
Abstract
Chlamydia pneumoniae is a respiratory tract pathogen but can also infect the central nervous system (CNS). Recently, the link between C. pneumoniae CNS infection and late-onset dementia has become increasingly evident. In mice, CNS infection has been shown to occur weeks to months after intranasal inoculation. By isolating live C. pneumoniae from tissues and using immunohistochemistry, we show that C. pneumoniae can infect the olfactory and trigeminal nerves, olfactory bulb and brain within 72 h in mice. C. pneumoniae infection also resulted in dysregulation of key pathways involved in Alzheimer’s disease pathogenesis at 7 and 28 days after inoculation. Interestingly, amyloid beta accumulations were also detected adjacent to the C. pneumoniae inclusions in the olfactory system. Furthermore, injury to the nasal epithelium resulted in increased peripheral nerve and olfactory bulb infection, but did not alter general CNS infection. In vitro, C. pneumoniae was able to infect peripheral nerve and CNS glia. In summary, the nerves extending between the nasal cavity and the brain constitute invasion paths by which C. pneumoniae can rapidly invade the CNS likely by surviving in glia and leading to Aβ deposition.
Similar content being viewed by others

Introduction
Chlamydia pneumoniae is a gram-negative respiratory pathogen, responsible for causing 5–20% of community-acquired pneumonia1,2. C. pneumoniae primarily infects the pulmonary and nasal mucosa, but has in recent years been linked to diseases distinct from the respiratory tract, such as atherosclerosis/coronary disease3, asthma4, inflammatory arthritis5, multiple sclerosis6 and, in particular, late-onset dementia (late-onset Alzheimer’s disease)7,8.
Several key studies have reported the presence of C. pneumoniae in post-mortem brains from late-onset dementia patients. In the late 1990s, it was shown that 90% of such post-mortem patient brains contained C. pneumoniae DNA, compared with only 5% of control age-matched brains9,10. More recently, C. pneumoniae DNA was found in 80% of patient brains, compared to 11% of control brains11. The presence of C. pneumoniae in brains from patients with late-onset dementia has also been shown using immunohistochemistry, where C. pneumoniae antigens were found in proximity to classical hallmarks of late-onset dementia pathology; senile plaques, amyloid beta (Aβ) deposits and cells containing neurofibrillary tangles in the cerebral cortex and hippocampus. It is now well known that Aβ is an antimicrobial peptide, released by neural cells in response to infectious agents12,13, so it is not surprising that the presence of bacteria in the brain can result in Aβ deposition reviewed in8. Viable C. pneumoniae bacteria have also been isolated from some post-mortem patient brains7. In contrast, however, other studies have failed to detect higher levels of C. pneumoniae in post-mortem brains from patients with late-onset dementia14, although the analytical methods have varied between studies7. Studies in mice have also suggested a link between C. pneumoniae and late-onset dementia. C. pneumoniae DNA, antigen and/or live bacteria have been detected in the brain of inoculated mice15,16,17,18, which resulted in Aβ deposition16,17 or altered appearance of Aβ deposits18. Importantly, the mice used in these studies were wild-type mice and not mouse models of Alzheimer’s disease, suggesting that C. pneumoniae can contribute to the neuropathologies associated with late-onset dementia.
Regardless of whether C. pneumoniae is a contributing factor to neurodegeneration, it is clear that this bacterium can infect the brain and potentially contributes to chronic CNS pathologies. To date, it remains unknown exactly how C. pneumoniae reaches the CNS. C. pneumoniae can infect lung macrophages, which migrate through the mucosal barrier and enter the blood; the bacteria can disseminate to vasculature by surviving intracellularly in blood monocytes, which can then cross the blood–brain barrier (BBB)19. It is also, however, possible that C. pneumoniae may enter the CNS via alternative routes. The nerves that extend between the nasal cavity and the brain, the olfactory and trigeminal nerves, have been shown to be a path for CNS infection by some infectious agents reviewed in20. These two nerves connect with the brain at the olfactory bulb and the brainstem, respectively. Interestingly, the olfactory bulb, entorhinal cortex and hippocampal formation (all olfactory structures), as well as the brainstem, are the CNS regions that exhibit the earliest signs of pathology in both late-onset dementia and familial Alzheimer’s disease21,22,23.
After intranasal inoculation in mice, C. pneumoniae antigens and/or infectious organisms have been detected in the olfactory mucosa and the olfactory bulb 1–4 months post intranasal inoculation16,17,18, and C. pneumoniae DNA in these tissues has been detected 1 week after inoculation18, strongly suggesting that the bacteria can infect the CNS via the olfactory nerve. Certain other bacteria, however, have been shown to very rapidly (within days) reach the CNS via the olfactory and/or trigeminal nerves reviewed in20. We recently demonstrated that Chlamydia muridarum can quickly (within two days) enter the CNS via these paths24, suggesting that C. pneumoniae may also rapidly infect the brain via the nerves.
Furthermore, it is unknown how soon Aβ starts to accumulate after C. pneumoniae inoculation. Whilst previous studies have shown that this occurs after months16,17,18, if the bacteria reach the CNS more rapidly, alterations in Aβ deposition may also occur sooner. In a transgenic mouse model of familial Alzheimer’s disease (5xFAD mice, which exhibit the human amyloid precursor protein and presenilin 1 transgenes with five mutations linked to Alzheimer’s disease), intracranial injection with Salmonella typhimurium resulted in a dramatic increase in Aβ deposition after only 48 h13. Whilst Aβ deposition is of course much more pronounced in Alzheimer’s disease mouse models, Aβ secretion in response to pathogens by (wild-type) neural cells can be rapid12.
Even though cranial nerves constitute a direct path by which microbes can access the brain, CNS infections are relatively rare, and only a small number of infectious agents are thought capable of accessing the brain via these paths. The nerves are well-protected physically and immunologically by the nasal epithelium which exhibits powerful innate and adaptive immune system components. Together with the associated nasopharynx-associated lymphoid tissue (NALT), the epithelium constitutes the first defence against microbes25. Injuries to the nasal epithelium are, however, relatively common26 and may expose the underlying cranial nerves to infection. Experimental injuries to the nasal epithelium of mice has been shown to increase the risk of bacterial invasion of the olfactory nerve and bulb by some bacteria27,28. Most microbes are, however, likely eliminated by phagocytic glia, olfactory ensheathing cells (OECs) and trigeminal Schwann cells (TgSCs), should they penetrate the epithelium and reach the nerves29,30,31. The glia limitans layer between the peripheral nerves and brain, populated by astrocytes, constitutes a further immunological barrier against CNS infection32,33. Whilst it is largely unknown why certain infectious agents can infect the CNS via cranial nerves, one key mechanism is thought to be the ability of these pathogens to infect and survive in OECs, TgSCs and astrocytes, as well as in microglia (the main phagocytes inside CNS tissue)20,27,28,34,35.
Chlamydiae are obligate intracellular bacteria with a unique biphasic life-cycle reviewed in36. Outside of host cells, Chlamydiae exist as infectious, biologically inactive elementary bodies (EBs), which exhibit strong resistance to environmental stress. C. pneumoniae EBs can become internalized into host cells, including many phagocytes1,37,38. The EBs are resistant to endosomal/lysosomal degradation, and inside the host cell transform into reticulate bodies (RBs). RBs replicate in inclusions (modified cellular vacuoles), which expand in size as the bacteria replicate. After approximately 72 h (in cell culture), the RBs transform into EBs, which are released by cell lysis and can infect new cells (exit via extrusion of membrane-bound compartments can also occur39). Chlamydiae can also persistently infect cells40 which is likely relevant for the link to chronic diseases1. Persistent Chlamydia infection can last for many years, and the persistent Chlamydia bacteria can re-activate41,42.
In the current study, we investigated whether C. pneumoniae could rapidly (3–7 days after intranasal inoculation) invade the CNS via the olfactory and/or trigeminal nerves in mice and if this resulted in any alterations in Aβ deposition in nerve/CNS tissue. Furthermore, we investigated whether C. pneumoniae could infect and survive in cultured primary mouse OECs, TgSCs, astrocytes and microglia. We also determined whether C. pneumoniae infection had any role in the regulation of Alzheimer's disease gene expression over the longer term.
Material and methods
Bacterial strains
Chlamydia pneumoniae AR39 (ATCC 53592) is a human pharyngeal isolate and was propagated in Hep-2 cells (sourced from the ATCC CCL-23). C. pneumoniae stocks were harvested in sucrose phosphate glutamate (SPG) and aliquots were stored at – 80 °C for future experiments. The infectious yield of C. pneumoniae was determined by counting inclusion forming units (IFU) in HEp-2 cells.
Animals
7–8 week old female BALB/c mice were sourced from Animal Resource Centre (ARC, Murdoch, Western Australia) and were intranasally inoculated (under anaesthesia with isofluorane 1.5–2%) with either 10 µL of PBS (phosphate buffer saline) as vehicle control (N = 5) or C. pneumoniae (1 × 106 IFU [inclusion forming units] per mice, N = 12), delivered as a 5 µL droplet per nostril. Mice were then sacrificed 1, 3, 7 and 28 days post intranasal inoculation by asphyxiation with rising carbon dioxide.
Nasal epithelium injury model
In this study, we also used the methimazole injury model to investigate whether epithelial injury can increase the risk of C. pneumoniae invasion of the olfactory/trigeminal nerves and brain. For this purpose, 7–8 week old female BALB/c mice were injected with a single dose of methimazole (50 mg/kg, 10 mg/mL in PBS) or vehicle (PBS only), according to our previously published protocol27,29. Three days after methimazole injection, animals were intranasally inoculated with C. pneumoniae (N = 12) or vehicle (N = 5) as outlined above.
