
Abstract
Until recently, Shigella and enteroinvasive Escherichia coli were thought to be primate-restricted pathogens. The base of their pathogenicity is the type 3 secretion system (T3SS) encoded by the pINV virulence plasmid, which facilitates host cell invasion and subsequent proliferation. A large family of T3SS effectors, E3 ubiquitin-ligases encoded by the ipaH genes, have a key role in the Shigella pathogenicity through the modulation of cellular ubiquitination that degrades host proteins. However, recent genomic studies identified ipaH genes in the genomes of Escherichia marmotae, a potential marmot pathogen, and an E. coli extracted from fecal samples of bovine calves, suggesting that non-human hosts may also be infected by these strains, potentially pathogenic to humans. We performed a comparative genomic study of the functional repertoires in the ipaH gene family in Shigella and enteroinvasive Escherichia from human and predicted non-human hosts. We found that fewer than half of Shigella genomes had a complete set of ipaH genes, with frequent gene losses and duplications that were not consistent with the species tree and nomenclature. Non-human host IpaH proteins had a diverse set of substrate-binding domains and, in contrast to the Shigella proteins, two variants of the NEL C-terminal domain. Inconsistencies between strains phylogeny and composition of effectors indicate horizontal gene transfer between E. coli adapted to different hosts. These results provide a framework for understanding of ipaH-mediated host-pathogens interactions and suggest a need for a genomic study of fecal samples from diseased animals.
Similar content being viewed by others


Introduction
Shigellosis is a widespread human intestinal infection disease. Its causative agent, Shigella, is one of Escherichia coli pathovars, but the genus name is maintained due to medical importance1,2. Based on the symptoms and molecular features of the infection, the Shigella genus has been classified into four species3. However, these Shigella species are not monophyletic and have arisen independently from different non-pathogenic E. coli by acquiring a large plasmid that encodes a substantial number of virulence genes1. Shigella and enteroinvasive E. coli (EIEC) enter epithelial cells of the colon, multiply within them, and move between adjacent cells4. Both Shigella and EIEC become invasive by acquiering a pINV plasmid with essential virulence determinants, including genes encoding the type III secretion system (T3SS)4. Furthermore, the pathogens’ genomes feature other genomic markers of adaptation to the intracellular lifestyle, such as chromosomal pathogenicity islands, accumulation of mobile elements, and lack of genes coding for bacterial motility or lactose fermentation5. Because EIEC retain the ability to live outside the host cells and their genomes harbor significantly less mobile elements and pseudogenes, EIEC are believed to be the precursors for Shigella lineages6.
Encoded in the “entry region” of pINV, the T3SS proteins have a range of diverse functions, being structural proteins, chaperones that protect Shigella and EIEC virulence proteins from aggregation and degradation, and effector proteins that are secreted into the host cell and selectively bind particular host proteins to regulate the host biological activity7. Numerous pathogenic bacteria affect the ubiquitination pathway of the host. In Shigella, novel E3 ubiquitin-ligases encoded by the ipaH genes modulate cellular ubiquitination leading to the degradation of host proteins8. The IpaH proteins are comprised of two domains, the highly conserved, novel E3 ligase (NEL) C-terminal domain that binds ubiquitin, and the variable leucine-rich repeat-containing (LRR) N-terminal domain that binds various human proteins hence providing the substrate specificity9. The IpaH proteins are thought to trigger cell death and to modulate host inflammatory-related signals during bacterial infection; however, the substrate specificity of many IpaH proteins remains uncertain10,11.
Expression of the ipaH genes can be regulated by several transcription factors. MxiE, a transcription activator encoded in the “entry” region of pINV, regulates the intracellular expression of genes encoding numerous factors secreted by the type III secretion system, including OspB, OspC1, OspE2, OspF, VirA, and IpaH12. Two plasmid-encoded virulence transcription factors, VirF and VirB, are known to turn on the Shigella virulence by activating major determinants, and thus may also control the ipaH genes13. Both the virF and virB genes have sites for thermal sensing, and at 30 °C both of them are negatively controlled by the global transcriptional silencer H-NS13,14. Upon invasion of the host organism, H-NS detaches from DNA, switching on the virulence cascades. H-NS normally binds A/T-rich elements making bridges or loops that affect transcription from the target promoters15,16. The regulatory regions of many virulence genes in Shigella have A + T rich tracks and H-NS-bound A + T tracks are common features of mobile elements or prophages, and may be a footprint of a recent horizontal gene transfer17.
