
Abstract
A hallmark of ribosomal RNA (rRNA) are 2′-O-methyl groups that are introduced sequence specifically by box C/D small nucleolar RNAs (snoRNAs) in ribonucleoprotein particles. Most data on this chemical modification and its impact on RNA folding and stability are derived from organisms of the Opisthokonta supergroup. Using bioinformatics and RNA-seq data, we identify 30 novel box C/D snoRNAs in Dictyostelium discoideum, many of which are differentially expressed during the multicellular development of the amoeba. By applying RiboMeth-seq, we find 49 positions in the 17S and 26S rRNA 2′-O-methylated. Several of these nucleotides are substoichiometrically modified, with one displaying dynamic modification levels during development. Using homology-based models for the D. discoideum rRNA secondary structures, we localize many modified nucleotides in the vicinity of the ribosomal A, P and E sites. For most modified positions, a guiding box C/D snoRNA could be identified, allowing to determine idiosyncratic features of the snoRNA/rRNA interactions in the amoeba. Our data from D. discoideum represents the first evidence for ribosome heterogeneity in the Amoebozoa supergroup, allowing to suggest that it is a common feature of all eukaryotes.
Similar content being viewed by others

Introduction
Early on, the peptidyltransferase reaction of the ribosome was shown to be resistant to protein degradative treatment1. This first indication for rRNA as the catalytic entity in protein biosynthesis, rather than proteins, was subsequently confirmed by ground-breaking and highly decorated crystallographic work2,3,4. Maturation of ribosomes is amongst the most complex cellular processes and requires about 200 facilitating proteins, as reviewed recently5. Amongst many other processes, the introduction of post-transcriptional, covalent modifications in rRNA is of utmost importance for ribosome biogenesis and function, as summarized in Ref.6. The most prominent nucleotide modifications in rRNA are 2′-O-ribose methylation (2′-O-Me) and pseudouridylation (Ψ) that are introduced site-specifically. These modifications are thought to be important for RNA folding, ribosome stability and translational fidelity7,8,9. In recent years, a specialization of ribosomes in response to environmental changes and/or developmental processes has been suggested, with substoichiometric chemical modifications being implicated as a major source of ribosome heterogeneity6,10. As such, examples for fractional rRNA modifications are found in various species, including Saccharomyces cerevisiae, where 18 positions are modified in less than 85% of the ribosomal population11, and also approximately a third of the 2′-O-Me positions in rRNA of Homo sapiens are found hypomodified12. Recently, altered 2′-O-Me levels were also discovered during the development of Danio rerio13. Functionally, ribosome heterogeneity has been proposed to constitute a fine-tuning mechanism for translational activity of an unknown subset of mRNAs14,15.
Ribose methylations and pseudouridylations in eukaryotes are introduced in rRNA site-specifically by small nucleolar ribonucleoprotein particles (snoRNPs), as summarized recently6,10,16. They come in two flavors: H/ACA snoRNPs catalyze the conversion of uridine to Ψ, while box C/D snoRNPs introduce methyl groups at the 2′-hydroxyl of ribose residues17,18. For each class of snoRNPs, a conserved and distinct set of four proteins form the catalytic complex, of which dyskerin in the H/ACA snoRNPs isomerizes uridine19, while fibrillarin in box C/D snoRNPs acts as the methyltransferase on 2′-hydroxyl groups20. The rRNA target positions are defined by the individual snoRNA components of the RNPs. For both classes, specific base pairing patterns define the nucleotide to be modified. In the following, we briefly summarize the interaction of box C/D snoRNAs with rRNA and refer for H/ACA snoRNAs to two recent excellent reviews6,10. Box C/D snoRNAs possess conserved box C (5′-RUGAUGA-3′) and box D (5′-CUGA-3′) motifs that are essential for their structure, function and biogenesis (Fig. 1), as well as less conserved box C′ and box D′ motifs21,22,23. Nucleotides of the box C and box D motifs interact with each other, forming a kink-turn, and similar, but weaker interactions may also occur between nucleotides of the box C′ and box D′ motifs. The intramolecular base pairs of box C and box D motifs are essential for snoRNA processing and the snoRNP structure. Immediately upstream of the D and/or D′ box are antisense elements that base pair with the rRNA target and thereby direct fibrillarin to its site of action. In an archaeal model for box C/D sRNPs, the substrate-binding channel of the complex accommodates 10 base pairs of the snoRNA/rRNA duplex24. Methylation occurs at the 2′-hydroxyl of the nucleotide base paired to the 5th nucleotide upstream of the D or D′ box in an S-adenosylmethionine (SAM)-dependent reaction20,25,26. Since both, the D and D′ box can potentially guide 2′-O-Me with their antisense elements, these snoRNAs can guide in principle two sites in rRNA. Furthermore, a reorientation of the box C/D snoRNA inside the snoRNP complex might lead to alternative base pairing, increasing the target number of a given snoRNA even further27,28.

Features of box C/D snoRNAs. Conserved residues of boxes C and D are shown. They interact to form a functionally important k-turn by means of trans Hoogsteen/sugar-edge A•G base pairs, shown in conventional Leontis–Westhof symbols75. The guiding sequences (“antisense element”; green) is upstream of the D box with methylation occurring in rRNA at the position pairing to the 5th nucleotide upstream of the D box (indicated with a red asterisk). Base pairing with rRNA (beige) is schematically shown. Boxes C′ and D′ are usually less well conserved (indicated by small lettering). The separate antisense sequence upstream of box D′, allows guidance to a further methylation site.
While a substantial amount of data on rRNA modifications by snoRNPs is available from organisms of the evolutionary supergroups of Opisthokonta and Archaeplastida11,12,13,29,30, information for the Amoebozoa supergroup remains scarce. Dictyostelium discoideum is a well-established model organism and arguably the best studied organism of the Amoebozoa31. A wide spectrum of experimental tools has been established for the amoeba32, and these are frequently used to study mechanisms governing cell motility, autophagy, social evolution (reviewed in Ref.33), mobile genetic elements34, and their domestication by the RNA interference (RNAi) machinery35,36,37. D. discoideum single cells usually propagate by mitotic division; upon starvation, however, a complex developmental process is initiated, in which about 100,000 cells aggregate to form a multicellular mobile slug after 16 h, resulting in a fruiting body within 24 h38. This allows to study fundamental developmental processes in the amoeba.
In most metazoans, the genes for the rRNAs are organized in rDNA clusters, an arrangement that is thought to facilitate efficient rRNA transcription. Such rDNA clusters exist also in D. discoideum, however, they are not encoded in chromosomes but localized on extrachromosomal elements39,40. Each nucleus contains about 100 copies of these elements of 88 kb, that each feature two rRNA transcription units organized as palindromes41. A first model for the processing of rRNAs from the primary 37S transcript in the amoeba has been proposed, and sequences of the mature rRNAs in D. discoideum were determined experimentally42. Earlier work has identified several box C/D snoRNAs in D. discoideum, and verified a function in rRNA 2′-O-methylation43. The study employed a shotgun cloning approach to identify novel non-coding RNAs (ncRNAs) in D. discoideum. This work led also to the discovery of the functionally important Class I RNAs, which recently were shown to be involved in the evolution of multicellularity in Dictyostelia44. Next to these, sequencing of cloned fragments yielded 17 box C/D and one box H/ACA snoRNA(s) in D. discoideum, besides other ncRNAs.
Owing to these observations, we set out here to elucidate the global 2′-O-Me pattern(s) in the amoeba. Employing RiboMeth-seq (RMS)11, we created a comprehensive map of the 2′-O-Me sites in Dictyostelium’s 17S and 26S rRNAs. We thereby positioned methylated residues in functional important parts of the rRNAs, for which we have determined sequence homology-based models of their secondary structures. Further, we also have identified bioinformatically and validated experimentally additional box C/D snoRNAs with which we can at large explain methylated rRNA positions in the amoeba.
Methods
Cell culture and growth conditions of D. discoideum
The D. discoideum strains AX245 and ∆drnB46 were cultivated in HL5 medium containing 50 μg/mL ampicillin, 250 ng/mL amphotericin, 500 U/mL penicillin/streptomycin at 22 °C in shaking suspension.
Filter development of D. discoideum
Filter development was performed using 5 × 108 of axenically grown D. discoideum cells pelleted for 5 min at 500×g and washed three times with Sørensen buffer [2 mM Na2HPO4, 15 mM KH2PO4, (pH 6.7)]. The pellet was resuspended in Sørensen buffer and transferred in a 6-cm dish containing two layers of Whatman® paper topped off with a nitrocellulose membrane. After 16 h, the slugs were harvested by washing the nitrocellulose membrane with Sørensen buffer and spun down by centrifugation at 500×g for 5 min. RNA was isolated from the resulting pellet.
Resources for RNA-seq datasets
RNA-seq datasets of AX2 and ∆drnB in axenic growth and slug stage of development were acquired from the sequence read archive (https://www.ncbi.nlm.nih.gov/sra) and used for RNA-seq validation of box C/D snoRNA candidates and expression analysis. Accession numbers of the utilized data sets can be found in Supplementary Table S1. Sample preparation and sequencing was described in Liao et al.47.