Tissue collection
Heads and tissues including the olfactory mucosa (containing the olfactory nerve fascicles), olfactory bulb, trigeminal nerve and the brain (the remainder of the brain after removal of olfactory bulbs) were collected from euthanized mice, 1, 3, 7 days and 28 days post inoculation, for bacterial load determination and histology.
Organ load assay
Chlamydia pneumoniae IFUs were detected by direct inoculation of tissue homogenate onto HEp-2 cells which were seeded on 96-well plates with 4000 cells/well. After 72 h, the C. pneumoniae inclusions in the entire wells were visualized by confocal microscopy and the numbers of IFUs isolated from the homogenates (IFU/mL) were determined (see workflow in Fig. 1).

Schematics illustrating the process for quantifying the amount of viable infectious C. pneumoniae present in various mouse tissues. (A) Mice were first intranasally inoculated with C. pneumoniae (i), some with epithelial injury and some without. Following either 24 h, 3 days or 7 days or 28 days post inoculation, selected tissues were collected and homogenised in tubes (ii). Homogenates were serially diluted onto HEp-2 cells and incubated (iii). Cells were fixed and immunolabelled for C. pneumoniae inclusions, which were counted and the number of inclusion-forming units (IFUs) per mL of homogenate was determined. Data were then compiled into organ (tissue) load graphs (see Fig. 2). (B) Microscopic image showing a sagittal tissue section of a mouse brain. Cell nuclei/DNA are shown in blue (DAPI stain). Key anatomical locations include the nasal cavity (NC), olfactory epithelium (OE), olfactory bulb (OB), trigeminal nerve (Tg; not visible so approximate location is shown by white dotted line) and the brain. Scale bar in µm.
Chlamydial PCR
DNA was extracted from whole blood 2, 3 and 4 days after infection using the Qiagen DNeasy blood and tissue kit according the manufacturer’s instructions. The quantitative Real-time PCR was carried out by using Platinum SYBR Green qPCR SuperMix-UDG (ThemoFischer cat# 11733038). C. pneumoniae was detected using 16S rRNA primers (Forward: 5′-CTCAACCCCAAGTCAGCATT-3′and Reverse: 5′-CTACGCATTTCACCGCTACA-3′. The cycling program was 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C, and a final dissociation stage. C. pneumoniae DNA was used as a positive control, and a no-template control was also included.
Tissue preparation and sectioning
Heads were fixed in 4% paraformaldehyde (PFA) in PBS overnight at 4 °C, followed by decalcification in 20% ethylenediaminetetraacetic acid (EDTA) for 4 weeks. The heads were then embedded in optimal cutting temperature (OCT) medium and frozen. Sagittal sections (30 µm) were cut using a cryostat (Leica CM1860).
Immunohistochemistry
Immunohistochemistry was performed as previously described27,43,44. Specimens were incubated with goat anti-C. trachomatis/C. pneumoniae (this antibody is used to detect both of these Chlamydia species; Abcam ab20929; 1:400) and/or rabbit anti-Aβ peptide (Abcam ab201060,1:500). Secondary antibodies were donkey anti-goat Alexa Fluor 488 (Abcam ab150129 1:400), donkey anti-rabbit 647 (Invitrogen A31573; 1:500). Antibodies were diluted in blocking buffer (2% bovine serum albumin with 0.3% Triton X-100 in PBS). Cryostat sections were first incubated with blocking buffer for 60 min at room temperature, followed by overnight incubation with primary antibodies at 4 °C. Sections were washed for 3 × 5 min, then incubated with secondary antibodies for 1 h. Cell nuclei were stained with 4′6-diamidino-2-pheylindole (DAPI).
Primary glia cell culture
Olfactory ensheathing cells (OECs), trigeminal Schwann cells (TgSCs), astrocytes and microglia were used in this study. OECs and TgSCs were prepared from postnatal day 7 (P7) S100ß-DsRed transgenic mice; we have previously generated and described this transgenic mouse line and the cell isolation method45. Astrocytes and microglia were prepared from postnatal day 3 (P3) S100ß-DsRed transgenic mice following a previous published protocol46. S100ß-DsRed transgenic reporter mice were used due to the expression of DsRed fluorescent protein which is driven by the human S100ß promoter, such that glial cells including OECs, Schwann cells, astrocytes and microglia express DsRed protein and facilitate easy visualisation and identification in culture. The entire brain cell population was isolated from the brain tissue by enzymatic digestion and mechanical dissociation using Neural Tissue Dissociation Kit with GentleMACS (Miltenyi Biotec, 130-093-231). The cell pellet consisting of a mixture of all brain cells was further subjected to magnetic cells sorting for microglia enrichment using CD11b/c microbeads (Miltenyi Biotec, 130-093-636) or for astrocytes using anti-GLAST (ACSA-1) microbeads kit (Miltenyi Biotec, 130–095-825) according to manufacturer’s protocol. The different glial preparations were separately plated in plastic 24-well plates and maintained in glial medium containing Dulbecco's Modified Eagle Medium with 10% foetal bovine serum (FBS), G5 supplement (Gibco), gentamycin (Gibco, 50 mg/mL) and l-glutamine (200 μM) at 37 °C with 5% CO2 for 5 days. Cells were replated into T-25 flasks and allowed to proliferate to ~ 80% confluency. Primary glial cultures with approximately 70–80% purity was used in the experiments.
In vitro infection of primary glial cells
Dilutions of C. pneumoniae bacteria were prepared in Dulbecco’s phosphate buffered saline (DPBS). OECs, TgSCs, astrocytes and microglia were seeded at the density of 4000 cells/well in 96-well plate (Costar) in glial medium. After 24 h, bacteria (multiplicity of infection (MOI): 1:1) were added and incubated with cells for 72 h. Following the infection, the cells were then rinsed in 1 × DPBS and were fixed for 20 min in 4% PFA in DPBS. Subsequently, cells were washed and incubated in blocking buffer for 1 h. Cells were then incubated with the following primary antibodies overnight at 4 °C; goat anti-C. pneumoniae/Chlamydia trachomatis (Abcam, ab20929; 1:400) and rabbit anti-glial fibrillary acidic protein (GFAP) antibody (Thermofisher Scientific, PA5-16291; 1:200) or rabbit anti-ionized calcium-binding adaptor molecule 1 (IBA1) microglia (Abcam, ab178847; 1:100). The following day, cells were washed with DPBS and incubated with secondary antibody donkey anti-goat Alexa Fluor 488 (Thermofisher Scientific, A11055; 1:400) and goat anti-rabbit 647 (ThermoFisher Scientific, A32733; 1:400) for 1 h. Nuclei were stained with DAPI. Hep-2 cells were visualized by CellMask Orange Plasma membrane stain (Thermofisher Scientific, C100455; 1:10,000).
Viability assay for glial cells
Primary glial cells were infected with C. pneumoniae as described above. Cultures were harvested after 72 h in SPG with 5 mM l-glutamine and stored at − 80 °C. Culture plates were thawed and probe sonicated for 10 s (Sonics Vibra-Cell VCX 130, amp 1). Cell lysates (containing bacteria) were collected and serially diluted on a monolayer of HEp-2 cells and, 72 h later, washed with PBS and fixed with 4% PFA. Following immunocytochemistry, the infectious yield C. pneumoniae was determined by counting of the inclusion forming units (IFU) ml−1 in Hep-2 cells.
RNA extraction and nanostring nCounter gene expression analysis
7–8 week old female BALB/c mice were infected as previously described. Mice were then sacrificed at 7 and 28 days post intranasal inoculation by asphyxiation with rising carbon dioxide. RNA from brain lysate (the remainder of the brain after removal of olfactory bulbs) of 7 and 28 days post C. pneumoniae inoculation and control mice (N = 3 for all groups) was extracted using Maxwell® RSC simplyRNA tissue kits (Promega, AS1340) using the manufacturer’s protocol. RNA was eluted in 50 µL of nuclease-free water and quality/quantity of RNA was assessed. Following RNA elution, gene expression analysis was undertaken using the NanoString nCounter analysis system (NanoString Technologies, Seattle, WA) using the commercially available nCounter Alzheimer’s disease panel kit (Cat number: XT-CSO-MAD1-12). The Alzheimer’s disease panel contains 23 neurodegeneration pathways, targeting 770 genes including 10 internal reference/housekeeping genes. A master mix was made following manufacturer’s protocol with 70ul hybridisation buffer added to Reporter probes. Individual reactions for each sample were made with 8 μL master mix, 5 μL extracted RNA diluted to 125 ng and 2 μL Capture probe. Each reaction was hybridised in a thermal cycler (Eppendorf) at 65 °C for 20 h. Samples were processed on the NanoString Prep Station and the target-probe complex was immobilised onto the analysis cartridge. Cartridges were scanned by the nCounter Digital Analyser for digital counting of molecular barcodes corresponding to each target at 555 fields of view.
Image capture
Images were captured using Nikon confocal microscope and Olympus FV3000 laser scanning confocal microscope. Three-dimensional reconstructions were made using Imaris × 64 (Version 7.4.2). For comparison between groups, the same image capture settings, laser intensity and focal depths were used. Images were colour balanced uniformly across the field of view using Adobe Photoshop Creative Cloud 2019 (20.0.4) and compiled into panels using Adobe Illustrator Creative Cloud 2019 (23.0.3).
Statistical analysis
Data are shown as means ± SEM. Statistical significance was analyzed using either a two-way ANOVA with Bonferroni’s post hoc test or a one-way ANOVA with Tukey’s post hoc test. Statistical analysis was performed using GraphPad Prism 9.0 software, and statistical significance was set at p < 0.05.
