
Abstract
The commensal microbes of the skin have a significant impact on dermal physiology and pathophysiology. Racial and geographical differences in the skin microbiome are suggested and may play a role in the sensitivity to dermatological disorders, including infectious diseases. However, little is known about the skin microbiome profiles of people living in Central Africa, where severe tropical infectious diseases impose a burden on the inhabitants. This study provided the skin profiles of healthy Cameroonians in different body sites and compared them to healthy Japanese participants. The skin microbiome of Cameroonians was distinguishable from that of Japanese in all skin sites examined in this study. For example, Micrococcus was predominantly found in skin samples of Cameroonians but mostly absent in Japanese skin samples. Instead, the relative abundance of Cutibacterium species was significantly higher in healthy Japanese. Principal coordinate analysis of beta diversity showed that the skin microbiome of Cameroonians formed different clusters from Japanese, suggesting a substantial difference in the microbiome profiles between participants of both countries. In addition, the alpha diversity in skin microbes was higher in Cameroonians than Japanese participants. These data may offer insights into the determinant factors responsible for the distinctness of the skin microbiome of people living in Central Africa and Asia.
Similar content being viewed by others


Introduction
Skin is one of the most crucial protective features available in humans. With diverse anatomical and physiological functions, starting from barrier function to thermostat regulation, the skin plays an essential role in a human’s life1. At present, the skin has been regarded as one of the largest microbial ecosystems found in the body, possibly due to direct contact and prolonged exposure to the dynamic external environment2,3,4,5,6,7. Indeed, the skin’s unique physiological and topographical features provide various distinct yet suitable niches for diverse types of microbial communities8. Although much focus has been given, the skin remains one of the less-charted territories in the field of microbiome investigation9,10,11,12,13. Nevertheless, the increase of sophisticated research tools, such as the utilization of next-generation sequencing and powerful bioinformatics applications, has opened the possibilities to carry out some exciting yet unexplored ideas in this field.
Skin microbial communities are influenced by many factors. Some of them depend on the interpersonal variation of host factors found in the human skin. Such variations may include a distinct set of topographical features and different temperatures, pH values, and sebum content14. These niche-specific physiological differences influence resident bacteria and fungi9,10,15; oily surfaces, such as the forehead, support lipophilic bacteria that differ from dry, low biomass sites, such as the forearm. The question arises: What bacteria must grow in some skin regions to have a healthy normal life? Solving this challenging puzzle would offer great relevance to the comprehensive study of the human microbiome.
Skin microbiome and dermatology-related studies are closely connected9,10,14. Some studies have suggested the role of the skin microbiome in developing certain dermatological disorders, such as acne, atopic dermatitis, psoriasis, rosacea, and seborrheic dermatitis2. Besides, dysbiosis of the skin microbiome has been implicated in the progressiveness of certain dermatological diseases. For example, leprosy and alteration of the skin microbiota composition in response to diseases and therapeutical interventions (such as systemic antimicrobials preparations) have been recently suggested16. To this end, it is interesting to see whether the composition of the skin microbiome in individuals of different countries or even different races will determine their susceptibilities to certain infectious diseases.
Several aspects, such as physiological and topographical features, have been suggested to influence the establishment of skin microbiome at individual levels6,14,15. Also, early reports have suggested that race17 and skin body sites18 are essential factors to consider in the skin microbiome profile. However, although the essentiality of these two factors is likely, information on them remains scarce. This study cataloged the skin profile of healthy Cameroonians and healthy Japanese in different body sites to support such an endeavor. Some studies have covered the skin microbiome of healthy Japanese19,20,21. However, to the authors’ knowledge, this study is the first to characterize the skin microbiome profile in three skin sites of healthy Cameroonians and compare the results to the ones obtained from healthy Japanese. This is the first comparative report describing the skin microbiome profile of people living in Central Africa (the west coast of the African continent) and the Asian population. These results may provide fundamental insights into the determinant factors responsible for the establishment of skin microbiota.
Results
Distinctive differences in the skin microbes between Cameroonians and Japanese
Dynamic variations in the human skin microbiome have been suggested to be associated with host factors (including age, diet, body sites, and sex) and environmental conditions (such as geographical location and climate)2,5. This study examined the relative abundance and prevalence of microbes in skin swabs collected from healthy participants of two different countries: Cameroon (n = 27) and Japan (n = 21). The demographic information was described in Table 1.