Although naturally Shigella was thought to be a primate-specific pathogen, experiments showed that it can infect other animals, yet with lower efficacy18,19. Recently, Shigella-like T3SS and associated effectors were found in Escherichia marmotae, a potential invasive pathogen of marmots20, which was also shown to be able to invade human cells20. Shigella marker genes were also found in isolates obtained from the excrement of bovine calves with diarrhea, although genome-wide data was lacking21.
Here, we applied a computational approach to predict whether some Escherichia may also be an infectious agent of non-human hosts, which, therefore, may serve as a reservoir of human pathogens and virulence genes. For that, we performed a comparative genomic analysis of the ipaH genes in Shigella, EIEC strains, and putatively invasive Escherichia species extracted from non-human hosts. We classified and compared members of the ipaH gene family based on domain sequence similarity, genomic location, and positioning of regulatory elements in upstream gene regions. Furthermore, for Shigella lineages we reconstructed the evolution of the ipaH genes on the species phylogenetic tree revealing multiple gene losses, paralogizations, and horizontal gene transfer.
Methods
Dataset of genomes
We downloaded 130 complete genomes of Shigella available in GenBank22 as of November 2020 and three complete genomes of enteroinvasive Escherichia coli (Supplementary Table S1). Additionally we downloaded all Escherichia assemblies extracted from non-human hosts that contained BLAST23 hits of the NEL-domain of Shigella IpaH (Supplementary Table S2).
Identification of the ipaH genes
Using pBLAST search of the NEL-domain (PDB: Shigella flexneri Effector IpaH1880 5KH1 https://www.rcsb.org/structure/5KH1), we found 445 protein sequences belonging to the E3 ubiquitin-ligase family. Then we clustered the sequences using CD-hit24 with a threshold of 90% aa identity and performed additional tBLASTn search of representative sequences from each cluster. It allowed us to add 419 sequences including non-annotated genes and pseudogenes. In total, we found and classified 864 ipaH sequences (Supplementary Table S3). The ipaH genes in non-human Escherichia were found using the same pipeline and collected in Supplementary Table S4.
Heatmaps
Heatmaps for sequence similarity were drawn using R packages seqinr, RColorBrewer, and gplots.
Phylogenetic tree
For construction of the Shigella species tree, we used the PanACoTA tool25. It annotates coding regions, finds orthologous groups, and constructs the phylogenetic tree for a concatenated alignment of single-copy common genes. The orthologous groups were constructed with a threshold of 80% aa identity, the phylogenetic tree was constructed with the IQ-TREE 2 module26. The tree was visualised using online iTOL27.
Annotation of regulatory elements in upstreams
Alignments of the ipaH regulatory regions were constructed with the Pro-Coffee tool28, additional promoters were mapped with the PlatProm algorithm29. The VirF binding sites were predicted manually based on phylogenetic footprinting of known binding regions. A + T tracks were classified as tracks if six or more A or T were present at the same time.
Modeling and visualization of protein structures
The three-dimensional structures of the IpaH proteins from Escherichia marmotae were modeled using the Swiss-Model program30 employing PDB: 5KH1.1 as the template. As a visualization tool, the UCSF Chimera software was used31.
Results
Validation of genome assemblies
We analysed 130 Shigella genomes including 46 S. flexneri, 25 S. dysenteriae, 19 S. boydii, 39 S. sonnei, and one unclassified Shigella strain (Supplementary Table S1). We used two criteria to validate the Shigella annotation, the presence of the ipaH genes and other components of T3SS (Table 1). As the T3SS markers we used the mxiC, mxiE, mxiG, virB, virF, spa15, spa32, spa40, ipgA, ipgB, ipgD, apaA, ipaB, ipaC, ipaD, mxiH, icsB genes. For three assemblies, we have found neither ipaH nor T3SS hits. These samples were extracted from soil, stream sediment, and Antarctic lichen so we classified them as non-invasive E. coli and excluded them from the analysis. Additionally, we checked that non-invasive E. coli strains did not have any of these virulence determinants using the set of 414 E. coli + Shigella genomes from32. In 17 assemblies, the plasmids were absent but we found chromosomal ipaH genes. 37 assemblies comprised plasmids but none of them held the components of T3SS. These results may be explained by elimination of the plasmids during cultivation33. Only 64 assemblies contained all essential virulence elements.