In silico identification and validation of box C/D snoRNA candidates
The genomic sequences were retrieved from Dictybase (www.dictybase.org) and the sequences of the 17S and 26S rRNA42 were retrieved from GenBank (www.ncbi.nlm.nih.gov/genbank/; Accession numbers are listed in Supplementary Table S3). The identification of box C/D snoRNA candidates in D. discoideum was performed using snoScan v. 0.9.148 with threshold settings (− C 0 − D 0 − X 0) disabled. Candidates with a combined box C and box D score higher than 9 and a box C-D distance between 50 and 100 nt were selected for RNA-seq validation. Sequencing reads from the axenic AX2 dataset were aligned to the genomic coordinates ± 150 bp using bowtie v. 1.2.349 allowing for one mismatch. Box C/D snoRNAs were considered validated, if reads specifically matched the predicted loci and read coverage calculated with BEDTools coverage v. 2.29.250 indicated a distinct 5′ end, yielding an expression score of 15. Box C/D snoRNA candidates lacking expression or a distinct 5′ end received a penalty of − 15. All scores were combined into a classifier score containing C/D box scores, terminal stem score, Box C–D distance score, and the expression score (Supplementary Fig. S1). If a total classifier score of 29 or higher was achieved, the candidate was considered to be an expressed bona fide box C/D snoRNA and kept for further analyses and assignment to the predicted ribosomal 2′-O-Me pattern.
RNA-seq analysis of box C/D snoRNAs in development
Reads were aligned using bowtie v. 1.2.349 allowing for one mismatch and counted with featureCounts v. 2.0.051. Between-sample normalization was done by DEseq2 v. 1.29.652. P-values were adjusted using the false discovery rate (FDR) method. Principal component analysis was performed on DESeq2-normalized reads using R-stats v. 4.0.0 and visualized with R-ggplot2 v. 3.3.2. The heatmap of log2 fold-change of box C/D snoRNAs was generated using ComplexHeatmap v. 2.5.353.
Radiolabeling of DNA oligonucleotides
DNA oligonucleotides were purchased from Merck and are listed in Supplementary Table S2. For primer extension and northern blot analysis, 10 pmol oligonucleotide was 5′-end-labeled by incubation with 10 U T4 polynucleotide kinase (Fermentas) for 30 min at 37 °C in 50 mM Tris–HCl (pH 7.6), 10 mM MgCl2, 5 mM DTT, 100 µM spermidine, and 0.37 MBq [γ-32P]-ATP. The reaction was stopped at 80 °C for 5 min, the radiolabeled oligonucleotides were phenol/chloroform-extracted and purified using a Sephadex G50 (GE Healthcare) column.
RNA extraction
RNA was isolated from 2 × 107 axenically grown D. discoideum cells washed with pre-cooled Sørensen buffer [2 mM Na2HPO4, 15 mM KH2PO4, (pH 6.7)]. Cells were pelleted and resuspended in TRIzol reagent (Invitrogen) containing 10 mM EDTA (pH 8.0). RNA was extracted according to the manufacturer’s instructions. RNA concentration was determined spectrophotometrically.
Primer extension
For primer extension, a box C/D snoRNA-specific 5′-radiolabeled oligonucleotide was annealed to 4 µg RNA at 65 °C for 5 min and cooled for at least 1 min on ice. Upon annealing, 1× SuperScript IV buffer (ThermoFisher Scientific, Inc.), 1 mM dNTP mix, 5 µM DTT, 40 U RiboLock RNase Inhibitor (ThermoFisher Scientific, Inc.) and 50 U SuperScript IV Reverse Transcriptase (ThermoFisher Scientific, Inc.) were added. The reaction was incubated at 55 °C for 30 min and stopped at 85 °C for 5 min. Products were phenol/chloroform-extracted, recovered by ethanol precipitation and separated on a polyacrylamide gel (12% PAA, 20 mM MOPS, pH 7.0, 7 M Urea) for 3 h at 25 mA.
Northern Blot analysis
For the detection of snoRNAs, 20 μg of total RNA was separated by gel electrophoresis on a 12% polyacrylamide gel (20 mm MOPS, pH 7.0, 7 M urea). The RNA was transferred to a nylon membrane (Amersham Biosciences HybondTM-NX) by electroblotting for 30 min at 20 V. Blotted RNA was crosslinked by 0.5 J/cm2 UV illumination. Blots were probed overnight with 5′-radiolabelled DNA oligonucleotides in Church buffer (1 mM EDTA, 7% (w/v) SDS, 1% (w/v) BSA in 0.5 M Pi buffer, pH 7.2). Probed Blots were washed two times for 20 min with each 2×, 1×, and 0.5× SSC buffer (20× SSC: 3 M NaCl, 0.3 M trisodium citrate, pH 7.0). Hybridization with an oligonucleotide complementary to tRNAUUC was used as a loading control.
RiboMeth-seq
The RiboMeth-seq analysis was performed in triplicates with barcoded adapters according to previously described protocols11,54. In brief, 10 µg RNA from each sample was degraded by alkaline for 6 min at 90 °C and the 20–40 nt fraction was excised and purified from a 10% urea polyacrylamide gel. A modified Arabidopsis tRNA ligase was used to ligate adaptors to the library fragments, and sequencing was carried out on the Ion Proton sequencing platform. The reads were mapped to rRNAs (GenBank: FR733593.1, FR733594.1, FR733597.1, FR733595.1) using Bowtie255 and scored for read-end counts. RMS scores representing “fraction methylated” were calculated as described previously (“score C”) in Ref.11 and barcode correction was applied when necessary56. The commercial RNA oligonucleotides used as 3′adaptors were found to be slightly heterogeneous in length, which can cause a fractional shift in the 3′-read-end count, if the 3′-library fragment nucleotide is identical to the expected 5′-end of the oligonucleotide. As the experiments were made in triplicate with barcodes carrying different 5′-ends, such errors were easily detected, and a manual correction was made at a few sites to counter the effect by excluding the 3′-read-end counts from the analysis.
Prediction of rRNA secondary structure
To locate the predicted 2′-O-Me sites in the mature rRNA, we predicted the secondary structure by comparative analysis with the LSU and SSU rRNAs of A. thaliana, C. elegans, H. sapiens, and D. melanogaster. For that purpose, we retrieved the corresponding SSU and LSU rRNA sequences for these organisms from GenBank (Supplementary Table S3). We aligned the sequences to the 17S and 26S rRNA of D. discoideum using MUSCLE57 in the ClustalW output format and inferred the secondary structure by homology manually. The resulting secondary structure diagrams were drawn using RNAviz v. 2.0.358. Due to the high conservation of the ribosomal core elements and experimental evidence of the tRNA site locations in other species, the nucleotides predicted in the A, P, and E sites of D. discoideum were inferred by sequence homology.
Mapping of predicted snoRNA candidates to the rRNA 2′-O-Me pattern
Mapping of box C/D snoRNAs to the predicted 2′-O-Me sites was performed using RNAhybrid59. 10 nt upstream and downstream of the 2′-O-Me sites were used as target sites against the full-length sequences of the box C/D snoRNAs. Selection of the likely correct duplex was achieved using the following criteria: (I) 2′-O-Me site is located at the 5th base paired nucleotide upstream of a D or D′ box and (II) a box C/D snoRNA/rRNA duplex length of minimum 7 bp with (III) a maximum of 1 mismatch. Conservation of box C and box D motifs was visualized using WebLogo v. 3.760. Calculation of the predicted duplex’ minimum free energy (MFE) in kcal/mol was performed using RNAduplex v. 2.4.1561. Box C/D snoRNAs that were not mapped to any predicted 2′-O-Me sites but were validated by RNA-seq, were classified as orphans.
Results
Identification and validation of 30 novel box C/D snoRNAs in the genome of D. discoideum
The number of 17 box C/D snoRNAs (Fig. 1) identified in D. discoideum prior to this study is relatively small for normally-sized rRNA sequences42 compared to orthologous RNAs found in other species62. Therefore, we set out here to search for additional box C/D snoRNAs in the amoeba. To this end, we employed an in silico-approach for the identification of novel box C/D snoRNAs by using the probabilistic model-dependent search tool snoScan48, which we combined with RNA-seq analyses. The sizes of previously described box C/D snoRNAs of D. discoideum range between 66 and 113 nt, with box C-D distances between 50 and 97 nt43. We searched accordingly first with snoScan in the genome of D. discoideum (available at www.dictybase.org) for sequences containing box C and box D motifs with a box C-D distance between 50 to 100 nt. Since inverted repeats at the 5′ and 3′ ends were not observed before43, we did not pre-require the presence of a terminal stem structure for a classification as a bona fide box C/D snoRNA. Using these settings, we identified 577 box C/D snoRNA candidates in the genome of D. discoideum (data not shown), including the set described before43. To refine our search, we next addressed the expression of these candidates in publicly available RNA-seq data of the axenic AX2 wild type strain, deposited in duplicate47 at the sequence read archive (https://www.ncbi.nlm.nih.gov/sra). Specifically, we mapped reads to the genomic loci of the candidates and selected only those sequences that exceeded a read count of 100 and were not part of a longer transcript, as indicated by a distinct 5′ end. Both, the lack of specific RNA-seq reads or of a distinct 5′ end, were penalized (‘expression score’, Supplementary Fig. S1). Sequences scoring 29 or higher in the classifier score (Supplementary Table S4) were classified as bona fide box C/D snoRNAs. This routine allowed us to identify 47 box C/D snoRNAs in D. discoideum, of which 30 are novel43. For the amoeba, box C/D snoRNA gene clusters have been described43 and primary transcripts of such clusters are often processed by an RNase III before exonucleolytic processing can occur63,64,65. We therefore included the knock-out strain of the nucleolar RNase III DrnB46,66,67 in the following analyses. Initially, we carried out primer extension experiments on RNA isolated from axenically grown or developed AX2 and ∆drnB cells. This resulted for the majority of the snoRNAs in a single signal at the predicted size (Supplementary Fig. S2), indicating that they have homogeneous 5′-ends. Their genomic locations are listed in Supplementary Table S5, allowing to characterize next the properties of box C/D snoRNA genes in D. discoideum.
