
Abstract
To assess severity markers and outcomes of patients with systemic sclerosis (SSc) with or without pulmonary arterial hypertension (PAH-SSc/non-PAH-SSc), and the impact of interstitial lung disease (ILD) on PAH-SSc. Non-PAH-SSc patients from the Spanish SSc registry and PAH-SSc patients from the Spanish PAH registry were included. A total of 364 PAH-SSc and 1589 non-PAH-SSc patients were included. PAH-SSc patients had worse NYHA-functional class (NYHA-FC), worse forced vital capacity (FVC) (81.2 ± 20.6% vs 93.6 ± 20.6%, P < 0.001), worse tricuspid annular plane systolic excursion (TAPSE) (17.4 ± 5.2 mm vs 19.9 ± 6.7 mm, P < 0.001), higher incidence of pericardial effusion (30% vs 5.2%, P < 0.001) and similar prevalence of ILD (41.8% vs. 44.9%). In individuals with PAH-SSc, ILD was associated with worse hemodynamics and pulmonary function tests (PFT). Up-front combination therapy was used in 59.8% and 61.7% of patients with and without ILD, respectively. Five-year transplant-free survival rate was 41.1% in PAH-SSc patients and 93.9% in non-PAH-SSc patients (P < 0.001). Global survival of PAH-SSc patients was not affected by ILD regardless its severity. The multivariate survival analysis in PAH-SSc patients confirmed age at diagnosis, worse NYHA-FC, increased PVR, reduced DLCO, and lower management with up-front combination therapy as major risk factors. In conclusion, in PAH-SSc cohort risk of death was greatly increased by clinical, PFT, and hemodynamic factors, whereas it was decreased by up-front combination therapy. Concomitant ILD worsened hemodynamics and PFT in PAH-SSc but not survival regardless of FVC impairment.
Similar content being viewed by others


Introduction
Systemic sclerosis (SSc) is a rare systemic autoimmune disease characterized by fibrosis of the skin and internal organs and vasculopathy1,2]. Pulmonary hypertension (PH)-of which pulmonary arterial hypertension (PAH) is the most frequent form in SSc- and interstitial lung disease (ILD) are the two leading contributing causes of early death3. When associated with connective tissue diseases (CTD) like SSc, PAH is classified as Group 1.4 of the PH classification4. Both PH and ILD may coexist5,6,7, and when ILD is significant PH is classified as Group 3. However, this classification is challenging and difficult to incorporate into clinical practice with SSc patients. Prevalence of PAH in SSc varies across studies between 5 and 19%8,9,10,11,12. Prevalence of clinically relevant ILD is higher with a range from 16 to 47% depending on the definition used13,14,15.
Prognosis of SSc-associated PAH is poor with an annual mortality rate of ~ 30% vs. ~ 10% for the idiopathic form of PAH (IPAH) despite similar hemodynamic features3,16,17. Response to PAH therapy is also worse16,18. The mortality rate attributable to ILD in SSc patients is ~ 33%3. Survival rate is significantly shortened when both pulmonary complications coexist6,7,19. Nonetheless, studies on mortality associated with PAH and/or ILD in SSc patients are limited by the reduced size of the populations; thus, nationwide registries are useful in these cases.
RESCLE (Registro de ESCLErodermia) is the Spanish registry of SSc patients, and has been running since 200620. The prevalence of PAH confirmed by right heart catheterization (RHC) in this registry is ~ 4%21,22. REHAP (Registro Español de Hipertensión Arterial Pulmonar) is the Spanish registry of patients with PAH, and was created in 200723. The prevalence of SSc-associated PAH was 9.2%23. The objective of this study was to assess the clinical characteristics and prognosis of patients with SSc with or without PAH (PAH-SSc/non-PAH-SSc), and the impact of ILD on PAH-SSc by analyzing both nationwide cohorts.
Results
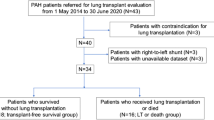
At the time of study inclusion, there were 1996 patients enrolled in the RESCLE and 3409 in the REHAP registries. Of these, 1589 (79.6%) RESCLE patients did not have a PAH diagnosis (non-PAH-SSc) and 364 (10.7%) REHAP patients had SSc (PAH-SSc) confirmed on RHC. RHC was only performed in 58 non-PAH-SSc patients ruling out this complication, and specifically in 6 out of 38 patients with sPAP > 40 mmHg by echocardiography. These were the populations analyzed. Autoantibody specificities were available in non-PAH-SSc patients, 687/1413 (48.6%) had anti-centromere antibody, 259/1386 (18.7%) had anti-topoisomerase I antibody and 42/353 (11.9%) had anti-RNA polymerase III antibody.
