
Abstract
A specific labeling strategy for bioactive molecules is presented for eptifibatide (integrilin) an antiplatelet aggregation inhibitor, which derives from the disintegrin protein barbourin in the venom of certain rattlesnakes. By specifically labeling the disulfide bridge this molecule becomes accessible for the nuclear spin hyperpolarization method of parahydrogen induced polarization (PHIP). The PHIP-label was synthesized and inserted into the disulfide bridge of eptifibatide via reduction of the peptide and insertion by a double Michael addition under physiological conditions. This procedure is universally applicable for disulfide-containing biomolecules and preserves their tertiary structure with a minimum of change. HPLC and MS spectra prove the successful insertion of the label. 1H-PHIP-NMR experiments yield a factor of over 1000 as lower limit for the enhancement factor. These results demonstrate the high potential of the labeling strategy for the introduction of site selective PHIP-labels into biomolecules’ disulfide bonds.
Similar content being viewed by others
Introduction
Besides analytical methods such as circular dichromatic spectroscopy, X-ray crystallography (XRD) or small-angle neutron scattering (SANS) nuclear magnetic resonance (NMR) spectroscopy is the method of choice for most scientists, when structural questions on biomolecules, signaling pathways or biological networks arise1,2. Unfortunately, the inherent limitation of NMR analysis is the low sensitivity and a distinct signal broadening in case of macromolecular systems3. Therefore, hyperpolarization techniques such as spin exchange optical pumping (SEOP)4, dynamic nuclear polarization (DNP)5, (parahydrogen induced polarization (PHIP)6,7 or SABRE (signal amplification by reversible exchange)8,9 gain increased interest in overcoming the low sensitivity of NMR. These methods differ in the source of polarization, which provides the signal enhancement on the investigated nuclei10,11,12,13. In case of biological structure analysis PHIP-measurements seem to be the most promising method, due to its inexpensive technical set up and easy measurement conditions14. Besides a multiple bond, on which a hydrogenation catalyst can hydrogenate, PHIP does not impose any special conditions on the substrate. There is no need of high or low temperature, no additional radical matrix, no microwave, laser radiation or the necessity of expensive hardware, etc.
One prerequisite for the structural investigation of biological entities, e.g. peptides via PHIP is the implementation of unsaturated moieties, which can be hydrogenated by para-H215.
To chemically functionalize natural proteins, the binding method and also the label itself must satisfy a number of requirements. In addition to biocompatibility, they must be stable and biologically safe. This implies that the label must have a high selectivity and reactivity towards a certain binding site, while preserving the tertiary structure16.
One approach is the use of tags (epitopes) to functionalize proteins17. Unfortunately, these labels are rather large, which can influence the molecule’s structural features. Furthermore, these tags bind specifically to a particular amino acid sequence. That’s why the target system must be known precisely in advance17,18. Besides this enzymatic approach, non-canonical amino acids that contain different functional groups (ethynyl-19, azido-20, halogen-21), can be incorporated into the peptide’s sequence and further utilized12,15,22,23,24,25,26,27,28,29,30,31,32,33. However, this process is exceptionally labor-intensive and proceeds with only low yields34.
To overcome these issues, the post modification of peptides with PHIP-relevant fragments by small and selective labels is the most promising approach. Among the numerous possibilities that are already described in literature35,36,37,38,39, we decided to adopt the bifunctional cross-link strategy based on acylated allylic sulfones, which was first described by Liberatore and Brocchini40,41. They selectively modified disulfide bridges that are frequently present in therapeutically relevant proteins as binding sites, e.g. antibodies41 or somatostatin16. Those disulfide bonds are reduced and expanded by three carbon atoms, which can carry additional functional elements. The decisive advantage of this method is that the integrity of the protein structure isn’t compromised42. Weil et al. took advantage of this concept and presented a toolbox of allylsulfone intercalators presenting various functional groups such as boronic acid, azide or rhodamine as imaging probe43. Moreover, they prepared the ethynyl-containing intercalator 2, which we utilized for our PHIP experiments (Scheme 1).