Gene expression data was processed using the Advanced Analysis Module in the nSolver Analysis Software version 4.0 from NanoString Technologies (NanoString Technologies, WA, USA). Quality control was assessed, and the data was analysed using Rosalind software (partner open-source software). Normalised data were generated by the software followed by fold change and p value. p value was adjusted using Benjamini–Hochberg method of estimating false discovery rate. Venn diagram was generated using an open-source software (http://bioinformatics.psb.ugent.be/webtools/Venn) normalised to day 7 and day 28 non-infected control. Principal component analysis (PCA) and volcano plot was generated using Graphpad prism 9.0. Hierarchical clustering was generated using Morpheus software with Pearson correlation and average linkage method across the samples for the most significant genes (adjusted p value < 0.05). Molecular process was generated from REACTOME database linked to Rosalind software with cut-off set at p value < 0.05. Pathway profile score was generated from nSolver Analysis Software using the 23 neurodegenerative pathways mentioned previously.
Ethics and biosafety
The experimental procedures used in the study were conducted with the approval of the Griffith University Biosafety Committee (NLRD/09/15_var7) and the Griffith University Animal Ethics Committee (MSC/08/18/AEC) in accordance with guidelines of the Australian Commonwealth Office of Gene Technology Regulator and the National Health and Medical Research Council of Australia. All the animal experiments in this study are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Results
Chlamydia pneumoniae infects the olfactory mucosa, olfactory bulb and cerebral cortex within 3 days after intranasal inoculation
The key aim of the current study was to determine whether C. pneumoniae could invade the brain via the olfactory/trigeminal nerve routes in the shorter term (≤ 1 week) after intranasal exposure, as has been shown for C. muridarum24. We intranasally inoculated adult mice with C. pneumoniae, then 3 days, 7 days and 28 days later mice were sacrificed. The 3 day time-point was chosen as C. pneumoniae has a life-cycle of approximately 72 h1,37. We then determined whether infectious (viable, inclusion-forming) C. pneumoniae could be isolated from the following homogenized mouse tissues: (1) olfactory mucosa (consisting of the neuroepithelium, underlying lamina propria and the many nerve fascicles that constitute the olfactory nerve), (2) olfactory bulb, (3) trigeminal nerve and (4) brain (beyond the olfactory bulb), as outlined in Fig. 1. Tissue homogenates were diluted onto HEp-2 cells and inclusion-forming units per mL of tissue homogenate were determined after 72 h.
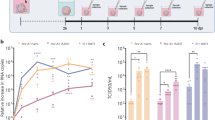
At 3 and 7 day time-points, infectious C. pneumoniae were isolated from all four tissues (Fig. 2A). No C. pneumoniae were detected in tissue lysate from control (vehicle-inoculated) mice (n = 2). For the olfactory bulb, the number of IFUs was significantly higher at 3 days than 7 days post inoculation (p.i.) (Fig. 2A,B), whereas the reverse occurred for the trigeminal nerve (Fig. 2A,B non-injury). At 28 days, low levels of infectious C. pneumoniae were isolated only from trigeminal nerve p.i. (not shown). We also determined whether C. pneumoniae was present in the blood 2, 3 and 4 days post intranasal inoculation using PCR. Based on the curves, all samples showed an absence of C. pneumoniae in blood (Fig. 2C). Note: the injury results presented in Fig. 2A,B are reported in the section “Injury to the nasal epithelium increases peripheral infection”.

C. pneumoniae infects the nasal peripheral nerves and brain in mice with or without pre-injured nasal epithelium after intranasal inoculation. (A,B) Graph showing the amounts of C. pneumoniae IFUs isolated from various tissues of mice with or without pre-injured olfactory epithelium at 3 and 7 days post C. pneumoniae inoculation. Infectious C. pneumoniae organisms were isolated from the olfactory mucosa, olfactory bulb, trigeminal nerve and brain (n = 9/group). No C. pneumoniae was isolated from various tissues of control mice (vehicle only, n = 2). Data are shown as the mean number of inclusions ± SEM, n = 9/group. *p ≤ 0.05, **p ≤ 0.01 ***p ≤ 0.001, two-way ANOVA with Bonferroni’s post hoc test. For comparisons between non-injury Day 3 versus Day 7: a-a′ p ≤ 0.01, b-b′ p ≤ 0.01; for comparisons between injury Day 3 versus Day 7: c–c′ p ≤ 0.01, d-d′ p ≤ 0.001, two-way ANOVA with Bonferroni’s post hoc test. (C) PCR amplification curves of blood for C. pneumoniae (Cpn) at 2, 3 and 4 days post intranasal inoculation. (+) shows the positive control in each graph. Samples tested include the control (vehicle only; N = 2), methimazole only (N = 2), methimazole + Cpn (N = 9) and Cpn only (N = 9).
We also analysed tissue sections from the olfactory nerve, olfactory bulb, trigeminal nerve and brain (beyond the olfactory bulb) for the presence of C. pneumoniae using immunohistochemistry. In addition to the 3 days, 7 days and 28 days mice, we also examined mice that had been sacrificed only 24 h after intranasal inoculation. 24 h after inoculation C. pneumoniae was detected within the olfactory mucosa and olfactory bulb and infectious C. pneumoniae were isolated from both the olfactory mucosa and olfactory bulb (Supplementary Fig. 1). However, as the bacteria at 24 h was likely to be from the inoculum we did not analyse this tissue further. At later time-points which are sufficient for at least one life cycle, C. pneumoniae inclusions were detected in the olfactory nerve (Fig. 3A–C), glomerular layer of olfactory bulb (Fig. 3D–F) and trigeminal nerve (Fig. 3H–L) at both 3 and 7 days p.i. Within the olfactory bulb, C. pneumoniae was only detected within the nerve fibre layer and glomerular layer, with C. pneumoniae inclusion bodies being present inside OECs (Fig. 3G). C. pneumoniae was also detected in the glomerular layer of the olfactory bulb at 28 days (Fig. 5Q).

C. pneumoniae infects the nasal peripheral nerves and brain in mice after intranasal inoculation. Panels show images of tissue sections from control (vehicle only) and inoculated mice (C. pneumoniae) for both 3 days and 7 days post inoculation (p.i.). Images are representative ones from the olfactory nerve (ON) and olfactory bulb (OB) of 3 control mice, 3 C. pneumoniae-inoculated mice at 3 days p.i. and 3 inoculated mice at 7 days p.i. C. pneumoniae inclusions are shown in green (C. pneumoniae immunolabelling) and indicated by arrows. Nuclei/DNA is shown in blue (DAPI stain). Panels show maximum projection of confocal microscopy z-stacks. (A–C) Olfactory nerve (ON). (A) Control (vehicle only). (B) 3 days p.i. (C) 7 days p.i. (D–F) The glomerular layer (GL) of the OB. (D) control. (E) 3 days p.i. (F) 7 days p.i. (G) The GL at 7 days p.i., immunostained with anti-S100 antibodies. (H–L) Trigeminal nerve (Tg). (H) control. I: 3 days p.i. (J) 7 days p.i. (K,L) 3D reconstruction of panel J. C. pneumoniae inclusions. (K) Inclusions (green) are within the Tg and contain DNA (L; blue, DAPI stain). Scale bars in µm. For a low-power image showing the approximate localization of the images, see Fig. 1B.
Despite being able to isolate viable C. pneumoniae from the brain (beyond the bulb), we did not find definitive C. pneumoniae inclusions in brain tissue sections from these mice (not shown), suggesting that inclusions in brain tissue were too small or sparse to be confirmed by histology when screening tissue sections.
Injury to the nasal epithelium increases peripheral infection
It has previously been shown that experimental injury to the olfactory neuroepithelium facilitates invasion of the olfactory nerve and bulb by certain bacteria27,28. To investigate whether epithelial injury could also affect C. pneumoniae infection of nerves and brain, we used our well-established methimazole-mediated model of nasal epithelial injury. Methimazole causes degeneration of the nasal epithelium in rodents47. We have shown that this mode of injury leads to patchy, dispersed injuries to the epithelium, separated by normal epithelium; this constitutes a model better resembling “natural” nasal injuries than other models, such as chemical irrigation models27,29.
Mice were treated with methimazole, and 3 days later, when epithelial degeneration peaks48, the mice were inoculated intranasally with C. pneumoniae. The 3-day time-point was also chosen to limit any potential unrelated effects of methimazole, as methimazole at this stage has been largely cleared49. We examined mice that had been sacrificed only 24 h after intranasal inoculation. The methimazole treatment clearly damaged the epithelial layer (Fig. 4A–C) and C. pneumoniae was found within the lamina propria underlying the epithelial layer and in the nerve fibre layer of the olfactory bulb (Supplementary Fig. 2); as the bacteria is likely to be from the inoculum we did not analyse this tissue further.

C. pneumoniae infection of the olfactory nerve, olfactory bulb and brain after injury to the olfactory epithelium. Panels show confocal images (maximum projection of z-stacks, A–I) of tissue sections from vehicle control and C. pneumoniae-inoculated mice, all with pre-injured olfactory epithelium before inoculation/vehicle treatment. Images are representative from n = 3 animals per group. C. pneumoniae inclusions are shown in green (immunolabelling) with nuclei/DNA in blue (DAPI stain). (A–C) The olfactory nerve (ON) in (A) control mice and mice inoculated with C. pneumoniae, (B) 3 days and (C) 7 days post inoculation. (D–F) The glomerular layer (GL) of the olfactory bulb in (D) control mice and inoculated mice (E) 3 days and (F) 7 days after inoculation. (G) A 3D reconstruction of the C. pneumoniae inclusion in panel (F) (green; arrow); (H) the same 3D reconstruction as panel (G), showing only the DAPI staining (blue), where bacterial DNA within C. pneumoniae inclusion is distinct from host cell DNA (arrow pointing to bacterial DNA). (I) Image showing a C. pneumoniae inclusion in the olfactory piriform cortex seven days after inoculation (green; arrow); (J,K) a 3D reconstruction and render of the C. pneumoniae inclusion shown in (I), with C. pneumoniae in green (J) and bacterial DNA shown in K, arrow). Scale bars in µm.