Impact of PAH on SSc patients
Table 1 summarizes the baseline demographic, clinical, and echocardiography data of patients according to presence of PAH. Compared to non-PAH-SSc patients, PAH-SSc patients were older, had worse New York Heart Association functional class (NYHA FC) and pulmonary function tests (PFTs) (as assessed by % of predicted forced vital capacity (FVC) and diffusing capacity for carbon monoxide (DLCO)); more patients had FVC/DLCO ≥ 1.4 and even ≥ 1.6. Furthermore, mean systolic pulmonary artery pressure (sPAP) was greater in PAH-SSc patients, more patients presented sPAP > 40 mmHg, any grade or moderate-severe degree of tricuspid regurgitation, pericardial effusion, or lower tricuspid annular plane systolic excursion (TAPSE) values. No differences were observed in the prevalence of ILD. Regarding medical treatment, most PAH-SSc patients (62.6%) received up-front combination therapy while 15.9% non-PAH-SSc patients received specific vasodilators for peripheral vasculopathy. These differences did not change when the population was compared according to the presence or absence of ILD (online supplementary table II and III).
Over a median (interquartile range, IQR) follow-up of 2 (1–4) years, 186 (51.1%) PAH-SSc patients died, 14 (3.8%) underwent pulmonary transplantation, and 13 (0.2%) were lost to follow-up. Over a follow-up period of 5 (2–11) years, 185 (11.6%) non-PAH-SSc patients died and 196 (12.3%) were lost to follow-up. The most common causes of death were related to PAH (heart failure and sudden cardiac death) in PAH-SSc patients, while in non-PAH-SSc patients they were related to SSc in 24.3% of cases, malignancies in 17.8%, and others in 29.2% (online supplementary table IV). Kaplan–Meier curves and 1-, 3- and 5-year survival rates for PAH-SSc and non-PAH-SSc patients are shown in Fig. 1. The 5-year survival rate from PAH diagnosis was 41.1% in PAH-SSc patients and 93.9% in non-PAH-SSc patients from SSc diagnosis (P < 0.001).

Kaplan–Meier analysis of transplant-free survival in PAH-SSc patients compared with non-PAH-SSc.
Impact of ILD and the severity of FVC impairment on PAH-SSc
Of the 220 PAH-SSc patients who had high-resolution computed tomography (HRCT) scans, 92 (41.8%) had ILD. Patients’ characteristics are shown in Table 2. Compared with PAH-SSc without ILD patients, those with concomitant ILD had lower female proportion and more patients presented impaired PFTs, with FVC < 60% and DLCO ≤ 55%. The extent or specific ILD patterns were not available. Nevertheless, in order to estimate the severity according to Goh’s criteria, 48 out of 88 (54.5%) PAH-SSc with ILD patients had extensive disease taking into account FVC < 70%. Right atrial pressure (RAP), cardiac output (CO), cardiac index (CI), and mean pulmonary artery pressure (mPAP) were significantly lower. No significant differences were found on echocardiography, but mean TAPSE was lower. No differences were observed regarding treatment strategies in patients with concomitant ILD and those without. Up-front combination therapy was the most frequent treatment in both cases (59.8% of patients with ILD and 61.7% of patients without ILD). No differences in transplant-free survival were observed in PAH-SSc patients according to the presence of ILD (P = 0.444) (Fig. 2A). Nevertheless, 1-, 3- and 5-year transplant-free survival rates were 82.5%, 60.2%, and 35% vs. 84.1%, 58.9%, and 43.5%, respectively, with a tendency for shorter survival in PAH-SSc patients with concomitant ILD.

Kaplan–Meier analysis of transplant-free survival in patients with PAH-SSc according to (A) presence of ILD and (B) severity of the restrictive lung disease.