Figure 1 shows a comparison of the skin microbiome compositions between healthy Cameroonians and healthy Japanese in the three skin sites: forehead, right forearm, and the mid-upper back (Supplementary Fig. S1). The microbiome of the Cameroonian and Japanese skin samples can be assigned into more than 20 genera that mostly belonged in three phyla: Actinobacteria, Proteobacteria, and Firmicutes. At the genus level, most obtained sequences were Cutibacterium, Staphylococcus, Micrococcus, Corynebacterium, Kocuria, and, to a lesser extent, Janibacter and Streptococcus (Fig. 1 and Table 2).

Top 20 skin microbiome at various positions between Cameroonian and Japanese participants. CMR, Cameroonian; JPN, Japanese.
Of all, Staphylococcus was predominantly found in all three body sites of Cameroonians and Japanese. However, it is important to note that the relative abundance of Staphylococcus at the back of Japanese participants was lower than that of other skin sites examined in this study. In addition, although the Cutibacterium population was higher in all three skin sites of Japanese than Cameroonians, its presence was highly observed in samples recovered from sebaceous (forehead and back) sites, similar to the results of the previous study8.
Compared to their Japanese counterparts, there were some distinct features of the skin microbiome in the Cameroonian skin. Micrococcus was predominantly found in Cameroonian skin samples but mostly absent in Japanese skin samples. Likewise, further analysis indicated that Kocuria was present in all skin samples of healthy Cameroonians but observable at a low amount only in the forearm skin sample of healthy Japanese. In contrast, the low abundance of Neisseria and Enhydrobacter seemed more prevalent to be recovered from Japanese skin samples, mostly in the back and forearm, respectively, than that of Cameroonians. Overall, these findings implied the involvement of geographical- or race-related factors in the difference of skin microbiome of the two countries.
Little effect of sex on the differences in skin microbiome between the two countries
Sexual dimorphism in microbiome composition has been suggested22. To investigate whether sexual dimorphism is a determinant in the difference of skin microbe composition observed in this study, skin samples collected at the forehead, forearm, and the back of healthy men and women participants from Cameroon and Japan were analyzed (Supplementary Fig. S2). A two-way (sex × country) analysis of variance revealed that the country difference had much more effects on the difference in the relative abundance of major skin microbiota than the sex difference, as indicated by the larger partial η2 index in the “country” term of the analysis, although a few genera on the specific area (e.g., Cutibacterium on the forehead and back skin) showed significant differences according to sex (Supplementary Table S1). In sum, a certain distinctiveness in the skin microbiome observed in participants from the two countries could scarcely be explained by sexual dimorphism.
Distinguishable beta diversity and higher alpha diversity in Cameroonians
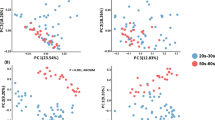
We have shown that the composition of skin microbiome in the Cameroonian people were apparently different from that in the Japanese people. This finding was further confirmed by the beta diversity analysis using the unweighted UniFrac distance. The principal coordinate analysis of beta diversity showed that the skin samples obtained from healthy Cameroonian participants formed apparently different clusters from healthy Japanese participants (Fig. 2), suggesting the difference of microbiome profiles between the two countries.

Beta diversity in reflection of the unweighted UniFrac distance. CMR, Cameroonian; JPN, Japanese; PC, principal coordinate.
Figure 3 shows the alpha diversity metrics, namely Chao1, the number of the observed operational taxonomic units (OTUs), Faith’s phylogenetic diversity, and Shannon index, of the skin microbiome between healthy Cameroonians and healthy Japanese in the three skin sites: forehead (Fig. 3a), forearm (Fig. 3b), and the back (Fig. 3c). The species richness and diversity of Cameroonians were higher than those of Japanese counterparts, as suggested by the significant increase in Chao1, observed OTUs, and Shannon index values on the forehead and the back skin (Fig. 3a, c). However, most, if not all, alpha diversity indices showed no significant differences between both groups on the forearm skin (Fig. 3b).