The box C/D snoRNA genes in D. discoideum
Usually, box C/D snoRNAs are encoded in intergenic regions or as part of introns in protein-coding genes, and in either set-up, they can be generated as mono- or poly-cistronic transcriptional units62. Aspegren et al.43 predicted four bi-cistronic transcriptional units of snoRNAs in D. discoideum and confirmed expression for several of them using RT-PCR. An analysis of the genomic location of the genes for our set of 47 box C/D snoRNAs revealed five additional clusters containing two box C/D snoRNAs and two clusters comprised of three box C/D snoRNAs (Supplementary Fig. S3). The genes for these box C/D snoRNAs appear equally spaced in the clusters. All box C/D snoRNA genes, in clusters or not, were found in intergenic regions, except CD38, which is encoded in an intron (Supplementary Table S5). The box C/D snoRNAs with a predicted target (see below) are encoded on all chromosomes without a noticeable pattern, but we observed that the majority of box C/D snoRNAs without a target are encoded on chromosome 4. The biological significance of this, if any, remains to be elucidated, and we cannot exclude that it is a random localization. Next, we set out to investigate the 2′-O-Me patterns in D. discoideum’s rRNAs, that would be guided by the encoded box C/D snoRNAs.
Dictyostelium discoideum 17S and 26S rRNAs have 49 high-confidence 2′-O-Me sites
To address 2′-O-Me in the 17S and 26S rRNA of D. discoideum, we employed RMS, a method introduced on yeast rRNA11, and subsequently used in several other organisms12,13,30. In brief, RMS is a next-gen sequencing-based method that relies on the cleavage-resistance of 2′-O-methylated nucleotides under alkaline conditions, resulting in an underrepresentation of read ends in fragmented RNA. The results are expressed as RMS scores, which represent the fraction of modified molecules at a given position. The method yields methylation stoichiometry comparable to RP-HPLC68. We generally considered sites with an RMS score > 0.75 as high-confidence 2′-O-Me sites.
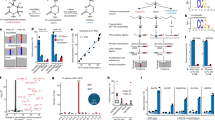
To investigate the global 2′-O-Me landscape in wild type Dictyostelium, we initially determined the RMS scores of rRNA isolated from axenic AX2 cells. During these experiments, we realized that one nucleotide (C784) was missing in the 17S reference sequence42, and its presence was independently confirmed by sequencing of a PCR product on total DNA. Using the criteria outlined above, we determined in total 17 and 32 positions with a 2′-O-Me moiety on the 17S rRNA and the 26S rRNA, respectively (Fig. 2A). Of these high-confidence sites, the majority appeared to be fully methylated. In axenically-grown AX2 cells, we identified 2 hypomethylated positions each in the 17S and 26S rRNAs. This indicates, to our knowledge for the first time, heterogeneity of the ribosome population in D. discoideum. Heterogeneity in rRNA modifications had been, however, reported previously for mouse, human, thale cress, and zebrafish12,13,29,30,69,70. In these studies, differences in the ribosome 2′-O-Me patterns between cultured cells and differentiated tissues, or during development have been described. Since D. discoideum undergoes development upon starvation, we set out next to elucidate any changes of the 2′-O-Me pattern in rRNAs of the slug stage of development in the AX2 wild type. The fractionally methylated positions in axenically-grown wild type cells were also substoichiometrically methylated during development, while the RMS score of most 2′-O-Me sites remained unchanged (Fig. 2A).

RiboMeth-seq analysis of the 17S and 26S in D. discoideum. RMS scores at 2′-O-Me sites on the 17S and 26S rRNA in axenic growth and development of AX2 (A) and ∆drnB (B) cells (n = 3).
If the nucleolar RNase III DrnB46,66,67 is involved in box C/D snoRNA maturation, a knockout strain of its gene might display altered RMS scores, which we investigated next. At large, the 2′-O-Me pattern of the AX2 strain, however, was also observed for axenic growth and development of the ∆drnB strain (Fig. 2B and Supplementary Fig. S4). Only one position, 26S-Am1463, exhibited a noticeable difference between the axenically-grown AX2 and ∆drnB strains (Supplementary Fig. S4C). This indicates that any effect that DrnB might have on the processing of box C/D snoRNA precursors does not manifest substantially in altered 2′-O-Me patterns. Similarly, that position 26S-Am1463 displayed different RMS scores between axenic growth and the slug stage in both, the AX2 and ∆drnB strains (Supplementary Fig. S4). The four 2′-O-methylated residues that we found either fractionally modified or changed in development had no orthologous modified sites in S. cerevisiae, H. sapiens, and A. thaliana (Table 1, and see below).
Secondary structure models for the small and large ribosomal subunits in Amoebae
As methylated rRNA positions are required for folding and structural stabilization of rRNAs, thereby contributing to ribosome function8, it was of interest to localize the 2′-O-methylated positions in the context of the rRNA structure of D. discoideum. A partial structure of the large ribosomal subunit of D. discoideum has been published recently71, but no high-resolution structural data is available for complete ribosomes from any species of the Amoebozoa. To obtain a model for the rRNA secondary structures, we employed homology modelling using sequences of species from the evolutionary supergroups of Opisthokonta and Archaeplastida[31]. In brief, we aligned the rRNAs from the amoeba with the corresponding small and large subunits’ (SSU and LSU, respectively) rRNA sequences from A. thaliana, Caenorhabditis elegans, Drosophila melanogaster, and H. sapiens (Supplementary Table S3). The inferred secondary structure models of the 17S and 26S (with the 5.8S) rRNAs of D. discoideum are shown in Figs. 3 and 4, respectively, and include the 2′-O-methylated positions.
Central parts of ribosomes from different species are structurally highly conserved and variation appears restricted to peripheral regions and the so-called expansion segments (ES)72, which often harbor species-specific sequences. This is exactly what the models for the amoebal rRNA structures display (Figs. 3 and 4). This holds particularly true for the conserved regions involved in the formation of A, P and E sites. Not surprisingly, the ES of D. discoideum, which are not covered in the aforementioned structure71, exhibited significant differences as compared to the ES in other species (exemplified for H. sapiens; Supplementary Table S6).
About half of the 2′-O methylated positions were found in the vicinity of nucleotides residing in the A, P and E sites, and the other half in other regions of the rRNAs (Figs. 3 and 4). These latter positions localized frequently to formally single stranded regions, or to nucleotides at the very beginning of helical stems. When comparing the 2′-O-Me patterns in wild type D. discoideum to those in S. cerevisiae, H. sapiens, and A. thaliana, we found 28 of the 2′-O-Me sites conserved in at least one of these organisms, and therefore, the other 21 sites are specific to D. discoideum (Table 1). Only one of these positions, Gm711 in the 26S rRNA, was found in an ES (Fig. 4), indicating that 2′-O-Me is largely restricted to the core of the ribosome in D. discoideum. Noteworthy, five of the 13 specific 2′-O-Me sites on the 26S rRNA were locating in domain 0, which has been shown in other species to coordinate folding of all other domains of the LSU rRNA, including the peptidyl transferase center (PTC)73.

Secondary structure of the 17S rRNA of D. discoideum with 2′-O-Me sites. The secondary structure of the 17S rRNA was inferred by homology and drawn using RNAviz (v. 2.0.3). The 2′-O-methylated nucleotides as identified by RiboMeth-seq are marked with an arrow and ‘M’ (red). Nucleotides located in the A, P, and E sites of the ribosome are indicated in pink. Helices (hx) are named to convention and expansion segments (ESx) are labeled with x: natural number.


Secondary structure of the 26S rRNA of D. discoideum with 2′-O-Me sites. The secondary structure of the 26S rRNA was inferred by homology and drawn using RNAviz (v. 2.0.3). The 2′-O-methylated nucleotides as identified by RiboMeth-seq are marked with an arrow and ‘M’ (red). Nucleotides located in the A, P, and E sites of the ribosome are indicated in pink. Due to the size of the 26S rRNA, the figure is split into the 5′ half and 3′ half. The predicted interaction with the 5.8S rRNA is shown at the 5′ end. Helices (Hx) are named to convention and expansion segments (ESx) are labeled.