We also characterised the ipaH genes in three available EIEC lineages from1. One strain (E. coli NCTC 9031) did not contain ipaH genes or T3SS genes, thus the strain was filtered out. Two other strains (E. coli CFSAN029787 and E. coli 8–3-Ti3) had pINV with genes of the T3SS system, and the ipaH genes on the chromosomes and plasmids (Supplementary Table S1).
Classification of the ipaH genes
There is no consistent nomenclature of the ipaH genes across Shigella strains and the number of the ipaH genes in a strain varies (see Table 1 in34), thus, we created a unifying classification of all ipaH gene family members. In 127 Shigella assemblies, we found 864 protein sequences belonging to the E3 ubiquitin-ligase family (see “Methods”, Supplementary Table S3). Based on sequence similarity of the recognition domains and the composition of regulatory elements in upstream regions, we divided all ipaH genes into nine classes (Fig. 1). Confirming this classification, proteins from different classes were also distinguishable by their length, the number of LRRs, and the length of conserved upstream regions (Table 2). Taking into account high sequence similarity of genes across Shigella, we used consensus ipaH sequences (Supplementary Table S5) from each class for gene annotation.

Heatmaps of the pairwise distances and the respective color keys for (a, c) the ipaH genes; (b, d) their upstream sequences in Shigella. Pairwise distances were calculated as \(\sqrt{\left(1-identity\right)}\).
Interestingly, the ipaH genes from classes #1–5 were present only in chromosomes while those from classes #6–9 were found only in plasmids. The only exception was a duplicated ipaH gene from class 5 in Shigella flexneri 1a strain 0228, where one copy was encoded in the chromosome and the other one in the plasmid. This assembly did not contain the pINV plasmid with T3SS genes, thus the observation might have been caused by miss-assembly. Genes from classes #4 and #8 had the highest protein sequence similarity while upstream regions were most similar for genes from classes #2 and #6.
Only 45% of the Shigella genomes hold a complete set of chromosomal ipaH genes and 20% genomes have a complete set of plasmid ipaH genes (for plasmids this is a lower-bound estimate as many assemblies lack plasmid sequences). Moreover, in many genomes ipaH classes #3, #5, and #9 were represented by more than one copy. Most ipaH copies were identical, the exception is two subclasses (#9a and #9b) that were distinguishable both by their gene and upstream sequences (Fig. 1). Subclass #9b was found in almost all Shigella flexneri genomes, so we hypothesized that the ipaH #9b copy had been acquired by the common ancestor of the S. flexneri branch.
Regulatory patterns in the ipaH upstream regions
In addition to the high level of sequence similarity in each ipaH class, the upstream regions of the genes were also highly conserved. Indeed, the upstream intergenic regions of different ipaH genes comprised 300–900 base pairs with identity of more than 90% in each class, except class #9 (see below). Interestingly, the similarity was high starting from the translation start codon to (and including) putative binding sites of transcription factor MxiE, especially in classes #2 and #6, suggesting a key role of MxiE in the regulation of ipaH transcription. Previously, the relative positioning of MxiE binding sites and transcription starts, as well as sequences of the MxiE box, − 10 box, and the spacer between them were used to classify the ipaH genes into eight regulatory classes34. Each class defined by our sequence similarity approach, except for class #9, corresponds to one of the regulatory classes (Fig. 2b). Indeed, each class has its unique regulatory pattern characterized not only by the MxiE-box positioning and the spacer sequence, but also by the presence of A + T rich tracks as possible targets for the interaction with VirF and H-NS. Specifically, classes #4, #5, and #7 possess both A- and T-rich tracks (see Fig. 2a for an example), classes #1 and #2 has mainly polyT-tracks, and class #3 has mainly polyA-tracks.

Regulatory elements in ipaH upstream regions (a) Alignment of the upstream region for selected representatives of the ipaH class #4. Representatives have been selected based on their sequences so that all sequence variants are presented. The putative MxiE box is indicated by the orange box, A/T tracks are in green boxes. The transcription start is indicated by the black arrow. (b) Comparison of the sequence-based ipaH classification with the classification based on positioning of the MxiE box, − 10 element, and the sequence of the spacer. Adapted from34. (c) Principal scheme of upstream regions of the ipaH classes #9a and #9b.