Alpha diversity of the forehead (A), forearm (B), and back (C) skin. CMR, Cameroonian; JPN, Japanese; OTU, operational taxonomic unit. *p < 0.05, **p < 0.01.
Discussion
This study analyzed the skin microbiome profile of healthy Cameroonians in three different body sites and compared them to healthy Japanese participants. Using the next generation sequencing (NGS) approach, whether geographical (to a greater extent, including racial difference) and topological features (three different skin sites: forehead, forearm, and the back) play distinctive roles in the human skin microbiome diversity was assessed. Consequently, the skin microbiome of Cameroonians was clearly distinguishable from that of Japanese individuals in all skin sites examined in this study.
This study identified a total collection of human skin microbiomes classified into more than 20 genera and spanned to at least three prominent phyla: Actinobacteria, Firmicutes, and Proteobacteria. Indeed, in agreement with this study, previous reports have demonstrated that the human skin microbiome is dominated by a similar microbiome profile4,8,23,24. Among the identified genera, Cutibacterium, Staphylococcus, Micrococcus, Corynebacterium, Kocuria, Janibacter, and Streptococcus were the most presented microbial genera on the skin of healthy participants from both countries (Fig. 1 and Table 2).
The relative abundance of Cutibacterium species (spp.) was significantly higher in healthy Japanese than healthy Cameroonians. This finding was in line with the skin microbiome studies in Asian population, which showed a high abundance of Cutibacterium spp. in healthy Chinese25, Korean26, and Japanese19. Three species of Cutibacterium (C. avidum, C. acnes, and C. granulosum) have been reported to colonize the skin surfaces of not only healthy individuals but also acne patients, possibly due to the attractiveness of these Cutibacterium species to the sebum-rich site of the skin27. This result implied that the Cameroonian skin environment might contain less sebum-rich ecological niches that support Cutibacterium spp. growth, although we do not have information regarding skin physiological functions due to inability to access to the equipment in Cameroon. Such a physiological assessment of the skin is warranted.
Staphylococcus was predominantly found in all three body sites of healthy Cameroonians and healthy Japanese. Staphylococcus spp. have been reported as the dominant and common bacterial colonizers of the skin28. This is in line with the result of this study in which the relative abundance of Staphylococcus spp. did not significantly different between Cameroonians and Japanese except on the back skin, likely due to much higher Cutibacterium spp. on the Japanese participants’ back skin (Fig. 1 and Table 2).
Micrococcus was predominantly found in skin samples of Cameroonians but mostly absent in Japanese skin samples. Micrococcus is a bacterial genus with a wide range of habitats, including dust and soil29,30, and has no difficulty growing in environments with little water or high-salt concentrations31,32. Likewise, Kocuria was abundantly present in all skin samples of healthy Cameroonians but only observable in the forearm skin samples of healthy Japanese. Although several species of Kocuria have been reported to inhabit the human skin and oral cavity33, Kocuria also has been reported to be widely spread in an environmental niche34,35. Taking these into account, the presence of Micrococcus and Kocuria in skin samples of Cameroonians might in part implicate plausible contact interaction of Cameroonians with the external natural ecological system, for example, farms, gardens, and beaches and/or dust-based environments that are plentiful in Cameroon on a regular basis. Several studies have also reported the effect of contact with soil, aquatic, and other environmental sources of microbiota on the skin microbiome36,37,38. However, due to the lack of accurate information on the lifestyle of the participants, we do not know the extent to which they interact with the natural environment. Further skin microbiome studies that take lifestyle into account are needed to show the influence of the external environment on the differences in the skin microbiome of the two countries.
The skin microbiome can be influenced by a number of external factors, such as lifestyle, hygiene, geographical and/or climate differences39,40,41. In addition, intrinsic factors such as aging, sex, and racial differences are also said to be responsible for skin microbial variability26,39,41,42. In this study, the Cameroonians and the Japanese people showed substantially different skin microbiome profiles; however, the question remains as to which factors may be relevant to such differences. A previous study by Harker, et al.43 revealed the link between axilla skin microbiome and a single nucleotide polymorphism of ABCC11 gene whose genotype varies according to racial differences44. In these studies, the G/G genotype which is common in African people led to significantly more Staphylococcus on the axillary skin, compared to Asian people who generally carry the A/A and G/A genotypes. These findings could possibly explain higher, albeit not significantly, Staphylococcus on the forearm and back skin of Cameroonian people in this study (Table 2). However, the study also showed that the G/G genotype was related to significantly less Corynebacterium, and Micrococcus was not affected by the ABCC11 genotypes, both of which contradict the results of this study (Table 2). Although the genetic variation is a possible contributor to the race-microbiome relationship45,46, we do not have information on genetic background of the participants and thus the relationship among race, genetic variation, and microbiome cannot be further discussed in this study. Genome-wide association studies46 can be beneficial to investigate the involvement of genetic factors in the distinct skin microbiome between Cameroon and Japan.