Mice were then sacrificed 3 and 7 days after inoculation, followed by determination of the amounts of viable C. pneumoniae (IFUs) in tissues (Fig. 2A,B), as well as immunohistochemistry of tissue sections (Fig. 4). Whilst described separately here for better clarity, these experiments were conducted simultaneously to the experiment groups described for Fig. 2 (so that methimazole-induced epithelial injury followed by C. pneumoniae inoculation could be compared to C. pneumoniae inoculation alone).
We compared the C. pneumoniae load (IFUs) between (1) mice inoculated with C. pneumoniae alone and (2) mice with pre-injured nasal epithelium. Epithelial injury resulted in an increased C. pneumoniae load in the olfactory mucosa (which includes the olfactory nerve; both at day 3 and day 7 post inoculation), olfactory bulb and trigeminal nerve (day 7 only) in comparison to mice without injury (Fig. 2A,B). However, epithelial injury did not result in a significant difference in C. pneumoniae load in the brain.
For both the pre-injured mouse group and the group that was not pre-injured, the bacterial load in the trigeminal nerve was higher on day 7 than day 3 post inoculation (Fig. 2A,B). In the pre-injured group, olfactory bulb infection also increased with time, whereas the opposite occurred in mice that were not pre-injured (Fig. 2A,B). Immunolabelling confirmed the presence of C. pneumoniae in the olfactory nerve (Fig. 4A–C) and bulb (Fig. 4D–H). Three-dimensional reconstructions provided clear visualisation of the inclusions within the glomerular layer, with the DNA of the bacteria being clearly distinct from the host cell DNA (Fig. 4G,H). In one mouse, in the pre-injured group, C. pneumoniae IB/s were also found in the olfactory piriform cortex, with three-dimensional reconstruction again showing the bacterial DNA being distinct from the host cell DNA (Fig. 4I–K).
Aβ is associated with regions of C. pneumoniae infection in the olfactory bulb
To determine whether C. pneumoniae inclusions were associated with Aβ deposits, we also immunolabeled the sections for Aβ. We found diffuse/patchy Aβ immunolabelling in olfactory nerve and bulb tissues from all animals, including control mice (Fig. 5A,B,E,F,I,J,P). However, we found that in tissues from inoculated mice, distinct Aβ deposits accumulated near C. pneumoniae inclusions. At 3 and 7 days after inoculation, we detected Aβ deposits near C. pneumoniae inclusions in the olfactory nerve (Fig. 5C,D,G,H). At 7 days, Aβ deposits near C. pneumoniae inclusions were detected in the glomerular layer of the olfactory bulb (Fig. 5K–N). The Aβ deposits were not detected in adjacent tissue regions where inclusions were not present (Fig. 5O). At 28 days, Aβ deposits continued to be detected near C. pneumoniae inclusions in the glomerular layer of the olfactory bulb (Fig. 5Q), while control uninfected mice exhibited diffuse Aβ deposits (Fig. 5P) which is similar to previous reports16,18,50. C. pneumoniae inclusions, as well as associated Aβ deposits, were detected sporadically within tissues. For this reason, we were not able to quantify the difference in Aβ levels between tissues from the different time-points after inoculation, as well as between inoculated and control animals. Correlating with the fact that we could not detect C. pneumoniae inclusions in the brain beyond the olfactory bulb, we also did not detect any evidence of distinct Aβ deposits in these areas (not shown).

C. pneumoniae is associated with Aβ peptide accumulation in both the olfactory nerve (ON) and the glomerular layer (GL) of the olfactory bulb (OB). Images show maximum projections of confocal microscopy z-stacks from vehicle control mice and C. pneumoniae-inoculated mice, 3, 7 and 28 days p.i. Images are representative ones from the ON or OB of 3 control and 3 inoculated mice. Immunolabelling shows Aβ peptide (anti-Aβ1-42) (red), C. pneumoniae (green) and DNA (DAPI, blue). (A–D) At 3 days, the ON of (A,B) a control mouse and (C,D) a C. pneumoniae-inoculated mouse. Arrows show the location of C. pneumoniae inclusions (green in C). Panels (B) and (D) show only Aβ immunolabelling (red). (E–H) Similarly, at 7 days the immunolabelling of the ON in (E,F) control and (E,F) inoculated mice. (I–L) At 7 days, the GL in (I,J) a control mouse and (K,L) a C. pneumoniae-inoculated mouse. Panels (J and L): Aβ immunolabelling only (red). Arrows indicate C. pneumoniae inclusions. (M) A 3D reconstruction of panel (K) showing Aβ (red) surrounding a C. pneumoniae inclusion (green). (N–O) Images of GL regions within the same tissue section of a C. pneumoniae-inoculated mouse, showing areas where C. pneumoniae inclusions are, or are not, localized. (N) A GL area were C. pneumoniae inclusions were detected. Inclusions (green) were associated with Aβ accumulation (red). (O) An adjacent GL region where inclusions were not detected. Only diffuse Aβ immunolabelling is seen (red). (P,Q) At 28 days, the GL of (P) a control mouse and (Q) a C. pneumoniae-inoculated mouse. Arrows indicate C. pneumoniae inclusions; Aβ (red). Scale bars in µm.
C. pneumoniae can infect primary glial cells
The capacity to infect and survive inside glial cells is thought to be a key mechanism for the ability of bacteria to invade the CNS via cranial nerves34,35. Therefore, we next examined whether glia from the olfactory and trigeminal nerves, olfactory bulb and brain could constitute host cells for C. pneumoniae infection. OECs, TgSCs, astrocytes and microglia were inoculated with C. pneumoniae (MOI: 1:1, i.e., 1 IFU/cell) for 72 h. For comparison and as a positive control, HEp-2 cells, which are highly susceptible to C. pneumoniae infection and in which the bacteria have strong capacity for intracellular survival51, were also included. Cells were either (1) fixed and immunolabelled for C. pneumoniae, or (2) lysed for determination of C. pneumoniae IFUs (viable, infectious organisms). Immunolabelling showed C. pneumoniae inclusions in all cell types (Fig. 6A–J). The HE-p2 cells had distinctly more pronounced inclusions compared to the other cells, with the DNA of the bacteria being strongly visible (Fig. 6J-J’). The different glia had similar levels of viable C. pneumoniae, but significantly lower amounts of viable C. pneumoniae were recovered from all the glia than from HEp-2 cells (Fig. 6K).

C. pneumoniae can infect glial cells from the olfactory and trigeminal nerves, olfactory bulb and brain. Cultured glia (OECs, TgSCs, astrocytes and microglia) from S100β-DsRed mice, in which glia express DsRed, were inoculated with C. pneumoniae (Cpn) for 72 h, along with HEp-2 cells. (A–J′) Maximum projection images from confocal microscopy z-stacks of control cells and C. pneumoniae-inoculated cells. Nuclei are stained with Hoechst (cyan). (A,C,E,G,I) Cells treated with cell culture medium alone (control; glia in red). (B,D,F,H,J) Cells inoculated with C. pneumoniae. Immunolabelling showed C. pneumoniae inclusions in the cells (green; arrows). (J-J′) Merged image shown in (J), C. pneumoniae inclusions alone shown in (J′). Scale bars in µm. (K) Graph showing the amounts of C. pneumoniae IFUs isolated from the cell cultures. The infectious yield of C. pneumoniae was significantly different between glia and HEp-2 cells (***p ≤ 0.001, one-way ANOVA with Tukey’s post hoc test). Data shown as the mean number of inclusions ± SEM.
C. pneumoniae infection modulates Alzheimer’s disease related gene expression
To investigate if C. pneumoniae infection had any role in the regulation of Alzheimer’s disease gene expression at the transcriptional level, we profiled 7 and day 28 day infected and non-infected mice brains using NanoString nCounter Alzheimer’s disease Panel. We first mapped the total number of genes which were up-regulated and down-regulated in day 7 and day 28 infected samples out of 760 genes which were all normalised to their respective non-infected samples. We found that a total of 514 genes were up-regulated in day 7 samples compared to 232 genes in day 28, out of which 81 genes were common to both and 433 (84.2%) were exclusively up-regulated in day 7 while 151 (65.1%) were exclusively up-regulated by day 28 (Fig. 7A). Interestingly, for genes that were down-regulated, 152 (64.7%) in day 7 and 433 (83.9%) genes in day 28 were exclusively down-regulated (Fig. 7B). We also investigated whether duration of infection had a role in differential gene expression (DGE) of Alzheimer’s disease genes. The normalised expression of all the 760 genes from day 7 and day 28 infected samples were used to construct correlation mapping using dimensional reduction technique (Principal component analysis—PCA). The maximum variance across the expressions was 43.03% (PC1 value) between the day 7 and day 28 infected samples (Fig. 7C). This analysis showed that the duration of infection had a definite role as the individual biological replicates from respective time points clustered together but away from each other as a group.

Differential gene expression (DGE) involved in Alzheimer’s disease for short (day 7) and long term (day 28) infection (n = 3/group). (A) Numbers and percentages of genes up-regulated for day 7 (pink) and day 28 (orange) infections normalised to their respective non-infected controls. (B) Numbers and percentages of downregulated genes for day 7 (pink) and day 28 (orange) infections normalised to their respective non-infected controls. (C) Principal component analysis of all the biological replicates of day 7 (pink) and day 28 infections (orange) for all gene expressions normalised to their respective controls. Axes show the percentage variation in each PC plots with maximum at 43.03% for PC1 followed by 26.31% for PC2. (D) Volcano plot of log twofold change of DGE of day 28 infection (normalised to day 7 infections) against –log 10 adjusted p value. Cut-offs for log twofold and –log 10 adjusted p value, set at ± 1.5 (± 0.585) and 0.5 (0.301), respectively. Blue dots represent down-regulated and red dots represent up-regulated genes. (E) Heat map of significant DGE plotted from (D) with hierarchical clustering on day 7 and day 28 infected replicates. Colour of the map is based on z-score from log twofold change for each gene (across rows). Blue represents down-regulated and red represents up-regulated genes. (F) Significant molecular processes (p < 0.05) in day 28 infection highlighted from REACTOME database with corresponding –log 10 p value. (G) Heat map of biological pathway profile scores compared between day 7 and day 28 infected samples from nSolver data for 23 pathways enlisted in Nanostring Alzheimer’s disease panel.