Features of CD RNAs in D. discoideum. (A) Examples of CD RNAs guiding 2′-O-Me at one or two rRNA positions. Single (top) and double (bottom) usage of D boxes of selected CD RNAs guiding positions in the 17S (left) and the 26S rRNA (right). Shown are CD RNA sequences (grey) with nucleotides involved in the formation of the k-turn (black). The guided part of the rRNA is shown in orange with the methylated residue highlighted in red. Intra- and intermolecular interactions are denoted for Watson–Crick (|) and G/U base pairing (*), as are the A/G and U/U base pairs (•) involved in the formation of the k-turn. (B) Conservation of C, C′, D and D′ box sequences shown with WebLogo60. (C) Distribution of CD RNAs using box D, D′ or both. Duplex lengths (in bp; D) and minimal free energies ΔG (in kcal/mol; E) of the interaction between CD RNA and the guided rRNA position.
The majority of 2′-O-Me sites in D. discoideum can be associated to box C/D snoRNAs
To identify snoRNA guides for the 2′-O-methylated sites, we employed next RNAhybrid, since snoScan alone was not able to predict all targets for our set of box C/D snoRNAs (Fig. 2B). This resulted in the prediction of 46/49 2′-O-Me sites with at least one, occasionally two box C/D snoRNA guides (Table 1). The snoRNAs guiding 2′-O methylation at these rRNA sites were named CDx (x = natural numbers; Supplementary Table S5). For the remaining 9 box C/D snoRNAs, we could not assign a 2′-O-Me site in either rRNA, and therefore we classified these sequences as orphans, and named them accordingly ORx (Supplementary Table S5). Seven of the CD RNAs can make use of both their D and D′ boxes to guide 2′-O-Me in one or both rRNAs (Tables 1 and Supplementary Table S7). For most positions targeted by these CD RNAs, no alternative guides were found. Rather, CD1 and CD19 have two targets each for their D′ boxes, additional to the targets of their D boxes (Supplementary Table S7). The majority of CD RNAs, however, is predicted to employ either its D or D′ box. Figure 5A displays examples for single and double usage of D boxes, shown exemplarily for one case each in the 17S and 26S rRNA. The predicted bimolecular interactions of the CD RNAs with their rRNA targets are shown in Supplementary Figs. S2 and S3 for D and D′ box guides, respectively. Earlier work had shown the functionality of box C/D snoRNA in guiding 2′-O-Me in D. discoideum by primer extension at a low dNTP concentration43.

Analysis of box C/D snoRNA expression in axenic growth and development of the AX2 and ∆drnB strains. (A) Principal component analysis (PCA) of data from RNA-seq on the indicated strains and conditions. Volcano plots of box C/D snoRNA expression changes in the slug stage of AX2 (B) and ∆drnB (C). Significantly up- or downregulated box C/D snoRNAs are labelled and colored green.
Features of box C/D snoRNAs and their interactions with rRNA
The box C/D snoRNAs in Dictyostelium are between 66 and 113 nt in length, with an average GC content of 32.2% and box C–D distances between 50 and 97 nt (Supplementary Table S5). The terminal stem often found in box C/D snoRNAs in other species (Fig. 1), is predicted by snoScan only in 25 of the 47 box C/D snoRNAs of D. discoideum (indicated with a positive TS score in Supplementary Table S4). In contrast, the box C and box D sequences forming the k-turn motif are highly conserved (Fig. 5B); in particular, the GA dinucleotides forming trans Hoogsteen/sugar-edge A•G base pairs are present in all CD RNAs selected by the described criteria (but not in all OR RNAs, see Supplementary Table S4). Furthermore, we found that almost all CD RNAs abide to the box D consensus sequence CUGA, with a small fraction of snoRNAs featuring an AUGA instead (Fig. 5B and Supplementary Table S4). Compared to these motifs, the box C′ and box D′ sequences show considerably more variation in Dictyostelium (Fig. 5B). Despite this, the majority of methylated positions is predicted to be guided by the D′ boxes of individual CD RNAs (Fig. 5C), similar to observations made for the human box C/D snoRNAs12. The lengths of the CD RNA/rRNA duplexes distributed around 11 bp within a range of 7–15 bp, with average minimal free energies (MFE) of − 13.9 kcal/mol (Fig. 5D,E). In these predicted CD RNA/rRNA interactions, we observe the frequent occurrence of G*U base pairs74, occasionally A/C base pairs75, and a single G/A mismatch (Supplementary Figs. S5 and S6). Only for the CD16/17S-G1589 duplex, we noticed that apparently the + 6 position is targeting, rather than the consensus + 5 position, as has also been observed before in other species13.
Box C/D snoRNAs accumulate differentially during development of D. discoideum
Our primer extension experiments (Supplementary Fig. S2) indicated no 5′-end size heterogeneity of box C/D snoRNAs in D. discoideum. In absence of an internal control, a correlation between band intensity and expression levels is difficult. Furthermore, we could not obtain a product for several snoRNAs, despite the use of several distinct primers in these experiments. Therefore, to obtain a more complete view on box C/D snoRNA accumulation, we retrieved RNA-seq datasets for AX2 and ∆drnB in axenic growth and in the slug stage of development from NCBI, which were originally deposited by Liao et al.47. As a first step, we performed a principal component analysis (PCA) of box C/D snoRNA expression on two biological replicates for each time point per strain. The analysis revealed global changes of box C/D snoRNA abundance in the development of the AX2 and ∆drnB strains (Fig. 6A), however, not between AX2 and ∆drnB. This is corroborated by comparative 2D plots of DESeq2-normalized reads of individual box C/D snoRNAs in the two strains and under the two growth conditions (Supplementary Fig. S7A). In a subsequent analysis of individual box C/D snoRNAs, we considered changes significant if an adjusted p-value < 0.05 and an at least 0.5-log2fold-change in RNA quantity was observed. Using these criteria, 22 box C/D snoRNAs were significantly up- or downregulated in the slug stage of development of AX2 (Fig. 6B,C and Supplementary Fig. S7B). In contrast to this and as seen before (Fig. 6A), we did not observe significant differences in the box C/D snoRNA between AX2 and ∆drnB except for OR9 and CD37, which were upregulated in the slug stage in ∆drnB, but not in AX2 (Fig. 6B,C and Supplementary Fig. S7B). For several box C/D snoRNAs we also performed Northern blot analyses (Supplementary Fig. S8) that confirmed at large the expression patterns seen by RNA-seq, in-line also with an earlier study employing Northern blotting on the 17 box C/D snoRNAs identified at the time43.
We wondered whether the changes that we observe in the 2′-O-Me patterns (Fig. 2) can be explained by differences in the accumulation of the guiding CD RNAs. This is clearly not the case, as a 2D plot of the DESeq2-normalized reads of CD RNAs against the RMS scores at all methylated sites revealed no correlation in axenic growth; rather, full and fractional methylation is observed independent of the CD RNAs’ abundance (Supplementary Fig. S7C). Furthermore, a 2D plot of the log2fold-change of the RMS score against the log2fold-change of CD RNA accumulation in the slug stage (Supplementary Fig. S7D) showed no differences. Thus, changes in the 2′-O-Me patterns can in general not be attributed to altered CD RNA amounts in the development of D. discoideum.
Discussion
Ribosome heterogeneity in Amoebozoa
In this study, we have investigated the 2′-O-Me landscape of D. discoideum’s rRNAs and associated box C/D snoRNAs. To our knowledge, this is the first comprehensive report on this topic for any species from the Amoebozoa, one of five eukaryotic evolutionary supergroups31. Using RMS11, we have identified 45 positions that are fully methylated in the rRNAs of the amoeba, and additionally 4 positions that exhibit a substoichiometric 2′-O-Me (Fig. 2 and Supplementary Fig. S4). This indicates that ribosome heterogeneity exists in Amoebozoa. Such variations in the chemical modification of nucleic acids making up the translation apparatus have been reported already for organisms from other evolutionary supergroups, in particular Opisthokonta11,12,13,15,30, but also in Archaeplastida29. With our data from a third evolutionary supergroup, the Amoebozoa, we suggest that ribosome heterogeneity represents a trait common to all eukaryotes.
Ribose methylation is thought to occur largely co-transcriptionally11,76. Thus, variation in the levels of this modification could be influenced by the rDNA organization. In D. discoideum, rRNAs are transcribed42 from extrachromosomal, palindromic elements39,40. Expression from extrachromosomal rDNA is rare, but described also, e.g., for D. rerio14. In the amoeba, clusters of the rDNA palindromes can condense into chromosome-like bodies41. This poses the question whether ribose methylation might be affected by limited accessibility for the snoRNPs to the nascent transcript. Our data indicates that the 2′-O-Me modification can be actually introduced equally well on rRNAs transcribed from extrachromosomal rDNA, as compared to chromosomally encoded transcripts.
A single 2′-O-methylated position, 26S-A1463, displayed altered RMS scores in the development of the amoeba and between the investigated strains (Fig. 2 and Supplementary Fig. S4). Such changes were also observed in the development of mouse30 and zebrafish13. Further, fractionally methylated sites in rRNA residues in cultured human cells became (close to) fully modified in differentiated tissues70. These aforementioned studies also all used RMS, as the preferred high-throughput analysis method of 2′-O-Me patterns, allowing for single nucleotide analysis in a quantitative manner, unlike alternative approaches. The advantages of RMS were also highlighted in a comparative study on rRNA from Trypanosoma brucei that further revealed 2′-O-Me patterns, which depended on the living conditions of the parasite77. Similar methodological advantages to RMS are also realized by the recently introduced and validated RiboMethSeq tool78,79 and the methylated positions reported here for the AX2 strain were at large confirmed independently using this method (Virginie Marchard and Yuri Motorin, personal communication).