Illustration of the Intercalation-hydrogenation strategy to boost signal intensity in NMR experiments using the ethynyl-containing label 2. Additionally, the biochemical relevant “K”GD-amino acid sequence ("Lys"-Gly-Asp) is highlighted. [We followed the usual nomenclature of the binding motif although it should read homo-R-G-D instead of K-G-D]48,49.
As a model system we chose the cyclic heptapeptide eptifibatide (Integrilin, 1), which is derived from a disintegrin protein44 found in the venom of rattlesnake (sistrurus miliarius barbouri), which is used as an antiplatelet aggregation inhibitor. The high potential of this drug is based on its cyclic structure and the "K"GD amino acid sequence (Lys-Gly-Asp)45,46,47.
Herein, we demonstrate by high performance liquid chromatography (HPLC) and electrospray ionization mass spectrometry (ESI–MS) that the intercalator 2 can be incorporated into the disulfide bridge of the peptide. Recently it has been shown that eptifibatide can be efficiently spin-labeled for hyperpolarization applications employing Dynamic Nuclear Polarization50. In the present paper we demonstrate that it is also feasible to employ derivatives of eptifibatide as efficient hyperpolarization sources in PHIP-NMR spectroscopy. In particular, we point out the high value of the label for PHIP-NMR spectroscopy by hydrogenation of the ethynyl fragment (Scheme 1).
Methods
All synthetic procedures, reagents, methods as well as spectroscopic parameters and spectra of the PHIP-measurements are reported in the Supporting Information.
Results and discussion
Synthesis and intercalation of label 2
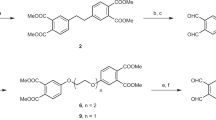
According to Scheme 2, we started our research by synthesizing the bissulfone label 2 following a published procedure to a similar compound51 with slight modifications. In the first step p-acetyl benzoic acid 5 was subjected to a double Mannich-type reaction. The crude reaction product was directly used in the following substitution reaction with p-thiocresol as a nucleophile yielding the bisulfide 6 which, after recrystallization was isolated in 63% yield. After oxidation of the sulfides by peracetic acid the corresponding bissulfone 7 was obtained in 76% yield. The implementation of the PHIP-relevant ethynyl-fragment was achieved by EDC-mediated (1-Ethyl-3-(3-dimethylamino-propyl)carbodiimide) amide-coupling with propargylamine. After purification by column chromatography the intercalator was separated from all hazardous impurities as observed by analytical HPLC and HPLC–MS (Fig. 1 and Supplementary Fig. 7) and was obtained as a mixture of bissulfone 2 and allylsulfone 8 with a yield of 43% and an overall yield of the 3-step synthesis of 21%. Considering the fact that the bissulfone 2 will be converted into the allylsulfone 8 in the intercalation reaction anyway, no further purification is needed. (For more information see Ref.43 and Supplementary Information).

Synthesis of the intercalator mixture 2 and 8.

HPLC traces of the labeling process shown in Scheme 3 (solvent gradient of ACN/H2O: 20/80 to 80/20, λ = 214 nm).
With the label mixture (2/8) in hand, the intercalation reaction proceeded in a two-step sequence. First, the mixture of 2 and 8 is converted completely to the reactive species 8 by dissolving the mixture in a slightly alkaline aqueous solution (pH = 7.8). At this point the reduced eptifibatide (after purification) was added and the resulting mixture stirred at room temperature until full consumption of the starting materials (Scheme 3).

Illustration of the intercalation process of the label 2/8 into the in advance reduced disulfide bond of eptifibatide (DTT: Dithiothreitol).
The insertion process of the unsaturated fragment into the disulfide bridge can be followed by HPLC (Fig. 1). The DTT-mediated (Dithiothreitol) reduction of eptifibatide results in a prolonged retention time of the resulting dithiol 9 compared to eptifibatide 1 with a closed disulfide bridge. Similarly, the insertion of the label 8 causes a delayed elution time for the modified peptide 3 relative to the dithiol 9. The HPLC-trace of the intercalator mixture 2 and 8 is shown to verify its complete consumption.