Next, we determined the significantly different genes with large changes in fold expression in infected samples of day 28 compared to day 7 in the form of a volcano plot. A volcano plot of the genes from day 28 infected samples using day 7 infected samples as a baseline and cut-offs at ± 1.5 on log twofold change and 0.5 on − log 10 adjusted p value was performed (Fig. 7D). A total of 53 genes were differentially regulated out of which 22 genes were down-regulated (blue) and 31 genes were up-regulated (red). We further explored these genes to investigate the relationship between the infected samples at the respective time points. A heatmap was constructed based on the z-score of the normalised gene expressions followed by hierarchical clustering using Pearson correlation method (Fig. 7E). We observed separate gene clustering of day 7 and day 28 infected mice. A total of 107 molecular processes were regulated (from REACTOME database using the Rosalind software) based on the DGE of day 28 infection compared to day 7. Out of these, 10 pathways were sorted based on their − log 10 p value with a cut-off at p < 0.05 (Fig. 7F). Interestingly, all these processes were down-regulated as the genes related to these processes were significantly down-regulated in day 28 infected samples.
We also used the pathway profile scores from nCounter to construct a heat map and compared the pathway modulation between day 7 and day 28 infected samples. A total of 23 pathways were compared which were already pre-selected in the Alzheimer’s disease Nanostring Panel (Fig. 7G). Most of the pathways were over-expressed in day 28 infected mice in comparison to day 7 infected mice. These trends also corroborated with the findings of the REACTOME database where most of the processes, especially ubiquitin-mediated protein degradation, were down-regulated reflecting the over-activation of “unfolded protein response” in day 28 infected mice pathway profile score.
In summary, these findings suggest that C. pneumoniae infection leads to a differential regulation of Alzheimer’s disease genes with long term infection (day 28) down-regulating most of the genes related to folding of proteins and aiding in misaggregation.
Discussion
In the current study, we showed that (1) C. pneumoniae rapidly infected both the olfactory and trigeminal nerves in mice, (2) C. pneumoniae entered the CNS via nerves within 24–72 h after intranasal inoculation and without concurrent blood infection, (3) injury to the nasal epithelium exacerbated peripheral nerve infection, but reduced brain infection, (4) C. pneumoniae inclusions in the olfactory nerve and bulb were associated with accumulations of Aβ, (5) the glial cells populating the olfactory/trigeminal nerves and brain supported C. pneumoniae replication, and (6) C. pneumoniae infection leads to differential regulation of Alzheimer’s disease related genes. Thus, C. pneumoniae can very rapidly spread from the periphery to the CNS via the nerves extending between the nasal cavity and the brain, without blood infection. To our knowledge, this study is the first report of Aβ deposition in response to C. pneumoniae infection of the primary olfactory nervous system, and the first time such rapid (72 h) deposition of Aβ in response to any bacterium in wild-type animals in vivo has been demonstrated.
The time-frame for infection of the CNS by C. pneumoniae was considerably faster than what has previously been shown (1 week–3 months16,17,18), which may be due to differences in the inoculation dose since we used a higher inoculation dose than two previous studies16,50 but lower than another18. Nevertheless, the time-frame is comparable to CNS invasion via cranial nerves by Burkholderia pseudomallei32,43,44,52, Streptococcus pneumoniae53, Neisseria meningitidis54, Listeria monocytogenes55 and now recently another Chlamydia species, C. muridarum24. The amoeba Naegleria fowleri56, as well as herpes simplex type virus type 1 (HSV-1)57, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)58,59,60 and other coronaviridae61 can also invade the CNS via these two paths (shown in humans and/or animals).
Within the olfactory bulb, C. pneumoniae inclusions were detected in OECs within the nerve fibre layer/glomerular layer. Another bacteria, Burkholderia pseudomallei also accumulated within the nerve fibre layer/glomerular layer after intranasal inoculation, suggesting that the glia limitans acts to restrict further progression of bacteria into the deeper regions. However, with C. pneumoniae while we could easily detect the inclusion bodies, the much smaller infectious elementary bodies would likely be missed in our analyses of the tissue sections; thus it is possible that elementary bodies were present deeper in the olfactory bulb. As inclusion bodies were detected in the olfactory piriform cortex, it suggests that C. pneumoniae did progress deeper into the olfactory bulb as previously reported15,16,18,50.
Injury to the nasal epithelium has been shown to increase infection of the olfactory nerve and bulb by B. pseudomallei27 and to allow the entry of S. aureus, which does not normally invade cranial nerves, to enter the olfactory bulb28. We therefore hypothesized that epithelial injury may lead to increased C. pneumoniae invasion of the olfactory/trigeminal nerves, olfactory bulb and remaining parts of the brain. We found that epithelial injury resulted in increased C. pneumoniae load in the olfactory mucosa (which contains the fascicles of the olfactory nerve), olfactory bulb and trigeminal nerve. In contrast, injury did not alter C. pneumoniae invasion of the brain after 7 days. We have previously observed a similar result for B. pseudomallei in some mice, in which the nasal infection in itself caused massive peripheral infection and destruction of the nasal epithelium (more pronounced than in our epithelial injury model used in the current study). In these mice, B. pseudomallei invasion of the CNS was negligible20. We then hypothesized that this may be because glia in the olfactory nerve and outer layers of the bulb responded to both the injury and bacteria, secreting large amounts pro-inflammatory factors which limited CNS infection; this may also be the case for C. pneumoniae infection in the current study.
The ability to infect glia is considered key for CNS invasion via the cranial nerve paths20,27,28,34,35. We here showed that C. pneumoniae could infect, survive in and replicate (form inclusions) within glia from the PNS (OECs and TgSCs) and the CNS (astrocytes and microglia). This is the first-time infection of OECs and TgSCs (or other Schwann cells) by C. pneumoniae has been reported, however, we have recently shown that C. muridarum can infect OECs and TgSCs24. Whilst C. pneumoniae infection of cultured primary astrocytes and microglia has not been described, infection of astrocyte and microglial cell lines has been demonstrated62,63,64,65,66. Most relevantly, however, C. pneumoniae antigens have been detected inside both astrocytes and microglia in post-mortem human brains9,11,67,68. OECs, Schwann cells and astrocytes are all innate immune cells which can respond to and phagocytose bacteria, and microglia (the macrophages of the CNS) are well characterized professional phagocytes31,69,70. The fact that C. pneumoniae can form inclusions in these cells suggest that the bacteria, at least to some extent, can overcome phagocytic destruction; this may be one important mechanism by which this bacterium can invade and establish long-term infection of the CNS.
We also detected localized deposition of Aβ adjacent to C. pneumoniae IBs and in the olfactory bulb after 7 days and 28 days post inoculation. Diffuse/scattered Aβ immunoreactivity was also present in these tissues of control mice, however, the co-localisation of Aβ deposits and C. pneumoniae inclusions in inoculated mice was clear and distinct. Previous studies have demonstrated Aβ deposits near C. pneumoniae-infected areas of the cerebral cortex 1–4 months post intranasal inoculation16. One study reported that whilst there were not necessarily more Aβ deposits in the cortex of C. pneumoniae-infected animals, Aβ deposits in infected animals were morphologically different from those in control animals18. A previous long-term study showed that C. pneumoniae infection of the cerebral cortex preceded the peak of Aβ deposition17. In combination with the findings of the current study, it appears that Aβ secretion occurs in response to the infection. One reason may be that Aβ is secreted as an antimicrobial agent12 but alternatively it may be secreted in response to infection because of pathway activation for the processing of the APP protein into Aβ which is then secreted; future work can clarify the secretion and role of Aβ in this context.
The secretion of Aβ may thus be a normal immune response to any microbe that may invade the nervous system, and if infection clears, the deposited Aβ can be cleared by phagocytic glia71. It is, however, possible that if bacteria are not cleared and instead become persistent or latent in neural cells, continued Aβ deposition may occur, contributing to late-onset dementia and/or accelerating Aβ deposition in familial Alzheimer’s disease7. In the case of C. pneumoniae, one study in wild-type mice demonstrated that Aβ deposits resulting from infection were subsequently cleared17, whilst another study showed that the deposits did not disappear over several months16.
It is interesting that we observed Aβ deposits in the olfactory nerve earlier than in the bulb, as one study in an Alzheimer’s disease mouse model (APP/PS1 mice) showed that the terminal end of the olfactory nerve within the nasal olfactory epithelium is the first nervous system area to exhibit Aβ deposition, which then progresses to the olfactory bulb and other CNS areas72. As the mice in that study were kept in a standard animal holding facility (not specific pathogen free), perhaps exposure to infectious agents may have contributed to this early, peripheral deposition of Aβ (which likely would be much more pronounced in an Alzheimer’s disease model than in wild-type mice).