Plasmid ipaH genes of class #9 were divided into two groups. Genes from class #9b had disrupted upstream regions due to a prophage insertion and thus did not appear to have regulatory elements typical for other ipaH classes (Fig. 2c). Also, no candidate promoters upstream of ipaH class #9b could be identified, suggesting that these genes may not be transcribed. The genes of class #9a also did not have an upstream MxiE box, however, they might be transcribed polycystronically with the ospE gene (Fig. 2c) utilizing its regulatory elements. The ipaH genes from class #9a were surrounded by multiple A + T-rich tracks typical for mobile elements or prophages17.
The upstream regions of different ipaH classes are not similar at any significant level, the only exceptions being classes #2 (chromosomal) and #6 (plasmid) that have a highly similar 150 bp fragment of the regulatory region between the MxiE box and the translation start codon (Fig. 2b).
Phyletic patterns of ipaH
We analyzed the phyletic patterns of ipaH in Shigella and EIEC strains (Fig. 3, Supplementary Table S3, Supplementary Fig. S1). The reconstructed phylogenetic tree was generally consistent with previous reconstructions1 and revealed five major Shigella clades with the tree topology not reflecting the species names. In our dataset, S. sonnei and S. flexneri were monophyletic (marked in yellow and violet in Fig. 3, respectively), S. boydii and S. dysenteriae were mixed in two distant clades (marked in orange and red in Fig. 3, respectively) and a set of S. dysenteriae strains formed the fifth clade (the green clade in Fig. 3). The phyletic patterns of the ipaH genes were highly mosaic. Nevertheless, we observed some clade-specific patterns. In particular, class #1 was rare in the orange clade, while class #3 was absent in the green clade.

Phyletic patterns of the ipaH genes in Shigella and EIEC. The coloring of the unrooted tree reflects major Shigella clades that putatively evolved from different non-pathogenic E. coli; two distant Shigella strains are shown in blue, the EIEC strains are shown in white. The presence of the ipaH genes is shown by dots whose color reflects the ipaH class (see the legend). The genes in classes #1–5 are located in chromosomes; the genes in classes #6–9, in plasmids. The genomes marked by the external blue arcs do not contain the T3SS genes.
The strains of EIEC did not cluster with the major Shigella clades or with each other (Fig. 3). The gene content and their genomic distribution was also consistent with polyphyletic origin of the EIEC strains. Specifically, Escherichia coli 8-3-Ti3 had a complete set of ipaH, while in Escherichia coli CFSAN029787, two chromosomal ipaH genes were missing. These genes were not distinguishable from Shigella effectors, and their location on chromosomes and plasmids was consistent with their class assignments. In Escherichia coli CFSAN029787, ipaH #1 and ipaH #3 had frameshifts, likely resulting in pseudogenization.
Interestingly, copies of ipaH genes were found in many Shigella genomes both on chromosomes and plasmids (Supplementary Table S3). We observed paralogs of ipaH #2, #4, #5 in the orange (boydii & dysenteriae) clade, only ipaH #4 in the green (dysenteriae) clade, and only ipaH #3 in the red (boydii & dysenteriae) clade (Supplementary Fig. S2a). Genomes of the violet (flexneri) clade had paralogs of the ipaH #3, #4, #5, #7, and #9 (Supplementary Fig. S2b), while genomes in the yellow (sonnei) clade did not have ipaH duplicates (Supplementary Fig. S2c). Surprisingly, none of ipaH paralogs were tandem repeats; in contrast, the copies located at some distance from each other and frequently surrounded by prophages and pseudogenes.
The ipaH repertoire in non-human Escherichia
We identified and compared the ipaH genes in pathogenic Escherichia spp. extracted from non-human hosts (Supplementary Table S4, Fig. 4). Previously, nine ipaH genes and two short ORFs containing fragments of ipaH genes were found in the genome of Escherichia marmotae HT073016, isolated from faecal samples of Marmota himalayana20. The authors reported automated annotation of eleven genes as ipaH: four on the pEM148 plasmid, five on the pEM76 plasmid, and two on the chromosome. According to our ipaH identification procedure (see “Methods”) we confirmed eight of these gene annotations and found one additional chromosomal gene. We excluded from the analysis short ORFs that did not contain the N-terminal domain assuming these to be mis-annotation or remains of pseudogenes.