Overall, the results suggested the presence of a diverse microbiome on the human skin. Although microbial diversity is distinguishable and similar species tend to reside in the same part of the skin of healthy subjects from both countries, the abundance of specific species is, to a larger extent, country (environment)-dependent and, to a lesser extent, skin site-dependent. With its limitations, the data suggested the possible relationship between the relative abundance of certain microbes with the geographical and topological features of the human skin. The inclusion of such information in a comprehensive analysis of skin microbes in healthy subjects may provide insights that would be beneficial in the discovery of safe and sound dermatological therapeutics to precisely cure and manage skin diseases.
Methods
Ethical considerations
This study was approved by the National Ethics Committee of Cameroon (approval no. 2018/06/1045/CE/CNERSH/SP), the hospital where the research was conducted, and the Medical Ethics Committee of Kanazawa University, where the NGS was performed (approval no. 894). All research was performed in accordance with the Declaration of Helsinki. Before inclusion, all participants were informed by a written document about the research, and written informed consent was obtained from all participants.
Study design and settings
This was a part of a cross-sectional study conducted in a human immunodeficiency virus (HIV)-clinic of the Efoulan District Hospital in Yaoundé, Cameroon, and in a single university in Japan. The hospital was selected for an existing collaboration between the hospital and the university research center. Prior to recruitment of participants, we first made a tentative inclusion list of possible candidates for the study with the inclusion criteria of being aged 21–65 years. Each candidate received an explanation of this study by the researcher, including the following request. The candidates were requested not to (1) take a bath or shower or (2) use emollients/creams on the sampling site, after midnight of the day of sampling. Pregnant women, those who had apparent skin disorders (e.g., atopic dermatitis, psoriasis, and Kaposi sarcoma) or other systemic diseases (e.g., HIV infection), those who did not consent to the study, and those who did not follow the instruction requested by the researchers were excluded.
Sample collection
Skin swabs were collected as described previously47. In brief, Puritan HydraFlock Sterile Flocked Swab (25–3306-H; Puritan Medical Products Co., ME, USA) was presoaked in normal saline (S5815; Teknova, CA, USA) with 0.1% Tween 20 (28353-14; Nacalai Tesque, Inc., Kyoto, Japan) solution. Swabbing was then performed in a 5 × 5 cm2 on designated positions. After swabbing, each swab head was cut and placed in a 1.5 mL microcentrifuge tube, carried in a cooler box with ice to prevent degradation of the samples, and stored at − 30 °C until DNA extraction.
DNA extraction
DNA from the swab head was performed as described previously47,48. Briefly, the swab heads were processed with the Kaneka Easy DNA Extraction Kit version 2 (KN-T110005; Kaneka Corp., Tokyo, Japan), followed by the enzymatic DNA extraction process with QIAamp DNA Mini Kit (51,304; Qiagen N.V., Venlo, The Netherlands). The extracted DNA samples were stored at − 30 °C until NGS preparation.
Controls
The swab without skin swabbing served as a negative control, and the ZymoBIOMICS Microbial Community Standard (D6300; Zymo Research Corp., Irvine, CA, USA) was used as a positive control (Supplementary Fig. 3). Little overlap between the observed skin microbiome (Fig. 1 and Table 2) and the negative control (Supplementary Fig. 3a) ruled out the possibility of contamination from the experimental materials (e.g., swabs and kits). The closeness of the theoretical and observed compositions of the positive control, especially the effective detection of gram-positive, skin-related Staphylococcus, implied the successful extraction and analysis of bacterial DNA (Supplementary Fig. 3b).