Chlamydia pneumoniae infection also resulted in up-regulation of key pathways involved in Alzheimer’s disease pathogenesis. The pathologic features of Alzheimer’s disease like activated microglia, production of inflammatory mediators and reactive oxygen species (ROS) were highly regulated in infected brain tissue at 28 days post inoculation as compared to 7 days post inoculation. Theses neuroinflammatory responses are considered a major driving factor in patients with neurodegeneration and Alzheimer’s disease pathology, which starts early in the course of the disease, prior to the formation of Aβ plaques in the brain73. Previous studies have shown that microglia and astrocytes act as host cells of C. pneumoniae in Alzheimer’s disease brain9. It has been shown that following infection, activated microglia and astrocytes secrete pro-inflammatory cytokines, including IL-1β, TNFα and IL-6 which are neurotoxic and may directly increase Aβ production via activation of β-secretase (BACE)66,74. BACE cleaves amyloid precursor protein and initiates the amyloid cascade. Microglia activation reduces the accumulation of Aβ in the brain by increasing its phagocytosis, clearance, and degradation75. However, the neuroinflammation associated with Alzheimer’s disease could be a double-edged sword because persistent microglia activation stimulated by the binding of microglia to Aβ can increase the production of inflammatory mediators and reactive oxygen species (ROS), which further amplifies the neuroinflammatory response causing chronic inflammation and neurodegeneration76.
Disturbance of endoplasmic reticulum (ER) function is emerging as a relevant factor driving neurodegeneration in Alzheimer’s disease77. Several reports have described manifestations of ER stress in post-mortem brain samples from Alzheimer’s disease patients78. Protein folding in the endoplasmic reticulum (ER) is an essential cell function and to safeguard protein production and ensure quality control, ER-stress triggers the activation of several biochemical pathways collectively referred to as the unfolded protein response (UPR). Chlamydia infection can induce cellular stress that impacts protein folding, thus inducing UPR activation however it is also proposed to modulate the UPR to promote their survival and replication79. Interestingly, we found UPR pathway being up-regulated in infected cortical tissues at 28 days post inoculation as compared to 7 days post inoculation. Intracellular pathogens like Chlamydia would benefit from UPR since increase in folding capacity and activation of lipid biosynthesis can sustain bacterial replication. However, if the ER stress due to infection is sustained and misfolded protein cannot be refolded or degraded, the cells can also directly increase Aβ production and associated neuroinflammation80. Conversely, Aβ oligomers have also been proposed to cause ER dysfunction leading to UPR mediated neurotoxicity and neuronal cell death77. We have also observed similar trends in our study where molecular pathways related to cell death like autophagy and apoptosis were up-regulated in cortical tissues at 28 days post inoculation.
In addition to considering key pathways, it is also useful to consider changes in individual gene expression. Long term C. pneumoniae infection (day 28) triggered down-regulation of most other key genes involved in AD pathogenesis. Most importantly there was downregulation of key protective heat shock protein (Hspa1b or Hsp70-2), associated with increased oxidative stress and initiation of AD pathology81. In addition, Bag2, a Bcl-2 associated co-chaperone gene which controls Hsp70 functionality was also downregulated leading to further failure of the system to protect cells from oxidative damage82. The long term infection also depressed the 26S proteasome ubiquitination system by downregulation of Psmd883 and Psmc684 leading to persistence of stress-induced protein aggregates. At a sub-cellular level, infection led to mitochondrial dysfunction evident by downregulation of Ndufa5 (a structural subunit of complex I)85 and Atp5j286. Nevertheless, all these gene modulations led to increased unfolded protein response, oxidative stress, and had higher disease association as evident by the biological processes heat map. In fact, the long-term infection was also associated with low expression of Cd2ap which has been previously associated with AD pathology aggravated by increased deposition of Aβ and Tau-induced neurotoxicity87.
In contrast to the downregulated genes, long-term infection was also associated with some repairing mechanisms, limiting the spread of further neuroinflammation. It led to higher expression of Cdk18 which is a cyclin-dependent kinase and usually functions to clear DNA damages88. Thus, mechanisms inducing chromatin modification, transcription and splicing were highly reduced in them. Amongst the heat shock proteins, we found Hspa4 to be upregulated which ensured that it maintained the disaggregating property of any misfolded proteins, thereby, preventing further damage and inducing tissue integrity89. Additionally, it also helped in sulfatase gene (Sumf2) maintenance which is associated with modulation of tissue homeostasis90. Interestingly, long term infection also induced expressions of Ezr91 and Cldn592 which are associated with maintenance of actin cytoskeletal structure, synapse and tight junctions, respectively. This ensured that long-term infection induced greater organisation of neuronal cytoskeleton and/or dendritic structure while also maintaining synaptic transmission and reuptake. Additionally, further protection was provided by increased Axl activity, a phagocytotic gene (autophagy) important in maintaining homeostatic levels of major AD related lipoprotein, ApoE93. Long term infection also induced Mbp gene, which maintained myelination and prevented further neuroinflammation94. In contrast, upregulation of Flt1, a key gene in vascular endothelial growth factor (VEGF) regulation was a key feature in long term infection which could reflect cognition impairment due to higher Aβ and Tau deposition in AD etiopathogenesis, as observed in a previous study95. As a result of increased growth factor signalling, angiogenesis was induced with increased trophic activity demonstrating ongoing inflammatory activity.
Conclusion
We have demonstrated the rapid invasion of the olfactory and trigeminal nerves, the olfactory bulb and brain (beyond the bulb) by C. pneumoniae in mice. In the olfactory nerve and bulb, C. pneumoniae inclusions were associated with localized deposits of Aβ, which appeared in the olfactory nerve earlier than in the bulb. We also showed that injury to the nasal epithelium led to increased bacterial load in the peripheral nerves and olfactory bulb, but did not increase load in the rest of the brain during the time course of our study. At the cellular level, we showed that C. pneumoniae can infect and replicate to form inclusion bodies within primary glial cells of the olfactory/trigeminal nerves and CNS. Of particular importance, C. pneumoniae infection led to the dysregulation of key pathways involved in Alzheimer’s disease pathogenesis up to 28 days after intranasal inoculation. From these results, we conclude that the nerves extending between the nasal cavity and the brain constitute invasion paths by which C. pneumoniae can rapidly invade the CNS and trigger genetic and molecular changes in the longer term which may contribute to the onset of Alzheimer’s disease pathogenesis.
References
Beagley, K. W., Huston, W. M., Hansbro, P. M. & Timms, P. Chlamydial infection of immune cells: Altered function and implications for disease. Crit. Rev. Immunol. 29, 275–305 (2009).
Hammerschlag, M. R. Chlamydia pneumoniae and the lung. Eur. Respir. J. 16, 1001–1007. https://doi.org/10.1183/09031936.00.16510010 (2000).
Filardo, S., Di Pietro, M., Farcomeni, A., Schiavoni, G. & Sessa, R. Chlamydia pneumoniae-mediated inflammation in atherosclerosis: A meta-analysis. Mediators Inflamm. 2015, 378658. https://doi.org/10.1155/2015/378658 (2015).
Webley, W. C. & Hahn, D. L. Infection-mediated asthma: Etiology, mechanisms and treatment options, with focus on Chlamydia pneumoniae and macrolides. Respir. Res. 18, 98. https://doi.org/10.1186/s12931-017-0584-z (2017).
Carter, J. D., Inman, R. D., Whittum-Hudson, J. & Hudson, A. P. Chlamydia and chronic arthritis. Ann. Med. 44, 784–792. https://doi.org/10.3109/07853890.2011.606830 (2012).
Furrows, S. J. et al. Chlamydophila pneumoniae infection of the central nervous system in patients with multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 75, 152–154 (2004).
Balin, B. J. et al. Chlamydia pneumoniae: An etiologic agent for late-onset dementia. Front. Aging Neurosci. 10, 302. https://doi.org/10.3389/fnagi.2018.00302 (2018).
Itzhaki, R. F. et al. Microbes and Alzheimer’s disease. J. Alzheimer’s Dis. 51, 979–984. https://doi.org/10.3233/JAD-160152 (2016).
Balin, B. J. et al. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med. Microbiol. Immunol. 187, 23–42. https://doi.org/10.1007/s004300050071 (1998).
Schumacher, H. R. Jr. et al. Lower prevalence of Chlamydia pneumoniae DNA compared with Chlamydia trachomatis DNA in synovial tissue of arthritis patients. Arthritis Rheum. 42, 1889–1893. https://doi.org/10.1002/1529-0131(199909)42:9%3c1889::AID-ANR13%3e3.0.CO;2-W (1999).
Gerard, H. C. et al. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol. Med. Microbiol. 48, 355–366. https://doi.org/10.1111/j.1574-695X.2006.00154.x (2006).
Soscia, S. J. et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5, e9505. https://doi.org/10.1371/journal.pone.0009505 (2010).
Kumar, D. K. et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 8, 340ra372. https://doi.org/10.1126/scitranslmed.aaf1059 (2016).
Ring, R. H. & Lyons, J. M. Failure to detect Chlamydia pneumoniae in the late-onset Alzheimer’s brain. J. Clin. Microbiol. 38, 2591–2594 (2000).
Little, C. S. et al. Age alterations in extent and severity of experimental in infection with Chlamydophila pneumoniae in BALB/c mice. Infect. Immun. 73, 1723–1734. https://doi.org/10.1128/IAI.73.3.1723-1734.2005 (2005).
Little, C. S., Hammond, C. J., MacIntyre, A., Balin, B. J. & Appelt, D. M. Chlamydia pneumoniae induces alzheimer’s-like amyloid plaques in brains of BALB/c mice. Neurobiol. Aging 25, 419–429. https://doi.org/10.1016/S0197-4580(03)00127-1 (2004).
Little, C. S. et al. Detection of bacterial antigens and Alzheimer’s-like pathology in the central nervous system of BALB/c mice following intranasal infection with a laboratory isolate of Chlamydia pneumoniae. Front. Aging Neurosci. 6, 304. https://doi.org/10.3389/fnagi.2014.00304 (2014).
van Boelen, E. et al. Detection of amyloid beta aggregates in the brain of BALB/c mice after Chlamydia pneumoniae infection. Acta Neuropathol. 114, 255–261. https://doi.org/10.1007/s00401-007-0252-3 (2007).