For the majority of 2′-O-methylated rRNA positions, we have bioinformatically identified suitable CD RNAs (Fig. 1, Table 1). A subset of 17 such molecules had been reported earlier43, and we have added here additional 21 novel box C/D snoRNAs with a target in rRNAs, plus nine without. Previously, small non-coding RNAs in the amoeba were all called DdR-x (x = natural number), for Dictyostelium discoideum RNA43. With a functional association, we now have decided to rename the box C/D snoRNAs with an rRNA target to CDx (x = natural number), and those without to ORx RNA (for orphan).
A secondary structure model for the ribosomal RNA in D. discoideum
For the localization of the 2′-O-methylated positions, we propose, additionally to the partial Cryo-EM structure of the nascent ribosome, here a complete model for the secondary structure of the large rRNAs in the amoeba (Figs. 3 and 4). This is based on a homology alignment of rRNA sequences from organisms of two evolutionary supergroups, the Opisthokonta and Archaeplastida31. In the rRNA models for the Amoebozoan D. discoideum, about half of the 2′-O-methylated nucleotides are found close to the A, P and E sites of the ribosome. The remainder localize either in formally single stranded regions or at the very beginning of helical stems where they presumably fulfil a stabilizing function or support rRNA folding. Our models of the D. discoideum rRNAs are greatly supported by the previously introduced Cryo-EM structure of the nascent 60S subunit of Dictyostelium71, that features parts of the proposed structural elements of the 26S rRNA (Fig. 4), while the ESs are not covered in this structure.
In D. discoideum, the 2′-O-methylated positions U3254 and G3255 on the 26S rRNA are orthologous to the methylated sites U2921 and G2922 in S. cerevisiae (Table 1). In yeast, Gm2922 is highly important for the docking of transfer RNAs (tRNA) in the A-site via base pairing with C75 in their CCA-tail80. This suggests that Gm3255 might fulfill the same function in Dictyostelium. U3254 is likely modified by the CD25 RNP (see also below), however, a guide for Gm3255 is missing (Table 1). Intriguingly, position G2922 in S. cerevisiae is modified by the SAM-dependent methyltransferase Spb1, independent of a box C/D snoRNA guide80. Dictyostelium’s genome encodes the homologous fsjC gene (http://dictybase.org/gene/DDB_G0284945), and by analogy we hypothesize that its gene product might fulfil the same function as Spb1 in yeast. We can, however, not exclude that the CD25 RNP might also introduce that methylation by using its + 6 position, in analogy to two D. rerio snoRNPs that guide neighbouring positions in the rRNAs13.
The box C/D snoRNA genes
Box C/D snoRNAs in D. discoideum are encoded in intergenic regions or as part of introns of protein-coding genes, and in either set-up, they can be generated from mono- or poly-cistronic transcriptional units62. The selected set of 38 CD RNAs and their encoding genes display overall features similar to those seen in the original 17 sequences43. We found all box C/D snoRNAs in intergenic regions except for CD38, which is encoded in an intron of DDB_G0283293 (Supplementary Table S5).
Aspegren et al.43 had reported three bi-cistronic transcriptional units of snoRNAs being expressed in D. discoideum. We identified seven additional clusters with two or three box C/D snoRNA genes (Supplementary Fig. S3). One of the tri-cistronic clusters (on chromosome 5; Supplementary Fig. S3), had been reported to contain CD16 and CD5, but the central CD23 gene had not been noticed at the time43. A primary transcript of that cluster was not observed, but for the other three originally reported bi-cistrons, primary transcripts had been shown43. The former observation might be explicable if the CD16–CD23–CD5 tri-cistron consists of independent mono- or bicistronic transcription units. In summary, box C/D snoRNAs in D. discoideum appear predominantly encoded in intergenic regions, half each as mono- and poly-cistrons.
Not only in D. discoideum, but also in other species with three-digit intron sizes, like A. thaliana, S. cerevisiae or Schizosaccharomyces pombe are box C/D snoRNAs largely encoded by independent genes (Supplementary Table S8). By contrast, in eukaryotes with larger introns such as D. melanogaster or H. sapiens, snoRNAs are more frequently encoded in the intervening sequences of protein-coding genes81. Neither the global abundance of introns in protein-coding genes, nor their frequency/gene appear to be correlated with an “intronization” of the box C/D snoRNA genes (Supplementary Table S8). Instead, their number appears increased in the analyzed multicellular organisms compared to those that can exist as unicellular species. In the evolutionary tree, the Amoebozoa with D. discoideum branched off after the split of the Archaeplastida (A. thaliana) and before the separation of the Opisthokonta encompassing as diverse organisms as D. melanogaster, H. sapiens, S. cerevisiae, or S. pombe31. This current situation might be explained by snoRNA numbers and their intronization having evolved after the split of the individual supergroups to meet the needs of the individual organism.
Interactions of CD RNAs with rRNAs in D. discoideum
A productive interaction between a box C/D snoRNA and its target has been suggested to require 7–20 base pairs, thereby allowing for G*U pairs and a few mismatches but excluding bulges82. However, only 10 base pairs actually fit in the substrate binding channel, as observed for an archaeal box C/D snoRNP24. Overall, the interactions that we are proposing for the CD RNA/rRNA pairs adhere to these rules (Supplementary Figs. S5 and S6). The minimum free energy for the formation of the duplexes (Fig. 5) is, however, considerably higher compared to H. sapiens12. At the same time, the lengths of the interactions do not differ as much. This discrepancy can be attributed to the frequent occurrence of G*U base pairs, the occasional presence of A/C base pairs, and a single G/A mismatch (see below) that are predicted in individual interaction pairs. G*U base pairs have been observed also in analogous pairs of other species12,13, and they can be isosteric to Watson–Crick base pairs74. However, their occurrence appears more frequent in the amoeba, and in the extreme case of the CD12/26S-U2580 interaction (Supplementary Fig. S6), 3/9 base pairs are G*U. In three predicted duplexes, we noted an A/C base pair that appeared to be confined to the 6th position upstream of the D box (CD7/26S-G711 and CD23/26S-C3292; Supplementary Fig. S5) or D′ box (CD28/17S-C1715; Supplementary Fig. S6). An A/C interaction can also substitute for a canonical Watson–Crick base pair, if the adenosine is protonated, i.e. A(+)/C75. Distinct from these is the single G/A mismatch seen in the CD29/17S-U1264 pair (Supplementary Fig. S5) that is likely to cause structural perturbations in the interaction, which possibly is counteracted by the overall 13 base pairs surrounding the mismatch. As had been observed before in zebrafish13, the methylated position 17S-G1589 appears to be guided by the + 6 position of CD16 (Supplementary Fig. S6). We noted that non-Watson–Crick interactions occur in all predicted pairs that result in a fractional, but also in some with complete methylation (Table 1 and Supplementary Figs. S5 and S6). However, the overall strength (or weakness) of the CD RNA/rRNA interaction in D. discoideum does not appear to correlate with the RMS score (Supplementary Fig. S9), similar to observations made in human cells12. The lower free energies observed for the resulting duplexes (Fig. 5E) might rather be explained by the lower optimal growth temperature of 21 °C of D. discoideum32, compared to yeast or humans. At this temperature, the inferred stabilities apparently warrant appropriate 2′-O-Me levels in the rRNAs in the amoeba (Fig. 2).
Features of the box C/D snoRNAs
The mature box C/D snoRNAs in D. discoideum exhibit generally established characteristics of this class of ncRNAs (Fig. 1). A stable terminal stem, however, is absent in about half of the mature box C/D snoRNAs (Supplementary Table S4). Such stems are considered important for the recognition by the box C/D snoRNA processing machinery21,22,26,64,83. In H. sapiens or Xenopus laevis, a lack of the terminal stem in mature snoRNAs appears to be compensated by self-complementary sequences in their precursors84,85. This allows for productive interactions with the processing machinery, upon which these sequences are thought to be removed21,22,26. Also in D. discoideum, complementary stretches can be found up- and downstream of some box C/D snoRNAs without a terminal stem (data not shown). Therefore, we speculate that these sequences might be present in presumed precursor molecules.
Dictyostelium discoideum CD RNAs are predicted to use the antisense elements associated with the weakly conserved D′ box sequences more frequently than those with the highly conserved D boxes (Fig. 5B,C). The latter form, together with the in D. discoideum equally conserved C boxes, the terminal k-turn structure (Fig. 1), which is essential for maturation and assembly of the box C/D snoRNP complexes86,87. To some extent similar, a preferred usage of the D′ boxes in guiding 2′-O-Me to rRNA targets has also been reported for H. sapiens and D. rerio12,13. These studies revealed that in humans, the box C′ and D′ sequences displayed a considerably stronger conservation than seen for the amoeba, while in zebrafish box D′ was also less conserved and box C′ appeared degenerated.