FVC was available in 88 out of 92 patients with PAH-SSc and ILD. Thirty-five (40%) of these patients had FVC < 60%. Patients’ characteristics are shown in supplementary table V. Patients with FVC < 60% were younger at diagnosis and had lower mean FVC/DLCO compared to their counterparts with FVC ≥ 60%. No differences were observed in gender, NYHA FC or 6-min walk test (6MWT), hemodynamics, biomarkers, electrocardiogram, or echocardiographic variables. Up-front combination therapy was the preferred approach in both cases (51.4% in FVC < 60% group and 66.0% in FVC ≥ 60% patients), although there was a trend towards lower use of up-front combination therapy and greater use of monotherapy in patients with FVC < 60% (P = 0.042). For PAH-SSc patients, no differences in survival were found regarding FVC impairment (FVC < 60% vs FVC ≥ 60% patients) (P = 0.167) (Fig. 2B). However, there was a numerical reduction at 1-, 3- and 5-year survival rates in patients with FVC < 60% compared with FVC ≥ 60% (76.8%, 51.2% and 27.0% vs. 85.0%, 68.5% and 42.3%, respectively), with a trend to shorter survival.
Univariate and multivariate survival analysis in PAH-SSc and non-PAH-SSc patients
For both populations, factors associated to transplant-free survival on univariate analysis are shown in Table 3.
The multivariate survival analysis in PAH-SSc patients identified age at diagnosis (hazard ratio (HR) 1.02 [95% CI 1.00–1.03]; P = 0.036), NYHA FC III-IV (HR 1.63 [95% CI 1.10–2.42]; P = 0.015), and pulmonary vascular resistance (PVR) (HR 2.41 [95% CI 1.37–4.25]; P = 0.002) as poor prognostic indicators, whereas DLCO per 10%—predicted increase (HR 0.87 [95% CI 0.78–0.97]; P = 0.009) and up-front combination therapy (HR 0.54 [95% CI 0.38–0.77]; P < 0.001) were the only factors associated to better prognosis (Table 4). The multivariate analysis in non-PAH-SSc patients showed that older age at diagnosis (HR 1.09 [95% CI 1.07–1.11]; P < 0.001) worsened prognosis, while FVC per 10%—predicted increase(HR 0.80 [95% CI 0.72–0.88]; P < 0.001) and DLCO per 10%—predicted increase (HR 0.92 [95% CI 0.85–1.00]; P = 0.048) were associated with greater survival.
Discussion
By linking two nationwide registries this study, the largest conducted to date, further highlights the huge impact that PAH has on SSc patients. Approximately half of the patients were diagnosed with ILD independently of the presence of PAH. In PAH-SSc patients, ILD was found to worsen PFTs and hemodynamics but did not have a direct significant impact on survival, regardless of the severity of the ventilatory restrictive pattern. Older age, worse NYHA FC stages, higher PVR, and reduced DLCO at the time of diagnosis were independently linked to poor prognosis. Current treatment strategies (i.e., greater use of up-front combination therapy) are likely to have had an impact on survival of PAH-SSc patients even when they experienced mild-moderate ILD.
PAH-SSc patients had higher sPAP and tricuspid regurgitation velocity, were older, had worse NYHA FC, lower DLCO and elevated FVC/DLCO ratio (> 1.6 and 1.4), and greater prevalence of pericardial effusion; all of them well known clinical features of SSc-associated PAH. The devastating effect of PAH on SSc was reflected by a reduction of nearly 60% in 5-year transplant-free survival (41.1% vs. 93.9% in non-PAH-SSc patients). Three-year survival rate of PAH-SSc patients in our study (59.1%) was similar to that reported by other registries9,19,24,25,26,27,28 and a meta-analysis performed by Lefevre et al.29. Only in the large prospective PHAROS study 3-year survival rate was higher (75%)30. This has been attributed to earlier diagnosis of PAH as reflected by the greater percentage of patients with NYHA FC I-II (59% vs. ~ 30% in the above-mentioned registries and in our series). Availability of new PAH-targeted therapies and treatment strategy changes during follow-up are also likely to affect survival. However, in the meta-analysis published by Lefevre et al.29 survival did not change between studies over time, and disease’s severity at baseline was the most important prognostic factor. Up to 60% of PAH-SSc patients in our series received up-front combination therapy in contrast to the 43% in the French registry and the 34% in the REVEAL registry, both contemporary24,27. Subsequent evidence endorsed up-front combination treatment31, and current guidelines recommend this strategy for most of the patients32. Even though the increased use of up-front combination therapy in our study was not correlated with better global survival compared to the registries mentioned above, this strategy was independently associated with greater survival in our cohort.