In parallel to the HPLC traces, the successful incorporation of the label can also be confirmed by ESI–MS. Analogous to the chromatograms shown above, the molecular ion peaks are shown, which indicate the increase in mass of the peptide due to the binding of the label. The measured mass spectra agree with the theoretically determined values (Fig. 2 and Supplementary Information).

Relevant sections of the ESI–MS spectra recorded during the intercalation process as described in Scheme 3. [(a) Eptifibatide, (b) Reduced eptifibatide, (c) Eptifibatide after label insertion, (d) intercalator 2/8].
Enhancement factor determination
To show the suitability of the label 2 for PHIP-NMR spectroscopy, the triple bond is hydrogenated with para-H2 using [Rh(dppb)COD)]BF4 as catalyst (Scheme 4).

Hydrogenation of the triple bond containing PHIP-label inserted in the disulfide bridge of eptifibatide 3 with para-H2.
For the determination of the enhancement factor several 1H-NMR spectra are acquired. In addition to the spectrum after complete hydrogenation (in thermal equilibrium structure 4), a spectrum showing the short-lived first hydrogenation step to the parahydrogen added N-allyl amide 10 was recorded (Fig. 3B). Based on this hydrogenation the PHIP activity of the ethynyl labeled eptifibatide 3 was proven and an enhancement factor determined was estimated (see Supplementary Information for calculation).

Comparison of the 1H-NMR spectra of (A) the thermally relaxed spectrum after full hydrogenation (4096 scans) and (B) the PHIP spectrum (single scan) after addition of para-H2 (enrichment of > 95%) (500 MHz, MeOH-d4, 298 K). The PHIP spectrum reveals hyperpolarized lines of the catalyst system (see Supplementary Fig. 10) and the strongly PHIP enhanced signal of the labeled eptifibatide, which is visible in the single scan spectrum with a good signal-to-noise ratio. The obtained enhancement factor, estimated from the insets for the signal at 5.17 ppm is above 1000 (see Supplementary Information for calculation).
Figure 3 shows the spectra of 3 in deuterated methanol under enhancement conditions (during hydrogenation with para-H2; Fig. 3B) and in thermal equilibrium after complete hydrogenation (Fig. 3A). The PHIP signals of the vinyl group are located at 5.95 and 5.17 ppm (terminal protons). The PHIP-signals with the low chemical shift and the complex multiplicity in the range from 4.38 to 1.54 ppm can be assigned to the 3 mg of the catalytically active complex Rh(dppb)(COD)BF4 contained in the sample (see Supplementary Information), the “pseudo” PHIP signal at 4.58 ppm is due to the PNL-Effect (partial negative line shape). This arises when molecular hydrogen enriched with para-H2 is solubilized in e.g., methanol interacting with an organometallic hydrogenation catalyst52.
The PHIP spectrum (Fig. 3B) is measured in a single scan. For the thermally relaxed spectrum (Fig. 3A), 4096 scans were required to observe a visible and evaluable signal of the hydrogenated label (the terminal methyl group of the fully hydrogenated product). The required high number of scans in the thermally relaxed spectrum is due to the low concentration of 0.2 mM of 3 in MeOH-d4.
Owing to the transient nature of the vinyl-intermediate, which is not visible in the thermally relaxed spectrum, the enhancement factor can be estimated only indirectly by comparing the integral ratios of the PHIP-spectrum (vinyl signal at 5.17 ppm) and the thermally relaxed spectrum (alkyl signal at 0.86 ppm), marked as insets in Fig. 3 and assigned in Scheme 4. Comparing these integrals, a factor of 1000 can be estimated as lower limit for the enhancement factor (details of the calculation can be found in Supplementary Information).
These results clearly prove that the insertion of label 2 was successful leading to a very efficient PHIP labelling. It has been demonstrated that PHIP-active, site selective labeled biomolecules can be synthesized using the approach described above. This PHIP labeling creates a very strong signal enhancement of the protons of the allyl-group. Such a signal enhancement can be employed for example for selective binding studies of the eptifibatide or similar molecules to a target protein15. Moreover this opens up the way to ULTRAFAST 2D-NMR of such complexes31.