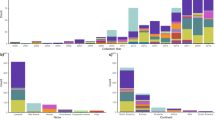
Composition of the ipaH genes in the Escherichia genomes from different hosts. Assemblies of marmot- and sheep-host Escherichia are complete, genomes of rat-host Escherichia are assembled as contigs. For Shigella, a genome with a complete set of the ipaH genes is shown.
In addition, we observed that Escherichia coli extracted from non-human hosts contained T3SS as well as ipaH genes. Specifically, two strains extracted from rat feces, Escherichia coli CFSAN092688 and Escherichia coli CFSAN085900 had six and three ipaH genes, respectively, and a strain from pooled sheep faecal samples, Escherichia coli RHB04-C17, had three ipaH genes.
Based on sequence similarity of recognition domains, we classified the IpaH proteins from non-human-host E. coli into nine classes (Fig. 5a,c). The level of sequence similarity between non-human hosts IpaH that belong to the same class is less than that for Shigella. Based on the location of the ipaH genes in completely assembled genomes of marmot- and sheep-host Escherichia, we assume that they retain their location in replicons.

Heatmap of the pairwise distances and the corresponding color keys of (a, c) the ipaH genes; (b, d) their upstream sequences in non-human-host Escherichia spp. Hosts are labeled according to the following principle: marmot is marked by red, rat is marked by dark green, sheep is marked by light green. Representative sequences of Shigella ipaH genes also were included in comparison, their labels marked in blue. Pairwise distances were calculated as \(\sqrt{\left(1-identity\right)}\).
Two ipaH classes #16 and #17, putatively in plasmids, were found in all non-human-host Escherichia spp. Putatively chromosomal ipaH class #14 were present in marmot-host and rat-host Escherichia spp.; in turn, the genomes of marmot- and sheep-host Escherichia spp. share ipaH class #10. Only one of non-human-host Escherichia ipaH genes (class #6) was present in Shigella, however the upstream sequences of ipaH class #6 in Shigella and Escherichia marmotae were significantly different (Fig. 5b,d). In particular, the regulatory regions of ipaH from non-human-host Escherichia spp. contain neither MxiE boxes, nor multiple A/T tracks.
The upstream regions of most ipaH classes were similar in rat-host and marmot-host E. coli, while the upstream regions in sheep-host E. coli genes and all ipaH upstream regions were unique. Classes #13 (in plasmid) and #14 (in chromosome) show gene-sequence and upstream-sequence similarity but have different numbers of LLRs, which indicates their evolution by gene duplication and subsequent deletions or tandem duplication of short genomic segments.
Surprisingly, in non-human hosts Escherichia spp., the C-terminal domain of IpaH proteins was not conserved (Supplementary Table S6). Non-human hosts IpaH classes #10, #11, #12 had a C-terminal domain similar to that in Shigella (92% aa identity), while the C-terminal domain of IpaH classes #13 through #17 was more diverged (75% aa identity) (Fig. 6). This observation also explains the results from20 that only a fraction of the identified ipaH genes were homologous to the ipaH of Shigella spp. Note that both variants are E. coli specific and distant from ubiquitin-ligase domains from other pathogens such as Salmonella, Yersinia, etc.

Alignment of the IpaH C-terminal domains from Shigella spp. and Escherichia marmotae. Consensus sequences are shown. The active site is marked in blue dots, alpha-helices are shown by orange frames. The differences between Shigella and both E. marmotae proteins are marked in red, the differences between two types of the C-terminal domains in E. marmotae are marked in green.
We mapped the amino acid substitutions between consensus sequences of C-terminal domains of IpaH from Shigella spp. and non-human-host Escherichia spp. on the three-dimensional structure of Shigella flexneri effector IpaH1880 (PDB: 5KH1) (Fig. 6, Supplementary Fig. S3). These differences were not clustered, nor did they affect the protein active site.
Discussion
Shigella spp. and enteroinvasive E. coli have a wide variety of IpaH effectors that play a significant role in invasion, modulation of inflammation, and host response3. Previously, several studies attempted to describe the ipaH gene family in Shigella aimed to compare representative strains from different Shigella species34,35. However, Shigella spp., and the known EIEC lineages, are paraphyletic with highly variable genomes. Therefore, a comprehensive comparative analysis combining all available genomic data was required to obtain a general picture of the gene family composition and evolution.