Seven CD RNAs of D. discoideum are predicted to utilize both antisense elements (Supplementary Table S7), with no paralogs or other box C/D snoRNAs known to be able to target the associated rRNA positions. At present, it is unknown, whether an interaction of both antisense elements with the target RNA(s) takes place simultaneously or sequentially. For S. cerevisiae, a simultaneous usage of both the antisense elements upstream the D and D′ boxes has been proposed, which might bring distant parts of the rRNA structure into proximity, thereby facilitating ribosomal maturation88,89. We wondered whether a similar situation might exist for “dual-use” CD RNAs in the amoeba. Since only a partial structure is available for the nascent 60S ribosomal subunit of Dictyostelium71, we inferred positions not included in that structure by homology to the human ribosome (PDB accession: 4UG0)90. Positions targeted by CD1, CD7 and CD19 (Supplementary Table S7) were not considered, as no orthologous methylated sites were found in other species (Table 1). CD25 of D. discoideum targets 17S-A612 and 26S-U3254 and the orthologous positions 18S-A668 and 28S-U4468 in the H. sapiens ribosome are around 100 Å apart, indicating sequential modification. Despite being distant in sequence, A1370 in helix H39 and G2952 in helix H80, which are both predicted targets of CD13 (Fig. 7A), lie only 16.7 Å apart in the available structure71 of the D. discoideum 60S subunit (Fig. 7B,C). That structure describes the large subunit at a late stage of maturation. It contains already helices H39 and H80, suggesting that the 2′-O-Me (not featured in the structure) must have taken place, as it requires the accessibility of the target sequences. We also cannot exclude that CD13 binds its targets after they reach proximity (Fig. 7). It is tempting to speculate, however, that the CD RNA actually might first spatially orient the target positions, then trigger their methylation, before the helices finally form. This would be supported by similar reports from S. cerevisiae88,89. Notably, in other species2, the orthologous nucleotides are part of the PTC, with G2952 being directly involved in the interaction with the CCA-tail of the tRNA residing in the ribosomal P site. The two predicted 26S rRNA targets of CD15 and CD19 (Supplementary Table S7) are so close that a simultaneous occupation of both positions would appear sterically challenging, if not impossible. On the other hand, it seems feasible that CD1 and CD8 might interact with their respective two predicted 17S positions (Supplementary Table S7) given their spacing. Thus, a simultaneous interaction with the two target sites appears unlikely for some of the “dual use” CD RNAs, but conceivable for others (CD1, CD8 or CD13).

A model on the function of CD13 in guiding 2′-O-Me at two positions in the 26S rRNA. (A) Binary secondary structure of CD13 bound to positions A1370 and G2952 in the 26S rRNA of D. discoideum. (B) Scheme of relevant structure parts of the nascent 60S ribosomal subunit of D. discoideum (PDB accession: 5AN9) determined at 3.3 Å resolution via cryo-EM71. Domain II is displayed in orange and domain V in green (cf. Fig. 4). (C) Close vicinity (16.7 Å) of nucleotide A1370 in helix H39 and nucleotide G2952 in helix H80 (both positions colored in blue).
Alternative functions of D. discoideum box C/D snoRNAs?
We noted that a substantial set of 22 box C/D snoRNAs are differentially accumulated in the development of the amoeba compared to axenic growth, however, without manifesting in altered 2′-O-Me levels at the targeted positions (Fig. 6). This indicates that the amounts of CD RNAs are under either condition sufficient to warrant the appropriate 2′-O-Me levels (Supplementary Fig. S7C). Changes in the level of individual CD RNAs during development of the amoeba had already been observed in northern blots, e.g. for CD9, CD13 or CD1543. This is similar to data from D. melanogaster91 and D. rerio13. In the absence of an influence on 2′-O-Me levels in the amoeba (Supplementary Fig. S7D), developmental changes of many box C/D snoRNAs might instead point towards other physiological roles. Established is an alternative function as small Cajal Body RNAs (scaRNAs), which are structurally similar to box C/D snoRNAs, carrying an additional CAB box motif, but guide the sequence-specific methylation of small nuclear RNAs (reviewed for example in Refs.16,92). Also, some box C/D snoRNAs are involved in the processing of precursor rRNA molecules in a variety of organisms (summarized in Ref.87). While 2′-O-Me in tRNA is usually introduced by specialized stand-alone methyltransferases, e.g. Ref.93, certain positions are also guided by specific box C/D snoRNAs (reviewed in Ref.16), either alone or together with a dedicated box C/D scaRNA, like in the case of the wobble cytidine 34 of human tRNAMet94. Further functions that are conceivable also for D. discoideum box C/D snoRNAs encompass rRNA acetylation88,95, regulation of 3′ pre-mRNA processing96,97 or even the generation of small, sno-derived RNAs that might have regulatory functions, as described for other organisms98,99,100. Future work will show whether these possible functions are realized in D. discoideum by any of the OR RNAs or those CD RNAs, in which one antisense sequence lacks an identified rRNA target.
Data availability
The nucleotide sequence of the 17S rRNA of D. discoideum reported in this paper has been included in the GenBankTM/EBI Data Bank entry with accession number OK576654. The RMS data have been deposited under the GEO accession GSE186560.
References
Noller, H. F., Hoffarth, V. & Zimniak, L. Unusual resistance of peptidyl transferase to protein extraction procedures. Science 256, 1416–1419 (1992).
Nissen, P., Hansen, J., Ban, N., Moore, P. B. & Steitz, T. A. The structural basis of ribosome activity in peptide bond synthesis. Science 289, 920–930. https://doi.org/10.1126/science.289.5481.920 (2000).
Schluenzen, F. et al. Structure of functionally activated small ribosomal subunit at 3.3 angstroms resolution. Cell 102, 615–623 (2000).
Wimberly, B. T. et al. Structure of the 30S ribosomal subunit. Nature 407, 327–339 (2000).
Bassler, J. & Hurt, E. Eukaryotic ribosome assembly. Annu. Rev. Biochem. 88, 281–306. https://doi.org/10.1146/annurev-biochem-013118-110817 (2019).
Sloan, K. E. et al. Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 14, 1138–1152. https://doi.org/10.1080/15476286.2016.1259781 (2017).
Decatur, W. A. & Fournier, M. J. rRNA modifications and ribosome function. Trends Biochem. Sci. 27, 344–351. https://doi.org/10.1016/s0968-0004(02)02109-6 (2002).
Polikanov, Y. S., Melnikov, S. V., Söll, D. & Steitz, T. A. Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat. Struct. Mol. Biol. 22, 342–344. https://doi.org/10.1038/nsmb.2992 (2015).
Liang, X.-H., Liu, Q. & Fournier, M. J. rRNA modifications in an intersubunit bridge of the ribosome strongly affect both ribosome biogenesis and activity. Mol. Cell 28, 965–977. https://doi.org/10.1016/j.molcel.2007.10.012 (2007).
Henras, A. K., Plisson-Chastang, C., Humbert, O., Romeo, Y. & Henry, Y. Synthesis, function, and heterogeneity of snoRNA-guided posttranscriptional nucleoside modifications in eukaryotic ribosomal RNAs. Enzymes 41, 169–213. https://doi.org/10.1016/bs.enz.2017.03.007 (2017).
Birkedal, U. et al. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew. Chem. Int. Ed. Engl. 54, 451–455. https://doi.org/10.1002/anie.201408362 (2015).
Krogh, N. et al. Profiling of 2′-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res. 44, 7884–7895. https://doi.org/10.1093/nar/gkw482 (2016).
Ramachandran, S. et al. The shift from early to late types of ribosomes in zebrafish development involves changes at a subset of rRNA 2′-O-Me sites. RNA https://doi.org/10.1261/rna.076760.120 (2020).
Locati, M. D. et al. Expression of distinct maternal and somatic 5.8S, 18S, and 28S rRNA types during zebrafish development. RNA 23, 1188–1199. https://doi.org/10.1261/rna.061515.117 (2017).
Georgeson, J. M. & Schwartz, S. The ribosome epitranscriptome: Inert—or a platform for functional plasticity?. RNA https://doi.org/10.1261/rna.078859.121 (2021).
Höfler, S. & Carlomagno, T. Structural and functional roles of 2′-O-ribose methylations and their enzymatic machinery across multiple classes of RNAs. Curr. Opin. Struct. Biol. 65, 42–50. https://doi.org/10.1016/j.sbi.2020.05.008 (2020).
Ganot, P., Bortolin, M. L. & Kiss, T. Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell 89, 799–809. https://doi.org/10.1016/s0092-8674(00)80263-9 (1997).
Kiss-László, Z., Henry, Y., Bachellerie, J. P., Caizergues-Ferrer, M. & Kiss, T. Site-specific ribose methylation of preribosomal RNA: A novel function for small nucleolar RNAs. Cell 85, 1077–1088. https://doi.org/10.1016/s0092-8674(00)81308-2 (1996).
Lafontaine, D. L., Bousquet-Antonelli, C., Henry, Y., Caizergues-Ferrer, M. & Tollervey, D. The box H + ACA snoRNAs carry Cbf5p, the putative rRNA pseudouridine synthase. Genes Dev. 12, 527–537. https://doi.org/10.1101/gad.12.4.527 (1998).
Tollervey, D., Lehtonen, H., Jansen, R., Kern, H. & Hurt, E. C. Temperature-sensitive mutations demonstrate roles for yeast fibrillarin in pre-rRNA processing, pre-rRNA methylation, and ribosome assembly. Cell 72, 443–457. https://doi.org/10.1016/0092-8674(93)90120-F (1993).