Conclusion
A bis-sulfone-based label was synthesized and its suitability for the site-selective insertion into the disulfide bridge of a bioactive peptide was demonstrated. The ethynyl-modified intercalator was obtained as a two-component mixture of the bissulfone 2 and its elimination product 8, where both components can participate in the labeling reaction because the active MICHAEL acceptor 8 is formed by elimination under the reaction conditions. As an example for the feasibility of the obtained unsaturated system as a substrate for PHIP labeling studies, the disulfide bridge of eptifibatide 1, a synthetic cyclic heptapeptide derived from a disintegrin protein from the venom of the rattlesnake Sistrurus miliarius barbourin, was chosen. After label insertion, it was possible to hyperpolarize the target molecule 3 by a hydrogenation reaction with para-H2. A factor of 1000 was estimated as a lower limit for the signal enhancement of the hyperpolarized protons of the newly formed vinyl group in comparison with the final formed alkyl group. This result paves the way for future applications of ethynyl-substituted systems like 2/8 to be successfully used for future PHIP-applications, utilizing the site selective opening of disulfide bridges in biologically active compounds.
Abbreviations
- NMR:
-
Nuclear magnetic resonance
- SEOP:
-
Spin exchange optical pumping
- DNP:
-
Dynamic nuclear polarization
- PHIP:
-
ParaHydrogen induced polarization
- SABRE:
-
Signal amplification by reversible exchange
- SE:
-
Signal enhancement
References
Lee, Y. et al. Hyperpolarized binding pocket nuclear overhauser effect for determination of competitive ligand binding. Angew. Chem. Int. Ed. 51(21), 5179–5182 (2012).
Pellecchia, M., Sem, D. S. & Wüthrich, K. NMR in drug discovery. Nat. Rev. Drug Discovery 1(3), 211–219 (2002).
Ardenkjaer-Larsen, J.-H. et al. Facing and overcoming sensitivity challenges in biomolecular NMR spectroscopy. Angew. Chem. Int. Ed. 54(32), 9162–9185 (2015).
Walker, T. G. & Happer, W. Spin-exchange optical pumping of noble-gas nuclei. Rev. Mod. Phys. 69(2), 629–642 (1997).
Ni, Q. Z. et al. High frequency dynamic nuclear polarization. Acc. Chem. Res. 46(9), 1933–1941 (2013).
Bowers, C. R. & Weitekamp, D. P. Parahydrogen and synthesis allow dramatically enhanced nuclear alignment. J. Am. Chem. Soc. 109(18), 5541–5542 (1987).
Eisenschmid, T. C. et al. Para hydrogen induced polarization in hydrogenation reactions. J. Am. Chem. Soc. 109(26), 8089–8091 (1987).
Atkinson, K. D. et al. Spontaneous transfer of parahydrogen derived spin order to pyridine at low magnetic field. J. Am. Chem. Soc. 131(37), 13362–13368 (2009).
Adams, R. W. et al. Reversible interactions with parahydrogen enhance NMR sensitivity by polarization transfer. Science 323(5922), 1708–1711 (2009).
Gutmann, T., Sellin, M., Breitzke, H., Stark, A. & Buntkowsky, G. Parahydrogen induced polarization in homogeneous phase—An example of how ionic liquids affect homogenization and thus activation of catalysts. Phys. Chem. Chem. Phys. 11(40), 9170–9175 (2009).
Trantzschel, T. et al. Parahydrogen induced polarization in face of keto–enol tautomerism: Proof of concept with hyperpolarized ethanol. Phys. Chem. Chem. Phys. 14(16), 5601–5604 (2012).
Ratajczyk, T. et al. NMR signal enhancement by effective SABRE labeling of oligopeptides. Chem. Eur. J. 21(36), 12616–12619 (2015).
Gutmann, T. et al. New investigations of technical rhodium and iridium catalysts in homogeneous phase employing parahydrogen induced polarization. Solid State Nucl. Magn. Reson. 40(2), 88–90 (2011).
Koptyug, I. V. Spin hyperpolarization in NMR to address enzymatic processes in vivo. Mendeleev Commun. 23(6), 299–312 (2013).
Sauer, G. et al. Effective PHIP labeling of bioactive peptides boosts the intensity of the NMR signal. Angew. Chem. Int. Ed. 53(47), 12941–12945 (2014).