Collecting a large set of ipaH genes, we classified them based on sequence similarity and unified their nomenclature while maintaining references to previously used gene names34,35. Although the sequences of most ipaH genes were highly conserved across strains, in class #9 (ipaH1.4) we detected paralog diversification that might indicate formation of a new ipaH class. Given the important role of this family in the Shigella virulence, a consistent gene annotation is of direct medical relevance. Our results suggest that using consensus ipaH sequences from each class for gene annotation is efficient, reduces errors in annotation, and might be useful for future studies of this gene family.
The presence of IpaH effectors is one of the markers used for Shigella serotyping36, however, less than a half of sequenced genomes had the entire set of the ipaH genes. Contrary to previous observations on smaller datasets32, none of the ipaH genes are common to all Shigella strains, and the phyletic patterns of the ipaH genes suggest numerous independent gene losses. While the targets of some IpaH proteins are unknown, some proteins have shown to affect the same pathway at different stages, working together to cause disease3. In this case, a complete set of IpaH would be functionally redundant and may not necessarily be preserved. Note that in case of bacterial isolates, elimination of plasmids and virulence factors in the course of cultivation may have led to the loss of plasmid classes prior to genome sequencing33.
The presence of non-tandem copies of ipaH genes with conserved upstream regions in many Shigella strains indicate the acquisition of DNA fragments with ipaH from the same source and functionality and specificity of ipaH upstream regions. Superficially, the chromosomal ipaH genes seem to have more A + T tracks and presumably more options for regulation than those located on the plasmid. Indeed, the virulence plasmids likely have resulted from multiple events of transmission and transposition, and may only hold elements absolutely necessary for fast switches between cell functional states.
We did not detect any consistent differences in the repertoire of the ipaH genes in the Shigella and EIEC pathotypes. Moreover, the regulatory patterns in the upstream regions were the same. As notation of Shigella and EIEC pathotypes is not strongly defined, it is still not clear whether these factors are responsible for the differences in the infectious dose and disease severity of Shigella/EIEC pathotypes.
Interestingly, the ipaH composition and regulatory patterns in non-human host derived Escherichia differed substantially from the human host derived strains. In total we detected eight new classes of the IpaH effectors in non-human host Escherichia spp. As the human-host Escherichia coli, they maintain their location in the chromosome or plasmids. Note that, although E. marmotae is an outgroup for E. coli clade20, rat-host and sheep-host E. coli contain similar IpaH effectors while the effectors in human-host E. coli is unique; the only one ipaH class (#6, ipaH9.8) was present both in Shigella spp. and Escherichia marmotae plasmids. Inconsistencies between strains phylogeny and composition of effectors indicate horizontal gene transfer between E. coli adapted to different hosts. In contrast to Shigella ipaH, the regulatory regions of ipaH from non-human-host Escherichia spp. contain neither MxiE boxes, nor multiple A/T tracks: the only example with two such tracks is ipaH class #16 from marmot and rat.
Surprisingly, in the IpaH proteins encoded in the Escherichia genomes from non-human hosts we found two diverse C-terminal domains. This observation may be explained by the acquisition of effectors horizontally as well as differentiation of their functional roles in non-human-hosts Escherichia.
The IpaH proteins are considered as a candidate target of antibiotics due to their Shigella specificity. The first strategy is to target the C-terminal domain as it is highly conserved among Shigella IpaH effectors37. However, IpaH can affect the antimicrobial activity of host proteins even in the absence of catalytic activity38. Thus, targeting N-domains may be more effective but this strategy requires understanding of the ipaH repertoire in specific strains. To date, approaches used for testing the presence of Shigella virulence factors do not distinguish the members of the IpaH family39, thus development of gene specific primers is required.
The ipaH variants of invasive Escherichia spp. from wildlife and domestic animals will require additional study as they may contribute to human-pathogen evolution. Notably, the annotation of the source of E. coli samples may be misleading. In particular, the E. coli genome extracted from a sample of sheep feces collected from the farm floor (BioSample: SAMN15147991) might be contaminated by bacteria from another host, such as a rat living on a farm. If this were the case, the new variant of the C-terminal domain of IpaH proteins may be rodent-specific. Extensive sampling and subsequent genomic sequencing of Escherichia spp. from different hosts will shed light on the specificity of the invasion system and IpaH effectors to the pathogens’ hosts.
Data availability
The datasets supporting the conclusions of this article are described in Supplementary Tables which are also available via the link https://github.com/zaryanichka/E3UbLigases.