Gieffers, J. et al. Phagocytes transmit Chlamydia pneumoniae from the lungs to the vasculature. Eur. Respir. J. 23, 506–510. https://doi.org/10.1183/09031936.04.00093304 (2004).
Dando, S. J. et al. Pathogens penetrating the central nervous system: Infection pathways and the cellular and molecular mechanisms of invasion. Clin. Microbiol. Rev. 27, 691–726. https://doi.org/10.1128/CMR.00118-13 (2014).
Mann, D. M., Tucker, C. M. & Yates, P. O. Alzheimer’s disease: An olfactory connection?. Mech. Ageing Dev. 42, 1–15. https://doi.org/10.1016/0047-6374(88)90058-9 (1988).
Murphy, C. Olfactory and other sensory impairments in Alzheimer disease. Nat. Rev. Neurol. 15, 11–24. https://doi.org/10.1038/s41582-018-0097-5 (2019).
Simic, G. et al. Does Alzheimer’s disease begin in the brainstem?. Neuropathol. Appl. Neurobiol. 35, 532–554. https://doi.org/10.1111/j.1365-2990.2009.01038.x (2009).
Nazareth, L. et al. Chlamydia muridarum can invade the central nervous system via the olfactory and trigeminal nerves and infect peripheral nerve glial cells. Front Cell Infect. Microbiol. 10, 607779. https://doi.org/10.3389/fcimb.2020.607779 (2020).
Wu, H. Y. & Russell, M. W. Nasal lymphoid tissue, intranasal immunization, and compartmentalization of the common mucosal immune system. Immunol. Res. 16, 187–201 (1997).
Menick, F. J. & Salibian, A. Primary intranasal lining injury cause, deformities, and treatment plan. Plast. Reconstr. Surg. 134, 1045–1056. https://doi.org/10.1097/PRS.0000000000000694 (2014).
Walkden, H. et al. Burkholderia pseudomallei invades the olfactory nerve and bulb after epithelial injury in mice and causes the formation of multinucleated giant glial cells in vitro. PLoS Negl. Trop. Dis. 14, e0008017. https://doi.org/10.1371/journal.pntd.0008017 (2020).
Herbert, R. P. et al. Cytokines and olfactory bulb microglia in response to bacterial challenge in the compromised primary olfactory pathway. J. Neuroinflamm. 9, 109. https://doi.org/10.1186/1742-2094-9-109 (2012).
Nazareth, L. et al. Olfactory ensheathing cells are the main phagocytic cells that remove axon debris during early development of the olfactory system. J. Comp. Neurol. 523, 479–494. https://doi.org/10.1002/cne.23694 (2015).
Nazareth, L. et al. Differing phagocytic capacities of accessory and main olfactory ensheathing cells and the implication for olfactory glia transplantation therapies. Mol. Cell Neurosci. 65, 92–101. https://doi.org/10.1016/j.mcn.2015.03.005 (2015).
Vincent, A. J., Choi-Lundberg, D. L., Harris, J. A., West, A. K. & Chuah, M. I. Bacteria and PAMPs activate nuclear factor kappaB and Gro production in a subset of olfactory ensheathing cells and astrocytes but not in Schwann cells. Glia 55, 905–916. https://doi.org/10.1002/glia.20512 (2007).
Nazareth, L. et al. Novel insights into the glia limitans of the olfactory nervous system. J. Comp. Neurol. 527, 1228–1244. https://doi.org/10.1002/cne.24618 (2019).
Jung, Y.-J. & Chung, W.-S. Phagocytic roles of glial cells in healthy and diseased brains. Biomol. Ther. (Seoul) 26, 350–357. https://doi.org/10.4062/biomolther.2017.133 (2018).
Macedo-Ramos, H. et al. Olfactory ensheathing cells as putative host cells for Streptococcus pneumoniae: Evidence of bacterial invasion via mannose receptor-mediated endocytosis. Neurosci. Res. 69, 308–313. https://doi.org/10.1016/j.neures.2010.12.015 (2011).
Delbaz, A. et al. Neisseria meningitidis induces pathology-associated cellular and molecular changes in trigeminal schwann cells. Infect. Immun. https://doi.org/10.1128/IAI.00955-19 (2020).
Abdelrahman, Y. M. & Belland, R. J. The chlamydial developmental cycle. FEMS Microbiol. Rev. 29, 949–959. https://doi.org/10.1016/j.femsre.2005.03.002 (2005).
Armitage, C. W., O’Meara, C. P. & Beagley, K. W. Chlamydia pneumoniae and Chlamydia trachomatis infection differentially modulates human dendritic cell line (MUTZ) differentiation and activation. Scand. J. Immunol. 82, 48–54. https://doi.org/10.1111/sji.12295 (2015).
Herweg, J. A. & Rudel, T. Interaction of Chlamydiae with human macrophages. FEBS J. 283, 608–618. https://doi.org/10.1111/febs.13609 (2016).
Zuck, M., Sherrid, A., Suchland, R., Ellis, T. & Hybiske, K. Conservation of extrusion as an exit mechanism for Chlamydia. Pathog. Dis. https://doi.org/10.1093/femspd/ftw093 (2016).
Airenne, S. et al. Chlamydia pneumoniae infection in human monocytes. Infect. Immun. 67, 1445–1449 (1999).
Dean, D., Suchland, R. J. & Stamm, W. E. Evidence for long-term cervical persistence of Chlamydia trachomatis by omp1 genotyping. J. Infect. Dis. 182, 909–916. https://doi.org/10.1086/315778 (2000).
Hammerschlag, M. R. et al. Persistent infection with Chlamydia pneumoniae following acute respiratory illness. Clin. Infect. Dis. 14, 178–182. https://doi.org/10.1093/clinids/14.1.178 (1992).
St John, J. A. et al. Burkholderia pseudomallei penetrates the brain via destruction of the olfactory and trigeminal nerves: Implications for the pathogenesis of neurological melioidosis. MBio 5, e00025. https://doi.org/10.1128/mBio.00025-14 (2014).
St John, J. A. et al. Burkholderia pseudomallei rapidly infects the brain stem and spinal cord via the trigeminal nerve after intranasal inoculation. Infect. Immun. 84, 2681–2688. https://doi.org/10.1128/IAI.00361-16 (2016).
Windus, L. C., Claxton, C., Allen, C. L., Key, B. & St John, J. A. Motile membrane protrusions regulate cell-cell adhesion and migration of olfactory ensheathing glia. Glia 55, 1708–1719. https://doi.org/10.1002/glia.20586 (2007).
Holt, L. M., Stoyanof, S. T. & Olsen, M. L. Magnetic cell sorting for in vivo and in vitro astrocyte, neuron, and microglia analysis. Curr. Protoc. Neurosci. 88, e71. https://doi.org/10.1002/cpns.71 (2019).
Brittebo, E. B. Metabolism-dependent toxicity of methimazole in the olfactory nasal mucosa. Pharmacol. Toxicol. 76, 76–79. https://doi.org/10.1111/j.1600-0773.1995.tb00107.x (1995).
Chehrehasa, F. et al. Olfactory glia enhance neonatal axon regeneration. Mol. Cell. Neurosci. 45, 277–288. https://doi.org/10.1016/j.mcn.2010.07.002 (2010).
Xie, F. et al. The tissue-specific toxicity of methimazole in the mouse olfactory mucosa is partly mediated through target-tissue metabolic activation by CYP2A5. Drug Metab. Dispos. Boil. Fate Chem. 39, 947–951. https://doi.org/10.1124/dmd.110.037895 (2011).
Little, C. S. et al. Detection of bacterial antigens and Alzheimer’s disease-like pathology in the central nervous system of BALB/c mice following intranasal infection with a laboratory isolate of Chlamydia pneumoniae. Front. Aging Neurosci. 6, 304. https://doi.org/10.3389/fnagi.2014.00304 (2014).
Wong, K. H., Skelton, S. K. & Chan, Y. K. Efficient culture of Chlamydia pneumoniae with cell lines derived from the human respiratory tract. J. Clin. Microbiol. 30, 1625–1630. https://doi.org/10.1128/JCM.30.7.1625-1630.1992 (1992).
Dando, S. J. et al. Burkholderia pseudomallei capsule exacerbates respiratory melioidosis but does not afford protection against antimicrobial signaling or bacterial killing in human olfactory ensheathing cells. Infect. Immun. 84, 1941–1956. https://doi.org/10.1128/IAI.01546-15 (2016).
Rake, G. The rapid invasion of the body through the olfactory mucosa. J. Exp. Med. 65, 303–315. https://doi.org/10.1084/jem.65.2.303 (1937).
Sjolinder, H. & Jonsson, A. B. Olfactory nerve—a novel invasion route of Neisseria meningitidis to reach the meninges. PLoS One 5, e14034. https://doi.org/10.1371/journal.pone.0014034 (2010).
Otter, A. & Blakemore, W. F. Observation on the presence of Listeria monocytogenes in axons. Acta Microbiol. Hung. 36, 125–131 (1989).
Jarolim, K. L., McCosh, J. K., Howard, M. J. & John, D. T. A light microscopy study of the migration of Naegleria fowleri from the nasal submucosa to the central nervous system during the early stage of primary Amebic meningoencephalitis in mice. J. Parasitol. 86, 50–55. https://doi.org/10.1645/0022-3395(2000)086[0050:ALMSOT]2.0.CO;2 (2000).
Tomlinson, A. H. & Esiri, M. M. Herpes simplex encephalitis. Immunohistological demonstration of spread of virus via olfactory pathways in mice. J. Neurol. Sci. 60, 473–484. https://doi.org/10.1016/0022-510x(83)90158-2 (1983).