Huang, G. M., Jarmolowski, A., Struck, J. C. & Fournier, M. J. Accumulation of U14 small nuclear RNA in Saccharomyces cerevisiae requires box C, box D, and a 5′, 3′ terminal stem. Mol. Cell Biol. 12, 4456–4463. https://doi.org/10.1128/mcb.12.10.4456 (1992).
Caffarelli, E. et al. Processing of the intron-encoded U16 and U18 snoRNAs: The conserved C and D boxes control both the processing reaction and the stability of the mature snoRNA. EMBO J. 15, 1121–1131 (1996).
Henras, A. K. et al. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol. Life Sci. 65, 2334–2359. https://doi.org/10.1007/s00018-008-8027-0 (2008).
Yang, Z., Lin, J. & Ye, K. Box C/D guide RNAs recognize a maximum of 10 nt of substrates. Proc. Natl. Acad. Sci. U. S. A. 113, 10878–10883. https://doi.org/10.1073/pnas.1604872113 (2016).
Wang, H., Boisvert, D., Kim, K. K., Kim, R. & Kim, S.-H. Crystal structure of a fibrillarin homologue from Methanococcus jannaschii, a hyperthermophile, at 1.6 Å resolution. EMBO J. 19, 317–323. https://doi.org/10.1093/emboj/19.3.317 (2000).
Bachellerie, J.-P. et al. Antisense snoRNAs: A family of nucleolar RNAs with long complementarities to rRNA. Trends Biochem. Sci. 20, 261–264. https://doi.org/10.1016/S0968-0004(00)89039-8 (1995).
van Nues, R. W. et al. Box C/D snoRNP catalysed methylation is aided by additional pre-rRNA base-pairing. EMBO J. 30, 2420–2430. https://doi.org/10.1038/emboj.2011.148 (2011).
van Nues, R. W. & Watkins, N. J. Unusual C′/D′ motifs enable box C/D snoRNPs to modify multiple sites in the same rRNA target region. Nucleic Acids Res. 45, 2016–2028. https://doi.org/10.1093/nar/gkw842 (2017).
Azevedo-Favory, J. et al. Mapping rRNA 2′-O-methylations and identification of C/D snoRNAs in Arabidopsis thaliana plants. RNA Biol. https://doi.org/10.1080/15476286.2020.1869892 (2021).
Hebras, J., Krogh, N., Marty, V., Nielsen, H. & Cavaillé, J. Developmental changes of rRNA ribose methylations in the mouse. RNA Biol. 17, 150–164. https://doi.org/10.1080/15476286.2019.1670598 (2020).
Adl, S. M. et al. The revised classification of eukaryotes. J. Eukaryot. Microbiol. 59, 429–493. https://doi.org/10.1111/j.1550-7408.2012.00644.x (2012).
Fey, P., Kowal, A. S., Gaudet, P., Pilcher, K. E. & Chisholm, R. L. Protocols for growth and development of Dictyostelium discoideum. Nat. Protoc. 2, 1307–1316. https://doi.org/10.1038/nprot.2007.178 (2007).
Bozzaro, S. The model organism Dictyostelium discoideum. Methods Mol. Biol. (Clifton, N.J.) 983, 17–37. https://doi.org/10.1007/978-1-62703-302-2_2 (2013).
Glöckner, G. et al. The complex repeats of Dictyostelium discoideum. Genome Res. 11, 585–594. https://doi.org/10.1101/gr.162201 (2001).
Wiegand, S. et al. The Dictyostelium discoideum RNA-dependent RNA polymerase RrpC silences the centromeric retrotransposon DIRS-1 post-transcriptionally and is required for the spreading of RNA silencing signals. Nucleic Acids Res. 42, 3330–3345. https://doi.org/10.1093/nar/gkt1337 (2014).
Schmith, A. et al. A host factor supports retrotransposition of the TRE5-A population in Dictyostelium cells by suppressing an Argonaute protein. Mob. DNA 6, 14. https://doi.org/10.1186/s13100-015-0045-5 (2015).
Malicki, M., Spaller, T., Winckler, T. & Hammann, C. DIRS retrotransposons amplify via linear, single-stranded cDNA intermediates. Nucleic Acids Res. 48, 4230–4243. https://doi.org/10.1093/nar/gkaa160 (2020).
Kessin, R. H. Dictyostelium—Evolution, Cell Biology and the Development of Multicellularity (Cambridge University Press, 2001).
Cockburn, A. F., Newkirk, M. J. & Firtel, R. A. Organization of the ribosomal RNA genes of Dictyostelium discoideum: Mapping of the nontranscribed spacer regions. Cell 9, 605–613 (1976).
Maizels, N. Dictyostelium 17S, 25S, and 5S rDNAs lie within a 38,000 base pair repeated unit. Cell 9, 431–438 (1976).
Sucgang, R. et al. Sequence and structure of the extrachromosomal palindrome encoding the ribosomal RNA genes in Dictyostelium. Nucleic Acids Res. 31, 2361–2368 (2003).
Boesler, C., Kruse, J., Soderbom, F. & Hammann, C. Sequence and generation of mature ribosomal RNA transcripts in Dictyostelium discoideum. J. Biol. Chem. 286, 17693–17703. https://doi.org/10.1074/jbc.M110.208306 (2011).
Aspegren, A., Hinas, A., Larsson, P., Larsson, A. & Soderbom, F. Novel non-coding RNAs in Dictyostelium discoideum and their expression during development. Nucleic Acids Res. 32, 4646–4656. https://doi.org/10.1093/nar/gkh804 (2004).
Kjellin, J. et al. Abundantly expressed class of noncoding RNAs conserved through the multicellular evolution of dictyostelid social amoebae. Genome Res. https://doi.org/10.1101/gr.272856.120 (2021).
Watts, D. J. & Ashworth, J. M. Growth of myxameobae of the cellular slime mould Dictyostelium discoideum in axenic culture. Biochem. J. 119, 171–174. https://doi.org/10.1042/bj1190171 (1970).
Avesson, L., Reimegård, J., Wagner, E. G. & Söderbom, F. MicroRNAs in Amoebozoa: deep sequencing of the small RNA population in the social amoeba Dictyostelium discoideum reveals developmentally regulated microRNAs. RNA 18, 1771–1782. https://doi.org/10.1261/rna.033175.112 (2012).
Liao, Z., Kjellin, J., Hoeppner, M. P., Grabherr, M. & Soderbom, F. Global characterization of the Dicer-like protein DrnB roles in miRNA biogenesis in the social amoeba Dictyostelium discoideum. RNA Biol. 15, 937–954. https://doi.org/10.1080/15476286.2018.1481697 (2018).
Lowe, T. M. & Eddy, S. R. A computational screen for methylation guide snoRNAs in yeast. Science 283, 1168–1171 (1999).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. https://doi.org/10.1186/gb-2009-10-3-r25 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. https://doi.org/10.1093/bioinformatics/btq033 (2010).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. https://doi.org/10.1093/bioinformatics/btt656 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. https://doi.org/10.1093/bioinformatics/btw313 (2016).
Krogh, N., Birkedal, U. & Nielsen, H. RiboMeth-seq: Profiling of 2′-O-Me in RNA. Methods Mol. Biol. 1562, 189–209. https://doi.org/10.1007/978-1-4939-6807-7_13 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Krogh, N., Kongsbak-Wismann, M., Geisler, C. & Nielsen, H. Substoichiometric ribose methylations in spliceosomal snRNAs. Org. Biomol. Chem. 15, 8872–8876. https://doi.org/10.1039/C7OB02317K (2017).
Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. https://doi.org/10.1093/nar/gkz268 (2019).
Rijk, P. D., Wuyts, J. & Wachter, R. D. RnaViz 2: An improved representation of RNA secondary structure. Bioinformatics 19, 299–300 (2003).
Rehmsmeier, M., Steffen, P., Hochsmann, M. & Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 10, 1507–1517. https://doi.org/10.1261/rna.5248604 (2004).
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190. https://doi.org/10.1101/gr.849004 (2004).
Lorenz, R. et al. ViennaRNA Package 2.0. Algorithms Mol. Biol. 6, 26. https://doi.org/10.1186/1748-7188-6-26 (2011).
Dieci, G., Preti, M. & Montanini, B. Eukaryotic snoRNAs: A paradigm for gene expression flexibility. Genomics 94, 83–88. https://doi.org/10.1016/j.ygeno.2009.05.002 (2009).
Petfalski, E., Dandekar, T., Henry, Y. & Tollervey, D. Processing of the precursors to small nucleolar RNAs and rRNAs requires common components. Mol. Cell Biol. 18, 1181–1189 (1998).
Qu, L.-H. et al. Seven novel methylation guide small nucleolar RNAs are processed from a common polycistronic transcript by Rat1p and RNase III in yeast. Mol. Cell. Biol. 19, 1144–1158. https://doi.org/10.1128/mcb.19.2.1144 (1999).
Chanfreau, G., Legrain, P. & Jacquier, A. Yeast RNase III as a key processing enzyme in small nucleolar RNAs metabolism11Edited by J. Karn. J. Mol. Biol. 284, 975–988. https://doi.org/10.1006/jmbi.1998.2237 (1998).