Abbreviations
- T3SS:
-
Type 3 secretion system
- pINV :
-
Plasmid of invasion
- NEL:
-
Novel E3-ubiquitin ligase
- LRR:
-
Leucine rich repeat
- EIEC:
-
Enteroinvasive Escherichia coli
- ORF:
-
Open reading frame
References
Hawkey, J., Monk, J. M., Billman-Jacobe, H., Palsson, B. & Holt, K. E. Impact of insertion sequences on convergent evolution of Shigella species. PLoS Genet. 16(7), e1008931 (2020).
Ranjbar, R. & Farahani, A. Shigella: Antibiotic-resistance mechanisms and new horizons for treatment. Infect. Drug Resist. 12, 3137–3167 (2019).
Mattock, E. & Blocker, A. J. How do the virulence factors of Shigella work together to cause disease?. Front. Cell Infect. Microbiol. 7, 64 (2017).
Pasqua, M. et al. The intriguing evolutionary journey of enteroinvasive E. coli (EIEC) toward pathogenicity. Front. Microbiol. 8, 2390 (2017).
Feng, Y., Chen, Z. & Liu, S.-L. Gene decay in Shigella as an incipient stage of host-adaptation. PLoS ONE 6(11), e27754 (2011).
van den Beld, M. J. C. & Reubsaet, F. A. G. Differentiation between Shigella, enteroinvasive Escherichia coli (EIEC) and noninvasive Escherichia coli. Eur. J. Clin. Microbiol. Infect. Dis. 31(6), 899–904 (2012).
Wagner, S. et al. Bacterial type III secretion systems: A complex device for the delivery of bacterial effector proteins into eukaryotic host cells. FEMS Microbiol. Lett. 365, 19. https://doi.org/10.1093/femsle/fny201 (2018).
Perrett, C. A., Lin, D.Y.-W. & Zhou, D. Interactions of bacterial proteins with host eukaryotic ubiquitin pathways. Front. Microbiol. 2, 143 (2011).
Keszei, A. F. A. & Sicheri, F. Mechanism of catalysis, E2 recognition, and autoinhibition for the IpaH family of bacterial E3 ubiquitin ligases. Proc. Natl. Acad. Sci. USA. 114(6), 1311–1316 (2017).
Singer, A. U. et al. Structure of the Shigella T3SS effector IpaH defines a new class of E3 ubiquitin ligases. Nat. Struct. Mol. Biol. 15(12), 1293–1301 (2008).
Maculins, T., Fiskin, E., Bhogaraju, S. & Dikic, I. Bacteria-host relationship: ubiquitin ligases as weapons of invasion. Cell Res. 26(4), 499–510 (2016).
Kane, C. D., Schuch, R., Day, W. A. Jr. & Maurelli, A. T. MxiE regulates intracellular expression of factors secreted by the Shigella flexneri 2a type III secretion system. J. Bacteriol. 184(16), 4409–4419 (2002).
Dorman, M. J. & Dorman, C. J. Regulatory hierarchies controlling virulence gene expression in Shigella flexneri and Vibrio cholerae. Front. Microbiol. 9, 2686 (2018).
Prosseda, G. et al. A role for H-NS in the regulation of the virF gene of Shigella and enteroinvasive Escherichia coli. Res. Microbiol. 149(1), 15–25 (1998).
Grainger, D. C. Structure and function of bacterial H-NS protein. Biochem. Soc. Trans. 44(6), 1561–1569 (2016).
Landick, R., Wade, J. T. & Grainger, D. C. H-NS and RNA polymerase: A love-hate relationship?. Curr. Opin. Microbiol. 24, 53–59 (2015).
Dorman, C. J. H-NS-like nucleoid-associated proteins, mobile genetic elements and horizontal gene transfer in bacteria. Plasmid 75, 1–11 (2014).
Shi, R. et al. Pathogenicity of Shigella in chickens. PLoS ONE 9(6), e100264 (2014).
Maurelli, A. T. et al. Shigella infection as observed in the experimentally inoculated domestic pig, Sus scrofa domestica. Microb. Pathog. 25(4), 189–196 (1998).
Liu, S. et al. Genomic and molecular characterisation of Escherichia marmotae from wild rodents in Qinghai-Tibet plateau as a potential pathogen. Sci. Rep. 9(1), 10619 (2019).