Meinhardt, J. et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. https://doi.org/10.1038/s41593-020-00758-5 (2020).
Brouwer, M. C., Ascione, T. & Pagliano, P. Neurologic aspects of covid-19: A concise review. Infez Med. 28, 42–45 (2020).
Gengler, I., Wang, J. C., Speth, M. M. & Sedaghat, A. R. Sinonasal pathophysiology of SARS-CoV-2 and COVID-19: A systematic review of the current evidence. Laryngosc. Investig. Otolaryngol. 5, 354–359. https://doi.org/10.1002/lio2.384 (2020).
Perlman, S., Jacobsen, G. & Afifi, A. Spread of a neurotropic murine coronavirus into the CNS via the trigeminal and olfactory nerves. Virology 170, 556–560. https://doi.org/10.1016/0042-6822(89)90446-7 (1989).
Dreses-Werringloer, U., Gerard, H. C., Whittum-Hudson, J. A. & Hudson, A. P. Chlamydophila (Chlamydia) pneumoniae infection of human astrocytes and microglia in culture displays an active, rather than a persistent, phenotype. Am. J. Med. Sci. 332, 168–174. https://doi.org/10.1097/00000441-200610000-00003 (2006).
Boelen, E. et al. Chlamydia pneumoniae infection of brain cells: An in vitro study. Neurobiol. Aging 28, 524–532. https://doi.org/10.1016/j.neurobiolaging.2006.02.014 (2007).
Boelen, E. et al. Inflammatory responses following Chlamydia pneumoniae infection of glial cells. Eur. J. Neurosci. 25, 753–760. https://doi.org/10.1111/j.1460-9568.2007.05339.x (2007).
Ikejima, H., Friedman, H. & Yamamoto, Y. Chlamydia pneumoniae infection of microglial cells in vitro: A model of microbial infection for neurological disease. J. Med. Microbiol. 55, 947–952. https://doi.org/10.1099/jmm.0.46348-0 (2006).
Al-Atrache, Z., Lopez, D. B., Hingley, S. T. & Appelt, D. M. Astrocytes infected with Chlamydia pneumoniae demonstrate altered expression and activity of secretases involved in the generation of β-amyloid found in Alzheimer disease. BMC Neurosci. 20, 6–6. https://doi.org/10.1186/s12868-019-0489-5 (2019).
Hammond, C. J. et al. Immunohistological detection of Chlamydia pneumoniae in the Alzheimer’s disease brain. BMC Neurosci. 11, 121. https://doi.org/10.1186/1471-2202-11-121 (2010).
Arking, E. J. et al. Ultrastructural analysis of Chlamydia pneumoniae in the Alzheimer’s brain. Pathogenesis (Amst) 1, 201–211 (1999).
Harris, J. A., West, A. K. & Chuah, M. I. Olfactory ensheathing cells: Nitric oxide production and innate immunity. Glia 57, 1848–1857. https://doi.org/10.1002/glia.20899 (2009).
Galloway, D. A., Phillips, A. E. M., Owen, D. R. J. & Moore, C. S. Phagocytosis in the brain: Homeostasis and disease. Front. Immunol. 10, 790. https://doi.org/10.3389/fimmu.2019.00790 (2019).
Ries, M. & Sastre, M. Mechanisms of abeta clearance and degradation by glial cells. Front. Aging Neurosci. 8, 160. https://doi.org/10.3389/fnagi.2016.00160 (2016).
Wu, N., Rao, X., Gao, Y., Wang, J. & Xu, F. Amyloid-beta deposition and olfactory dysfunction in an Alzheimer’s disease model. J. Alzheimer’s Dis. 37, 699–712. https://doi.org/10.3233/JAD-122443 (2013).
Kinney, J. W. et al. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y) 4, 575–590. https://doi.org/10.1016/j.trci.2018.06.014 (2018).
Alasmari, F., Alshammari, M. A., Alasmari, A. F., Alanazi, W. A. & Alhazzani, K. Neuroinflammatory cytokines induce amyloid beta neurotoxicity through modulating amyloid precursor protein levels/metabolism. Biomed. Res. Int. 3087475–3087475, 2018. https://doi.org/10.1155/2018/3087475 (2018).
Ries, M. & Sastre, M. Mechanisms of Aβ clearance and degradation by glial cells. Front. Aging Neurosci. https://doi.org/10.3389/fnagi.2016.00160 (2016).
Cheignon, C. et al. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464. https://doi.org/10.1016/j.redox.2017.10.014 (2018).
Hetz, C. & Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. https://doi.org/10.1038/nrn3689 (2014).
Hashimoto, S. & Saido, T. C. Critical review: Involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol. 8, 180024. https://doi.org/10.1098/rsob.180024 (2018).
Smith, J. A. Regulation of cytokine production by the unfolded protein response; implications for infection and autoimmunity. Front. Immunol. https://doi.org/10.3389/fimmu.2018.00422 (2018).
Wang, M. & Kaufman, R. J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335. https://doi.org/10.1038/nature17041 (2016).
Clarimón, J., Bertranpetit, J., Boada, M., Tàrraga, L. & Comas, D. HSP70-2 (HSPA1B) is associated with noncognitive symptoms in late-onset Alzheimer’s disease. J. Geriatr. Psychiatry Neurol. 16, 146–150 (2003).
Qin, L., Guo, J., Zheng, Q. & Zhang, H. BAG2 structure, function and involvement in disease. Cell. Mol. Biol. Lett. 21, 1–11 (2016).
Liu, H., Luo, K. & Luo, D. Guanosine monophosphate reductase 1 is a potential therapeutic target for Alzheimer’s disease. Sci. Rep. 8, 1–10 (2018).
Brooks, L. R. & Mias, G. I. Data-driven analysis of age, sex, and tissue effects on gene expression variability in Alzheimer’s disease. Front. Neurosci. 13, 392 (2019).
Peralta, S. et al. Partial complex I deficiency due to the CNS conditional ablation of Ndufa5 results in a mild chronic encephalopathy but no increase in oxidative damage. Hum. Mol. Genet. 23, 1399–1412 (2014).
Liang, W. S. et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. 105, 4441–4446 (2008).
Tao, Q.-Q., Chen, Y.-C. & Wu, Z.-Y. The role of CD2AP in the pathogenesis of Alzheimer’s disease. Aging Dis. 10, 901 (2019).
Barone, G. et al. Human CDK18 promotes replication stress signaling and genome stability. Nucleic Acids Res. 44, 8772–8785 (2016).
Koopman, M. B. & Rüdiger, S. G. Alzheimer cells on their way to derailment show selective changes in protein quality control network. Front. Mol. Biosci. 7, 25 (2020).
Roberts, R. O. et al. Decreased expression of sulfatase 2 in the brains of Alzheimer’s disease patients: Implications for regulation of neuronal cell signaling. J. Alzheimer’s Dis. Rep. 1, 115–124 (2017).
Lanke, V., Moolamalla, S., Roy, D. & Vinod, P. Integrative analysis of hippocampus gene expression profiles identifies network alterations in aging and Alzheimer’s disease. Front. Aging Neurosci. 10, 153 (2018).
Yamazaki, Y. et al. Selective loss of cortical endothelial tight junction proteins during Alzheimer’s disease progression. Brain 142, 1077–1092 (2019).
Zhao, W. et al. Axl receptor tyrosine kinase is a regulator of apolipoprotein E. Mol. Brain 13, 1–13 (2020).
Ou-Yang, M.-H. & Van Nostrand, W. E. The absence of myelin basic protein promotes neuroinflammation and reduces amyloid β-protein accumulation in Tg-5xFAD mice. J. Neuroinflamm. 10, 1–12 (2013).
Mahoney, E. R. et al. Brain expression of the vascular endothelial growth factor gene family in cognitive aging and alzheimer’s disease. Mol. Psychiatry 26, 888–896 (2021).
Acknowledgements
This work was supported by a Goda Foundation Grant to JE and JS, a Clem Jones Foundation Grant to JS and JE, an ARC Discovery Grant to JE, KB and JS (DP150104495), a Menzies Health Institute Queensland Capacity Grant to JE, AC and KB, and a Griffith University International postgraduate research scholarship (GUIPRS) to SB. We would also like to acknowledge Ms Sarah Hanson and the Systems Biology and Data Science Research Facility in the Menzies Health Inst QLD at Griffith University for their advice on the NanoString nCounter analysis.
Author information
Authors and Affiliations
Contributions
All authors planned and designed the experiments. Experiments were conducted by A.C., A.D., H.W., S.B., C.A., T.E., L.T., E.M., N.W. A.C., A.D., H.W., N.W., J.S. and J.E. analysed the data. AC drafted the manuscript. All authors reviewed and edited manuscript. J.E., J.S., K.B. funded the research. J.E. provided the overall supervision of the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chacko, A., Delbaz, A., Walkden, H. et al. Chlamydia pneumoniae can infect the central nervous system via the olfactory and trigeminal nerves and contributes to Alzheimer’s disease risk. Sci Rep 12, 2759 (2022). https://doi.org/10.1038/s41598-022-06749-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-06749-9
This article is cited by
-
Contribution of CNS and extra-CNS infections to neurodegeneration: a narrative review
Journal of Neuroinflammation (2024)
-
The endotoxin hypothesis of Alzheimer’s disease
Molecular Neurodegeneration (2024)
-
Current understanding of the Alzheimer’s disease-associated microbiome and therapeutic strategies
Experimental & Molecular Medicine (2024)
-
Olfactory immunology: the missing piece in airway and CNS defence
Nature Reviews Immunology (2024)
-
Smoking Impacts Alzheimer’s Disease Progression Through Oral Microbiota Modulation
Molecular Neurobiology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