Kruse, J. et al. The protein domains of the Dictyostelium microprocessor that are required for correct subcellular localization and for microRNA maturation. RNA Biol. 13, 1000–1010. https://doi.org/10.1080/15476286.2016.1212153 (2016).
Meier, D. et al. Analysis of the microprocessor in Dictyostelium: The role of RbdB, a dsRNA binding protein. PLoS Genet. 12, e1006057. https://doi.org/10.1371/journal.pgen.1006057 (2016).
Krogh, N. & Nielsen, H. Sequencing-based methods for detection and quantitation of ribose methylations in RNA. Methods 156, 5–15. https://doi.org/10.1016/j.ymeth.2018.11.017 (2019).
Erales, J. et al. Evidence for rRNA 2′-O-methylation plasticity: Control of intrinsic translational capabilities of human ribosomes. Proc. Natl. Acad. Sci. U. S. A. 114, 12934–12939. https://doi.org/10.1073/pnas.1707674114 (2017).
Krogh, N. et al. Profiling of ribose methylations in ribosomal RNA from diffuse large B-cell lymphoma patients for evaluation of ribosomes as drug targets. NAR Cancer. https://doi.org/10.1093/narcan/zcaa035 (2020).
Weis, F. et al. Mechanism of eIF6 release from the nascent 60S ribosomal subunit. Nat. Struct. Mol. Biol. 22, 914–919. https://doi.org/10.1038/nsmb.3112 (2015).
Ramesh, M. & Woolford, J. L. Jr. Eukaryote-specific rRNA expansion segments function in ribosome biogenesis. RNA 22, 1153–1162. https://doi.org/10.1261/rna.056705.116 (2016).
Petrov, A. S. et al. Secondary structures of rRNAs from all three domains of life. PLoS One 9, e88222. https://doi.org/10.1371/journal.pone.0088222 (2014).
Westhof, E., Yusupov, M. & Yusupova, G. The multiple flavors of GoU pairs in RNA. J. Mol. Recognit. 32, e2782. https://doi.org/10.1002/jmr.2782 (2019).
Leontis, N. B., Stombaugh, J. & Westhof, E. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 30, 3497–3531 (2002).
Kos, M. & Tollervey, D. Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol. Cell 37, 809–820. https://doi.org/10.1016/j.molcel.2010.02.024 (2010).
Rajan, K. S. et al. The large repertoire of 2′-O-methylation guided by C/D snoRNAs on Trypanosoma brucei rRNA. RNA Biol. 17, 1018–1039. https://doi.org/10.1080/15476286.2020.1750842 (2020).
Marchand, V., Blanloeil-Oillo, F., Helm, M. & Motorin, Y. Illumina-based RiboMethSeq approach for mapping of 2′-O-Me residues in RNA. Nucleic Acids Res. 44, e135. https://doi.org/10.1093/nar/gkw547 (2016).
Pichot, F. et al. Holistic optimization of bioinformatic analysis pipeline for detection and quantification of 2′-O-Methylations in RNA by RiboMethSeq. Front. Genet. 11, 38. https://doi.org/10.3389/fgene.2020.00038 (2020).
Lapeyre, B. & Purushothaman, S. K. Spb1p-directed formation of Gm2922 in the ribosome catalytic center occurs at a late processing stage. Mol. Cell 16, 663–669. https://doi.org/10.1016/j.molcel.2004.10.022 (2004).
Kufel, J. & Grzechnik, P. Small nucleolar RNAs tell a different tale. Trends Genet. 35, 104–117. https://doi.org/10.1016/j.tig.2018.11.005 (2019).
Chen, C. L., Perasso, R., Qu, L. H. & Amar, L. Exploration of pairing constraints identifies a 9 base-pair core within box C/D snoRNA-rRNA duplexes. J. Mol. Biol. 369, 771–783. https://doi.org/10.1016/j.jmb.2007.03.052 (2007).
Dunbar, D. A., Chen, A. A., Wormsley, S. & Baserga, S. J. The genes for small nucleolar RNAs in Trypanosoma brucei are organized in clusters and are transcribed as a polycistronic RNA. Nucleic Acids Res. 28, 2855–2861. https://doi.org/10.1093/nar/28.15.2855 (2000).
Darzacq, X. & Kiss, T. Processing of Intron-Encoded Box C/D small nucleolar RNAs lacking a 5′,3′-terminal stem structure. Mol. Cell. Biol. 20, 4522–4531 (2000).
Watkins, N. J., Leverette, R. D., Xia, L., Andrews, M. T. & Maxwell, E. S. Elements essential for processing intronic U14 snoRNA are located at the termini of the mature snoRNA sequence and include conserved nucleotide boxes C and D. RNA 2, 118–133 (1996).
Szewczak, L. B., DeGregorio, S. J., Strobel, S. A. & Steitz, J. A. Exclusive interaction of the 15.5 kD protein with the terminal box C/D motif of a methylation guide snoRNP. Chem. Biol. 9, 1095–1107. https://doi.org/10.1016/s1074-5521(02)00239-9 (2002).
Watkins, N. J. & Bohnsack, M. T. The box C/D and H/ACA snoRNPs: Key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip. Rev. RNA 3, 397–414. https://doi.org/10.1002/wrna.117 (2012).
Sharma, S. et al. Specialized box C/D snoRNPs act as antisense guides to target RNA base acetylation. PLoS Genet. 13, e1006804. https://doi.org/10.1371/journal.pgen.1006804 (2017).
Martin, R. et al. A pre-ribosomal RNA interaction network involving snoRNAs and the Rok1 helicase. RNA 20, 1173–1182. https://doi.org/10.1261/rna.044669.114 (2014).
Khatter, H., Myasnikov, A. G., Natchiar, S. K. & Klaholz, B. P. Structure of the human 80S ribosome. Nature 520, 640–645. https://doi.org/10.1038/nature14427 (2015).
Agrisani, A., Tafer, H., Stadler, P. F. & Furia, M. Unusual novel SnoRNA-like RNAs in Drosophila melanogaster. Noncoding RNA 1, 139–150. https://doi.org/10.3390/ncrna1020139 (2015).
Kiss, T. Biogenesis of small nuclear RNPs. J. Cell Sci. 117, 5949–5951. https://doi.org/10.1242/jcs.01487 (2004).
Guy, M. P. et al. Yeast Trm7 interacts with distinct proteins for critical modifications of the tRNAPhe anticodon loop. RNA 18, 1921–1933. https://doi.org/10.1261/rna.035287.112 (2012).
Vitali, P. & Kiss, T. Cooperative 2′-O-methylation of the wobble cytidine of human elongator tRNA(Met)(CAT) by a nucleolar and a Cajal body-specific box C/D RNP. Genes Dev. 33, 741–746. https://doi.org/10.1101/gad.326363.119 (2019).
Ito, S. et al. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J. Biol. Chem. 289, 35724–35730. https://doi.org/10.1074/jbc.C114.602698 (2014).
Shi, J., Huang, C., Huang, S. & Yao, C. snoRNAs associate with mRNA 3′processing complex: New wine in old bottles. RNA Biol. 15, 194–197. https://doi.org/10.1080/15476286.2017.1416278 (2018).
Huang, C. et al. A snoRNA modulates mRNA 3′ end processing and regulates the expression of a subset of mRNAs. Nucleic Acids Res. 45, 8647–8660. https://doi.org/10.1093/nar/gkx651 (2017).
Taft, R. J. et al. Small RNAs derived from snoRNAs. RNA (New York, N.Y.) 15, 1233–1240. https://doi.org/10.1261/rna.1528909 (2009).
Scott, M. S. & Ono, M. From snoRNA to miRNA: Dual function regulatory non-coding RNAs. Biochimie 93, 1987–1992. https://doi.org/10.1016/j.biochi.2011.05.026 (2011).
Bratkovic, T., Bozic, J. & Rogelj, B. Functional diversity of small nucleolar RNAs. Nucleic Acids Res. 48, 1627–1651. https://doi.org/10.1093/nar/gkz1140 (2020).
Acknowledgements
We thank our colleague Dr. Monica Hagedorn for discussion and helpful comments on the manuscript. We also acknowledge Drs. Virginie Marchard and Yuri Motorin for sharing unpublished data and Dr. Nicolai Krogh for support with the GEO submission. This work was supported by the DFG [HA3459/14] and by the DFG Priority Program 1784 Chemical Biology of Native Nucleic Acid Modifications [HA3459/17]. Funding for open access charge: Jacobs University funds and DFG [HA3459/17].
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
J.D. designed and carried out most experiments; U.B. generated and analyzed the RMS data; J.K. analyzed RNAseq data; K.P.J. established tools for and performed bioinformatic analyses; J.Z. performed initial snoRNA analyses; F.S., H.N. and C.H. supervised research. J.D., H.N. and C.H. wrote the paper. All authors read and proved the final version of this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Diesend, J., Birkedal, U., Kjellin, J. et al. Fractional 2′-O-methylation in the ribosomal RNA of Dictyostelium discoideum supports ribosome heterogeneity in Amoebozoa. Sci Rep 12, 1952 (2022). https://doi.org/10.1038/s41598-022-05447-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-05447-w
This article is cited by
-
Chromosome-level genome assembly and annotation of the social amoeba Dictyostelium firmibasis
Scientific Data (2024)
-
FUS regulates a subset of snoRNA expression and modulates the level of rRNA modifications
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