Approximately half of the patients with PAH presented ILD, with 40% showing a moderate-severe restrictive ventilatory pattern. PAH-SSc patients with ILD had lower FVC, DLCO, TAPSE, and CI. Recently, Chauvelot et al. analyzed 128 patients from the French prospective PH registry: 66 with SSc-PH-ILD and 62 with SSc-PAH33. Patients with SSc-PH-ILD had lower FVC and lower DLCO. Use of first-line PAH-specific therapies was similar in both groups and included endothelin receptor antagonists (80%), phosphodiesterase 5 inhibitors (13%), or a combination of both (6%). Only 3 patients received a prostacyclin analog as initial treatment.
In our study, PAH-SSc patients with ILD and FVC ≥ 60% presented with FVC/DLCO ≥ 1.6 more frequently, indicating a more prominent vascular involvement in this subgroup. The threshold determining the extension of ILD leading to one or another classification PH group is blurred and remains to be defined. Thus, when patients present with precapillary PH and mild ILD they are classified into PH Group 1, and when they have more severe ILD they are classified into PH Group 3 (PH-ILD). Despite this fact, a significant proportion of patients present with intermediate severity of ILD and with different degrees of PH, which make PH classification and the following treatment decision especially challenging.
In this cohort, the worse hemodynamics and pulmonary capacity at PAH-SSc diagnosis associated with concomitant ILD did not turn into worse transplant-free survival, although a trend to higher 5-year survival was observed in patients with lower FVC. Previous studies have reported increased mortality in patients with PAH-SSc and ILD15,29,34, but most of them merged PAH-SSc patients with mild ILD (PH group 1) with PH-ILD patients. As in our series, Volkmann et al. reported similar 3-year survival rates in PAH-SSc patients with and without ILD (50% and 60%, respectively)35, which were associated with early use of aggressive treatment (i.e., prostanoid therapy was used in 52% of patients with ILD). Recently, Young et al. have described a prospective cohort of 93 patients with ILD, identifying a PH prevalence of 29 (31.2%) with a 3-year survival of 91%36. Such optimal survival may be explained by the intensive PH screening program and the extensive use of vasodilator therapy (82.8% of the patients with PH). Conversely, the survival in the French registry was significantly shorter in patients with SSc-PH-ILD compared to those with SSc-PAH. In SSc-PH-ILD patients, the survival rates at 1, 2, and 3 years were 91.9%, 78.8%, and 58.5%, respectively, compared to 95.9%, 91.3%, and 78.6% in SSc-PAH patients (P = 0.04)33.
A more conservative treatment approach with higher use of PAH monotherapy at diagnosis was observed for patients with ILD and FVC < 60%. This was probably due to safety concerns associated with the use of PAH-targeted therapies in patients with PH-ILD18 as these latter patients have traditionally been excluded from PAH clinical trials37.
Facing the reality that we still cannot precisely classify PH-SSc when associated with ILD, there is increasing evidence suggesting that early use of pulmonary vasodilator treatment improves outcomes, and nationwide registries confirm a widespread off-label use of these drugs in real life, reflecting that treating PAH is a priority for clinicians irrespective of the severity of ILD. Our results reinforce this idea and indicate that treating PAH-SSc aggressively from onset improves outcomes regardless of the presence of ILD.
In PAH-SSc patients, prognostic factors identified in univariate survival analysis were similar to prior meta-analysis29, although lower FVC or DLCO, increased FVC/DLCO ratio ≥ 1.4, and lower use of up-front combination therapies at the time of PAH diagnosis were also identified as indicators of poorer survival. Interestingly, the presence of ILD or reduced FVC were not identified as risk factors in the multivariate analysis, whereas older age, worse NYHA FC, elevated PVR or reduced DLCO, and monotherapy at PAH diagnosis were associated to worse prognosis. Conversely, in the French PH registry only the presence of ILD, chronic kidney disease, and 6-min walk distance at baseline were associated with greater mortality33. Concerning the age at PAH diagnosis, the French national study conducted between 2006 and 2017, has described an improvement in survival in patients ≤ 70 years but not in older ones28. That may be explained by the higher proportion of patients that, in the later years, has been treated with pulmonary vasodilator up-front combination therapy both in the first 4 months (48.6% vs 25.6%) and throughout the study (64.3% vs 39%). Our results support the use of up-front combination therapy at early PAH diagnosis regardless of age in order to improve transplant-free survival.