Kuan, S. L., Wang, T. & Weil, T. Site-selective disulfide modification of proteins: Expanding diversity beyond the proteome. Chem. Eur. J. 22(48), 17112–17129 (2016).
Lotze, J., Reinhardt, U., Seitz, O. & Beck-Sickinger, A. G. Peptide-tags for site-specific protein labelling in vitro and in vivo. Mol. BioSyst. 12(6), 1731–1745 (2016).
Lin, M. Z. & Wang, L. Selective labeling of proteins with chemical probes in living cells. Physiology 23(3), 131–141 (2008).
Miyake-Stoner, S. J. et al. Probing protein folding using site-specifically encoded unnatural amino acids as FRET donors with tryptophan. Biochemistry 48(25), 5953–5962 (2009).
Zhu, S. et al. Genetically encoding a light switch in an ionotropic glutamate receptor reveals subunit-specific interfaces. Proc. Natl. Acad. Sci. U.S.A. 111(16), 6081–6086 (2014).
Jackson, J. C., Hammill, J. T. & Mehl, R. A. Site-specific incorporation of a (19)F-amino acid into proteins as an NMR probe for characterizing protein structure and reactivity. J. Am. Chem. Soc. 129(5), 1160–1166 (2007).
Glöggler, S. et al. Parahydrogen induced polarization of amino acids, peptides and deuterium–hydrogen gas. Phys. Chem. Chem. Phys. 13, 13759–13764 (2011).
Gruppi, F., Xu, X., Zhang, B., Tang, J. A. & Jerschow, A. Peptide hydrogenation and labeling with parahydrogen. Angew. Chem.-Int. Ed. 51, 11787–11790 (2012).
Soon, P. C. et al. Hyperpolarization of amino acid precursors to neurotransmitters with parahydrogen induced polarization. Chem. Commun. 49, 5304–5306 (2013).
Trantzschel, T. et al. Application of parahydrogen-induced polarization to unprotected dehydroamino carboxylic acids. Appl. Magn. Reson. 44(1–2), 267–278 (2013).
Korner, M. et al. PHIP-label: Parahydrogen-induced polarization in propargylglycine-containing synthetic oligopeptides. Chem. Commun. 49(71), 7839–7841 (2013).
Lego, D. et al. Parahydrogen-induced polarization of carboxylic acids: A pilot study of valproic acid and related structures. NMR Biomed. 27(7), 810–816 (2014).
Kiryutin, A. S. et al. A highly versatile automatized setup for quantitative measurements of PHIP enhancements. J. Magn. Reson. 285, 26–36 (2017).
Brodrecht, M. et al. Efficient building blocks for solid-phase peptide synthesis of spin labeled peptides for electron paramagnetic resonance and dynamic nuclear polarization applications. ChemPhysChem 20(11), 1475–1487 (2019).
Link, A. J., Mock, M. L. & Tirrell, D. A. Non-canonical amino acids in protein engineering. Curr. Opin. Biotechnol. 14(6), 603–609 (2003).
Kiryutin, A. S. et al. Ultrafast single-scan 2D NMR spectroscopic detection of a PHIP-hyperpolarized protease inhibitor. Chem. Eur. J. 25(16), 4025–4030 (2019).
Ratajczyk, T. et al. Magnetic resonance signal amplification by reversible exchange of selective PyFALGEA oligopeptide ligands towards epidermal growth factor receptors. ChemBioChem https://doi.org/10.1002/cbic.202000711 (2020).
Pravdivtsev, A. N., Buntkowsky, G., Duckett, S. B., Koptyug, I. V. & Hövener, J.-B. Parahydrogen-induced polarization of amino acids. Angew. Chem. Int. Ed. 60, 23496–23507 (2021).
Sletten, E. M. & Bertozzi, C. R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 48(38), 6974–6998 (2009).
Ban, H., Gavrilyuk, J. & Barbas, C. F. III. Tyrosine bioconjugation through aqueous ene-type reactions: A click-like reaction for tyrosine. J. Am. Chem. Soc. 132(5), 1523–1525 (2010).
