions with laboratory environment. Science 284(5420), 1670–1672 (1999).
Wahlsten, D. et al. Different data from different labs: Lessons from studies of gene-environment interaction. J. Nuerobiol. 54, 283–311 (2003).
Brownie, J. et al. The elimination of primer-dimer accumulation in PCR. Nucleic Acids Res. 25(16), 3235–3241 (1997).
Boncristiani, H. F. et al. In vitro infection of pupae with israeli acute paralysis virus suggests disturbance of transcriptional homeostasis in honey bees (Apis mellifera). PLoS ONE 8(9), e73429 (2013).
Acknowledgements
Thank you to the NCDA&CS and NCSU research stations for maintaining the sampling plots. Our most sincerest thanks to Tim Sit for providing invaluable advice and troubleshooting on lab processes. Thank you to our colleagues and members of the Tarpy Lab “lunch bunch” for their assistance in collecting samples in the field and/or processing samples in the laboratory, with special thanks to Ashley Rua, Chesney Banasik, Kaya Hamilton, Nicole Hanselman, Rachel Laminack, and especially Erin McDermott.
Funding
This work was supported by the NCSU Biology Graduate Program, Garden Club of America [Grant Number 566558–02697], Southern SARE [Grant Number GS19-215], and grants from the NC Department of Agriculture & Consumer Services.
Author information
Authors and Affiliations
Contributions
Both authors developed the experimental design, secured funding for the project, and analyzed the data. H.K.L. completed the field collection of samples, the lab processing of samples, wrote the main manuscript text, and prepared all figures. D.R.T. edited the manuscript text and figures. Both authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Levenson, H.K., Tarpy, D.R. Effects of planted pollinator habitat on pathogen prevalence and interspecific detection between bee species. Sci Rep 12, 7806 (2022). https://doi.org/10.1038/s41598-022-11734-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-11734-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