Several limitations have to be recognized in the interpretation of this study, some of them (e.g., only using variables common to both registries, not analyzing treatments during follow-up nor the last one reported) have been already noted. RHC was not performed for the selection of non-PAH-SSc patients due to it is an invasive procedure, and it is indicated after cautious doctor’s decision. ILD-targeted therapy was not available in PAH-SSc cohort that may also influence on survival of this patients. Both registries are voluntary, which leads to a lack of information on variables that may impact prognosis. To mitigate this limitation, multivariable analysis was carried out using only variables available in > 60% of patients.
Conclusion
The largest assessment ever of the impact of PAH on SSc confirms the very relevant clinical and prognostic repercussion of PAH on SSc. When associated with ILD, PAH-SSc presents with worse hemodynamic features and PFTs, but not poorer survival independently of ILD severity. Baseline treatment with pulmonary vasodilator up-front combination therapy was established in a majority of PAH-SSc patients regardless of the presence of ILD and was independently associated with longer survival.
Materials and methods
Patients
Study design, inclusion and exclusion criteria, and data collection of RESCLE and REHAP registries have been published elsewhere20,23. All methods were carried out in accordance with relevant guidelines and regulations and the study was approved by the Hospital Vall d’Hebron Institutional Review Board [PR(AMI)280/2018]. In brief, RESCLE is a voluntary nationwide registry of patients with SSc diagnosed on the 2013 ACR/EULAR criteria for SSc38 and/or on the modified LeRoy and Medsger classification criteria39. The onset of scleroderma was defined as the first symptom related to SSc including Raynaud’s phenomenon. Both prevalent and incident non-PAH-SSc patients from RESCLE registry were included in the analysis, and excluding PH-SSc patients. REHAP is also a voluntary nationwide registry designed to prospectively collect exhaustive information on the demographics, management, and outcome of patients newly and previously diagnosed with PAH by RHC23. PAH was defined as a mean pulmonary arterial pressure (mPAP) ≥ 25 mmHg at rest with a pulmonary artery wedge pressure ≤ 15 mmHg and pulmonary vascular resistances ≥ 3 Wood units at RHC32. For the purposes of this study, only prospectively recruited incident patients with SSc-associated PAH (PAH-SSc) from REHAP registry were included in this analysis.
Data collected
Baseline data from RESCLE and REHAP registries at the time of diagnosis of SSc and PAH respectively were collected. The following variables, considered potential risk factors for PAH in SSc40,41,42, were common to both registries and included in the analyses: (1) Demographics: age at the time of diagnosis (SSc or PAH) and gender24,41; (2) Clinical: New York Heart Association functional class (NYHA FC) and time since SSc diagnosis20,24,40,41 (only for RESCLE patients); (3) Pulmonary function test (PFT): predicted forced vital capacity (FVC %), predicted diffusing capacity for carbon monoxide (DLCO %)24,40,41 and the FVC%/DLCO% ratio > 1.643. We also evaluated a less restrictive cut-off value of 1.4, which has been associated to PH in patients with ILD44,45. ILD was defined as the presence of an interstitial pattern on high-resolution computed tomography (HRCT) in REHAP, and by HRCT or chest x-ray in RESCLE. Comparative analyses were performed only in patients with HRCT-confirmed ILD; (4) Echocardiography assessments: left ventricular ejection fraction (LVEF), systolic PAP (sPAP), degree of tricuspid regurgitation, pericardial effusion, and tricuspid annular plane systolic excursion (TAPSE)24; (5) Causes of death, which were homogenized as both registries had different approaches of capturing these data (online supplementary table I). For the definition of independent prognostic factors in PAH-SSc and to analyze the impact of ILD on PAH-SSc, we also selected prognostic variables including 6-min walk test (6MWT), hemodynamic parameters (cardiac output [CO], cardiac index [CI], mean pulmonary artery pressure [mPAP], pulmonary vascular resistance [PVR], right atrial pressure [RAP], and mixed venous oxygen saturation [SvO2]), and biomarkers (N-terminal pro B-type natriuretic peptide [NTproBNP] or B-type natriuretic peptide [BNP]).
Patient demographics, clinical variables, cardiac, and pulmonary assessments were prospectively recorded by participating physicians according to a standard protocol. Both registries required all patients to provide written informed consent in order to participate. The Institutional Review Boards of the participating hospitals approved the respective registries.