Bauer, D. M., Ahmed, I., Vigovskaya, A. & Fruk, L. Clickable tyrosine binding bifunctional linkers for preparation of DNA–protein conjugates. Bioconjug. Chem. 24(6), 1094–1101 (2013).
MacDonald, J. I., Munch, H. K., Moore, T. & Francis, M. B. One-step site-specific modification of native proteins with 2-pyridinecarboxyaldehydes. Nat. Chem. Biol. 11(5), 326–331 (2015).
Obermeyer, A. C., Jarman, J. B. & Francis, M. B. N-terminal modification of proteins with o-aminophenols. J. Am. Chem. Soc. 136(27), 9572–9579 (2014).
Shen, B.-Q. et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 30(2), 184 (2012).
Brocchini, S. et al. PEGylation of native disulfide bonds in proteins. Nat. Protoc. 1(5), 2241–2252 (2006).
Liberatore, F. A. et al. Site-directed chemical modification and crosslinking of a monoclonal antibody using equilibrium transfer alkylating crosslink reagents. Bioconjug. Chem. 1(1), 36–50 (1990).
Godwin, A. et al. Molecular dynamics simulations of proteins with chemically modified disulfide bonds. Theor. Chem. Acc. 117(2), 259–265 (2007).
Wang, T. et al. A disulfide intercalator toolbox for the site-directed modification of polypeptides. Chem. Eur. J. 21(1), 228–238 (2015).
Mary Ann, M., Elda, E. S., Alice, W., Carrie, P.-S. & John, C. P. Disintegrins. Curr. Drug Targets Cardiovasc. Hematol. Disord. 4(4), 327–355 (2004).
Phillips, D. R. & Scarborough, R. M. Clinical pharmacology of eptifibatide. Am. J. Cardiol. 80(4), 11B-20B (1997).
Scarborough, R. M. Development of eptifibatide. Am. Heart J. 138(6), 1093–1104 (1999).
Zeymer, U. & Wienbergen, H. A review of clinical trials with eptifibatide in cardiology. Cardiovasc. Drug Rev. 25(4), 301–315 (2007).
Hashemzadeh, M., Furukawa, M., Goldsberry, S. & Movahed, M. R. Chemical structures and mode of action of intravenous glycoprotein IIb/IIIa receptor blockers: A review. Exp. Clin. Cardiol. 13(4), 192–197 (2008).
Schror, K. & Weber, A. A. Comparative pharmacology of GP IIb/IIIa antagonists. J. Thromb. Thrombolysis 15(2), 71–80 (2003).
Herr, K. et al. A novel strategy for site selective spin-labeling to investigate bioactive entities by DNP and EPR spectroscopy. Sci. Rep. 11(1), 13714 (2021).
McDowall, L. & Stenzel, M. H. Disulfide bridge based conjugation of peptides to RAFT polymers. Polym. Chem. 5(5), 1772–1781 (2014).
Kiryutin, A. S. et al. Parahydrogen allows ultrasensitive indirect NMR detection of catalytic hydrogen complexes. J. Phys. Chem. C 121(18), 9879–9888 (2017).
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft DFG under contract Bu-911/22-2 is gratefully acknowledged.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
M.F. and K.H. performed the syntheses and the chemical characterization. F.T. and S.K: and L.W. performed the NMR and the PHIP enhanced NMR measurements. M.R. and M.B. surpervised the organic syntheses. G.B. conceived the project and supervised the NMR and PHIP experiments. All authors contributed to the writing and the preparation of the figures. All authors reviewed the manuscript and the revision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fleckenstein, M., Herr, K., Theiß, F. et al. A disintegrin derivative as a case study for PHIP labeling of disulfide bridged biomolecules. Sci Rep 12, 2337 (2022). https://doi.org/10.1038/s41598-022-06327-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-06327-z
This article is cited by
-
Parahydrogen-induced polarization allows 2000-fold signal enhancement in biologically active derivatives of the peptide-based drug octreotide
Scientific Reports (2023)
-
Parahydrogen-induced polarization enables the single-scan NMR detection of a 236 kDa biopolymer at nanomolar concentrations
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