Zhu, Z. et al. Virulence factors and molecular characteristics of Shigella flexneri isolated from calves with diarrhea. BMC Microbiol. 21(1), 214 (2021).
Benson, D. A. et al. GenBank. Nucleic Acids Res. 41, D36-42 (2013).
Madden, T. The BLAST Sequence Analysis Tool (National Center for Biotechnology Information, 2003).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28(23), 3150–3152 (2012).
Perrin, A. & Rocha, E. P. C. PanACoTA: A modular tool for massive microbial comparative genomics. NAR Genom. Bioinform. 3(1), 106 (2021).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37(5), 1530–1534 (2020).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44(W1), W242–W245 (2016).
Magis, C. et al. T-Coffee: Tree-based consistency objective function for alignment evaluation. Methods Mol. Biol. 1079, 117–129 (2014).
Shavkunov, K. S., Masulis, I. S., Tutukina, M. N., Deev, A. A. & Ozoline, O. N. Gains and unexpected lessons from genome-scale promoter mapping. Nucleic Acids Res. 37(15), 4919–4931 (2009).
Waterhouse, A. et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46(W1), W296-303 (2018).
Pettersen, E. F. et al. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 25(13), 1605–1612 (2004).
Seferbekova, Z. et al. High rates of genome rearrangements and pathogenicity of Shigella spp. Front. Microbiol. 12, 628622 (2021).
Sansonetti, P. J., Kopecko, D. J. & Formal, S. B. Shigella sonnei plasmids: Evidence that a large plasmid is necessary for virulence. Infect. Immun. 34(1), 75–83 (1981).
Bongrand, C., Sansonetti, P. J. & Parsot, C. Characterization of the promoter, MxiE box and 5’ UTR of genes controlled by the activity of the type III secretion apparatus in Shigella flexneri. PLoS ONE 7(3), e32862 (2012).
Ashida, H., Toyotome, T., Nagai, T. & Sasakawa, C. Shigella chromosomal IpaH proteins are secreted via the type III secretion system and act as effectors. Mol. Microbiol. 63(3), 680–693 (2007).
Wu, Y., Lau, H. K., Lee, T., Lau, D. K. & Payne, J. In silico serotyping based on whole-genome sequencing improves the accuracy of Shigella identification. Appl. Environ. Microbiol. 85, 7. https://doi.org/10.1128/AEM.00165-19 (2019).
Ashida, H. & Sasakawa, C. Shigella IpaH family effectors as a versatile model for studying pathogenic bacteria. Front. Cell Infect. Microbiol. 5, 100 (2015).
Ye, Y., Xiong, Y. & Huang, H. Substrate-binding destabilizes the hydrophobic cluster to relieve the autoinhibition of bacterial ubiquitin ligase IpaH9.8. Commun. Biol. 3(1), 752 (2020).
Aranda, K. R. S., Fagundes-Neto, U. & Scaletsky, I. C. A. Evaluation of multiplex PCRs for diagnosis of infection with diarrheagenic Escherichia coli and Shigella spp. J. Clin. Microbiol. 42(12), 5849–5853 (2004).
Acknowledgements
The project was initiated with Aygul Minnegalieva and Yulia Yakovleva at the Summer School of Molecular and Theoretical Biology (SMTB-2020), supported by the Zimin Foundation. We thank Inna Shapovalenko, Daria Abuzova, Elizaveta Kaminskaya, and Dmitriy Zvezdin for their contribution to the project during SMTB-2020. We also thank Peter Vlasov for fruitful discussions.
Funding
This study was supported by the Russian Foundation for Basic Research (RFBR), Grant # 20-54-14005 and Fonds zur Förderung der wissenschaftlichen Forschung (FWF), Grant # I5127-B. The work of OB is supported by the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie Grant Agreement No. 754411. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
O.O.B. conceived and designed the study. N.O.D. and M.N.T. analysed the data. M.S.G. and F.A.K. aided in interpreting the results. All authors wrote, read, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dranenko, N.O., Tutukina, M.N., Gelfand, M.S. et al. Chromosome-encoded IpaH ubiquitin ligases indicate non-human enteroinvasive Escherichia. Sci Rep 12, 6868 (2022). https://doi.org/10.1038/s41598-022-10827-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-10827-3
This article is cited by
-
The effects of antibiotic use on the dynamics of the microbiome and resistome in pigs
Animal Microbiome (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