Statistical analyses
Continuous variables were summarized as the mean ± SD or the median and interquartile range (IQR) as appropriate and compared using Student’s t-test or Mann–Whitney U test, respectively. Categorical variables were compared using the chi-square and Fisher’s exact tests as appropriate. P values < 0.05 (2-tailed) were considered significant. Bonferroni correction was applied in multiple comparisons. Patients lost to follow-up were censored on the day of their last visit. Time-to-event analyses were performed using the Kaplan–Meier method until date of lung transplantation or death. Transplant-free survival was estimated since the time of SSc diagnosis in non-PAH-SSc patients, and since PAH diagnosis in PAH-SSc patients. Factors associated with worse prognosis were identified using the Cox proportional hazards models. Variables collected in > 60% of patients that were found to be significant in univariate analysis (P < 0.05) were incorporated into a step-wise multivariate model.
Data availability
The data that support the findings of this study are available on request from the corresponding author.
Change history
28 July 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41598-022-17525-0
References
Denton, C. P. & Khanna, D. Systemic sclerosis. Lancet 390, 1685–1699. https://doi.org/10.1016/S0140-6736(17)30933-9 (2017).
Kowal-Bielecka, O. et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann. Rheum. Dis. 76, 1327–1339. https://doi.org/10.1136/annrheumdis-2016-209909 (2017).
Steen, V. D. & Medsger, T. A. Changes in causes of death in systemic sclerosis, 1972–2002. Ann. Rheum. Dis. 66, 940–944. https://doi.org/10.1136/ard.2006.066068 (2007).
Simonneau, G. et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. https://doi.org/10.1183/13993003.01913-2018 (2019).
Chang, B., Wigley, F. M., White, B. & Wise, R. A. Scleroderma patients with combined pulmonary hypertension and interstitial lung disease. J. Rheumatol. 30, 2398–2405 (2003).
Trad, S. et al. Pulmonary arterial hypertension is a major mortality factor in diffuse systemic sclerosis, independent of interstitial lung disease. Arthritis Rheum. 54, 184–191. https://doi.org/10.1002/art.21538 (2006).
Mathai, S. C. et al. Survival in pulmonary hypertension associated with the scleroderma spectrum of diseases: Impact of interstitial lung disease. Arthritis Rheum. 60, 569–577. https://doi.org/10.1002/art.24267 (2009).
Hachulla, E. et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: A French nationwide prospective multicenter study. Arthritis Rheum. 52, 3792–3800. https://doi.org/10.1002/art.21433 (2005).
Mukerjee, D. et al. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: Application of a registry approach. Ann. Rheum. Dis. 62, 1088–1093. https://doi.org/10.1136/ard.62.11.1088 (2003).
Hsu, V. M. et al. Development of pulmonary hypertension in a high-risk population with systemic sclerosis in the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma (PHAROS) cohort study. Semin. Arthritis Rheum. 44, 55–62. https://doi.org/10.1016/j.semarthrit.2014.03.002 (2014).
Avouac, J. et al. Prevalence of pulmonary hypertension in systemic sclerosis in European Caucasians and metaanalysis of 5 studies. J. Rheumatol. 37, 2290–2298. https://doi.org/10.3899/jrheum.100245 (2010).
Coghlan, J. G. et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: The DETECT study. Ann. Rheum. Dis. 73, 1340–1349. https://doi.org/10.1136/annrheumdis-2013-203301 (2014).
Hunzelmann, N. et al. The registry of the German Network for Systemic Scleroderma: Frequency of disease subsets and patterns of organ involvement. Rheumatology (Oxford) 47, 1185–1192. https://doi.org/10.1093/rheumatology/ken179 (2008).
Vonk, M. C. et al. Systemic sclerosis and its pulmonary complications in The Netherlands: An epidemiological study. Ann. Rheum. Dis. 68, 961–965. https://doi.org/10.1136/ard.2008.091710 (2009).
Michelfelder, M. et al. Interstitial lung disease increases mortality in systemic sclerosis patients with pulmonary arterial hypertension without affecting hemodynamics and exercise capacity. Clin. Rheumatol. 36, 381–390. https://doi.org/10.1007/s10067-016-3504-6 (2017).
Chaisson, N. F. & Hassoun, P. M. Systemic sclerosis-associated pulmonary arterial hypertension. Chest 144, 1346–1356. https://doi.org/10.1378/chest.12-2396 (2013).
Weatherald, J. et al. Screening for pulmonary arterial hypertension in systemic sclerosis. Eur. Respir. Rev. https://doi.org/10.1183/16000617.0023-2019 (2019).
Le Pavec, J. et al. Systemic sclerosis-related pulmonary hypertension associated with interstitial lung disease: Impact of pulmonary arterial hypertension therapies. Arthritis Rheum. 63, 2456–2464. https://doi.org/10.1002/art.30423 (2011).
Condliffe, R. et al. Connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era. Am. J. Respir. Crit. Care Med. 179, 151–157. https://doi.org/10.1164/rccm.200806-953OC (2009).
Alba, M. A. et al. Early- versus late-onset systemic sclerosis: Differences in clinical presentation and outcome in 1037 patients. Medicine (Baltimore) 93, 73–81. https://doi.org/10.1097/MD.0000000000000018 (2014).
Garcia-Hernandez, F. J. et al. Pulmonary hypertension in Spanish patients with systemic sclerosis. Data from the RESCLE registry. Clin. Rheumatol. 38, 1117–1124. https://doi.org/10.1007/s10067-018-4390-x (2019).
Pestana-Fernandez, M. et al. Long-term efficacy and safety of monotherapy versus combination therapy in systemic sclerosis-associated pulmonary arterial hypertension: A retrospective cohort study from the Nationwide Spanish Scleroderma Registry (RESCLE). J. Rheumatol. https://doi.org/10.3899/jrheum.180595 (2019).
Escribano-Subias, P. et al. Survival in pulmonary hypertension in Spain: Insights from the Spanish registry. Eur. Respir. J. 40, 596–603. https://doi.org/10.1183/09031936.00101211 (2012).
Chung, L. et al. Unique predictors of mortality in patients with pulmonary arterial hypertension associated with systemic sclerosis in the REVEAL registry. Chest 146, 1494–1504. https://doi.org/10.1378/chest.13-3014 (2014).
Hurdman, J. et al. ASPIRE registry: Assessing the Spectrum of Pulmonary hypertension Identified at a REferral centre. Eur. Respir. J. 39, 945–955. https://doi.org/10.1183/09031936.00078411 (2012).
Hoeper, M. M. et al. Mortality in pulmonary arterial hypertension: Prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur. Respir. J. 2017, 50. https://doi.org/10.1183/13993003.00740-2017 (2015).
Weatherald, J. et al. Haemodynamics and serial risk assessment in systemic sclerosis associated pulmonary arterial hypertension. Eur. Respir. J. https://doi.org/10.1183/13993003.00678-2018 (2018).
Hachulla, E. et al. Survival improved in patients aged ≤ 70 years with systemic sclerosis-associated pulmonary arterial hypertension during the period 2006 to 2017 in France. Chest https://doi.org/10.1016/j.chest.2019.10.045 (2006).
Lefevre, G. et al. Survival and prognostic factors in systemic sclerosis-associated pulmonary hypertension: A systematic review and meta-analysis. Arthritis Rheum. 65, 2412–2423. https://doi.org/10.1002/art.38029 (2013).
Kolstad, K. D., Li, S., Steen, V., Chung, L. & Investigators, P. Long-term outcomes in systemic sclerosis-associated pulmonary arterial hypertension from the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma Registry (PHAROS). Chest 154, 862–871. https://doi.org/10.1016/j.chest.2018.05.002 (2018).
Galiè, N. et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N. Engl. J. Med. 373, 834–844. https://doi.org/10.1056/NEJMoa1413687 (2015).
Galiè, N. et al. ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 37, 67–119. https://doi.org/10.1093/eurheartj/ehv317 (2016).
Chauvelot, L. et al. Hemodynamic response to treatment and outcomes in pulmonary hypertension associated with interstitial lung disease versus pulmonary arterial Hypertension in Systemic sclerosis: Data from a study identifying prognostic factors in pulmonary hypertension associated with interstitial lung disease. Arthritis Rheumatol. 73, 295–304. https://doi.org/10.1002/art.41512 (2021).
Lee, M. H. & Bull, T. M. The role of pulmonary arterial hypertension-targeted therapy in systemic sclerosis. F1000Res https://doi.org/10.12688/f1000research.20313.1 (2019).
Volkmann, E. R. et al. Improved transplant-free survival in patients with systemic sclerosis-associated pulmonary hypertension and interstitial lung disease. Arthritis Rheumatol. 66, 1900–1908. https://doi.org/10.1002/art.38623 (2014).
Young, A. et al. Prevalence, treatment, and outcomes of coexistent pulmonary hypertension and interstitial lung disease in systemic sclerosis. Arthritis Rheumatol. 71, 1339–1349. https://doi.org/10.1002/art.40862 (2019).
Nathan, S. D. et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur. Respir. J. https://doi.org/10.1183/13993003.01914-2018 (2019).
van den Hoogen, F. et al. 2013 classification criteria for systemic sclerosis: An American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 65, 2737–2747. https://doi.org/10.1002/art.38098 (2013).
LeRoy, E. C. & Medsger, T. A. Jr. Criteria for the classification of early systemic sclerosis. J. Rheumatol. 28, 1573–1576 (2001).
Valenzuela, A., Nandagopal, S., Steen, V. D. & Chung, L. Monitoring and diagnostic approaches for pulmonary arterial hypertension in patients with systemic sclerosis. Rheum. Dis. Clin. N. Am. 41, 489–506. https://doi.org/10.1016/j.rdc.2015.04.009 (2015).
Yaqub, A. & Chung, L. Epidemiology and risk factors for pulmonary hypertension in systemic sclerosis. Curr. Rheumatol. Rep. 15, 302. https://doi.org/10.1007/s11926-012-0302-2 (2013).
Morrisroe, K. et al. Risk factors for development of pulmonary arterial hypertension in Australian systemic sclerosis patients: Results from a large multicenter cohort study. BMC Pulm. Med. 16, 134. https://doi.org/10.1186/s12890-016-0296-z (2016).
Steen, V. & Medsger, T. A. Jr. Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement. Arthritis Rheum. 48, 516–522. https://doi.org/10.1002/art.10775 (2003).
Eid, D. & Mohamed-Hussein, A. A. R. Evaluation of FVC/DLCO ratio as a predictor for pulmonary hypertension in patients with interstitial lung diseases. Eur. Respir. J. 50, 25 (2017).
Steen, V. D., Graham, G., Conte, C., Owens, G. & Medsger, T. A. Jr. Isolated diffusing capacity reduction in systemic sclerosis. Arthritis Rheum. 35, 765–770. https://doi.org/10.1002/art.1780350709 (1992).
Acknowledgements
We express our gratitude to Merck Sharp & Dohme (MSD), GlaxoSmithKline (GSK), and Ferrer for supporting the REHAP Registry with an unrestricted educational Grant. We gratefully acknowledge all investigators who are part of the REHAP Registry (Appendix A) and the RESCLE Registry (Appendix B). We also thank the REHAP and RESCLE Registries Coordinating Center, S&H Medical Science Service, for their quality control, logistic and administrative support and especially Prof. Salvador Ortiz, PhD Universidad Autónoma de Madrid and Statistical Advisor S&H Medical Science, for the statistical analysis of the data presented in this paper. We also thank Beatriz Viejo, PhD for her assistance in the writing of the manuscript and editorial support.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Consortia
Contributions
A.G.C., M.L.M., P.E.S., C.P.S.-A. were involved in the study conception and design; A.G.-C., M.L.M., V.F.-P., B.S.G., D.C.-A., E.R.-L., M.R.-R., M.J.C.R., A.A., J.A.B.M., X.P.S., A.M.M., A.B.M.V., A.L.P., L.S.C., J.A.D.M., C.G.-E., T.M., N.O.-C., M.M.G., C.T.-V., I.B., P.E.S., C.P.S.-A. in acquisition of data; A.G.-C., M.L.M., P.E.S., C.P.S.-A. in analysis and interpretation of data; A.G.-C., M.L.M., M.R.-R., P.E.S., C.P.S-A. in drafting the article or revising it critically for important intellectual content. All authors gave final approval of the version submitted.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in the spelling of the author Joan Albert Barberá which was incorrectly given as Joan Albert Barberá Mir.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guillén-Del-Castillo, A., Meseguer, M.L., Fonollosa-Pla, V. et al. Impact of interstitial lung disease on the survival of systemic sclerosis with pulmonary arterial hypertension. Sci Rep 12, 5289 (2022). https://doi.org/10.1038/s41598-022-09353-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-09353-z
This article is cited by
-
Reproducibility of pulmonary function tests in patients with systemic sclerosis
Scientific Reports (2023)
-
Pulmonary hypertension in systemic sclerosis with usual interstitial pneumonia
Internal and Emergency Medicine (2023)
-
Clinical characteristics and survival of pulmonary arterial hypertension with or without interstitial lung disease in systemic sclerosis
Arthritis Research & Therapy (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