NGS
The extracted DNA was dedicated to the NGS analysis as described previously48. The DNA samples were first quantified for the copy number of the 16S ribosomal RNA (rRNA) gene for normalization with a quality check. The same amount of the 16S rRNA gene from each sample was then used to amplify the V3-V4 hypervariable region48,49,50. After indexing polymerase chain reaction with the Nextera XT Index Kit version 2 (FC-131-2001 to 2004; Illumina, Inc., San Diego, CA, USA), the resultant library was loaded onto the MiSeq System (SY-410-1003; Illumina) with MiSeq Reagent Kit version 3, 600 cycles (MS-102-3003; Illumina), and 15% PhiX Control v3 (FC-110-3001; Illumina) spike-in.
Sequence analysis
The raw fastq sequences were first quality-filtered by sickle51 and chimera-eliminated by usearch52,53. The high-quality, chimera-free sequences were then fed into the Qiime software version 1.9.154. For the calculation of the OTUs, a 97% threshold was used. The Silva database55 version 132 was used for taxonomic assignment. Beta diversity was calculated based on the unweighted UniFrac distance followed by principal coordinate analysis. Alpha diversity indices [Chao1, observed OTUs, Faith’s phylogenetic diversity, and Shannon index] were calculated by the “alpha_rarefaction.py” command of Qiime1. The samples were rarefied at 3000 depth of sequences, and the samples with less than 3000 sequences were discarded.
Statistical analysis
The data were expressed as means ± standard deviation, medians [interquartile range (IQR)], or n (%) where appropriate. The boxplot denotes the 25th, 50th, and 75th percentile boxes with 25th percentile − 1.5 × IQR to 75th percentile + 1.5 × IQR whiskers. For categorical variables, Fisher’s exact test was used. For continuous variables, Welch’s t-test or Mann–Whitney U test was used for two-group comparison where appropriate. A two-way analysis of variance with partial η2 values of effect size was used to take into account the two factors of sex and country differences in relative abundance. For multiple comparisons of bacterial abundance, Benjamini-Hochberg’s false discovery rate control was employed. The corrected p-values were denoted hereafter as q-values. p- or q-values < 0.05 was considered statistically significant. All statistical analyses were performed using R statistical software56 version 3.9.2.
Accession number
All raw sequences of the NGS analysis were deposited in DNA Data Bank of Japan (DDBJ; accession number is DRA011596).
References
Segre, J. A. Epidermal barrier formation and recovery in skin disorders. J. Clin. Invest. 116, 1150–1158. https://doi.org/10.1172/JCI28521 (2006).
Byrd, A. L., Belkaid, Y. & Segre, J. A. The human skin microbiome. Nat. Rev. Microbiol. 16, 143–155. https://doi.org/10.1038/nrmicro.2017.157 (2018).
Costello, E. K. et al. Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. https://doi.org/10.1126/science.1177486 (2009).
Gao, Z., Tseng, C. H., Pei, Z. & Blaser, M. J. Molecular analysis of human forearm superficial skin bacterial biota. Proc. Natl. Acad. Sci. U. S. A. 104, 2927–2932. https://doi.org/10.1073/pnas.0607077104 (2007).
Grice, E. A. & Segre, J. A. The skin microbiome. Nat. Rev. Microbiol. 9, 244–253. https://doi.org/10.1038/nrmicro2537 (2011).
Li, W. et al. Nested PCR-denaturing gradient gel electrophoresis analysis of human skin microbial diversity with age. Microbiol. Res. 169, 686–692. https://doi.org/10.1016/j.micres.2014.02.008 (2014).
Sanford, J. A. & Gallo, R. L. Functions of the skin microbiota in health and disease. Semin. Immunol. 25, 370–377. https://doi.org/10.1016/j.smim.2013.09.005 (2013).
Grice, E. A. et al. Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192. https://doi.org/10.1126/science.1171700 (2009).
Fredricks, D. N. Microbial ecology of human skin in health and disease. J. Investig. Dermatol. Symp. Proc. 6, 167–169. https://doi.org/10.1046/j.0022-202x.2001.00039.x (2001).
Lunjani, N., Hlela, C. & O’Mahony, L. Microbiome and skin biology. Curr. Opin. Allergy Clin. Immunol. 19, 328–333. https://doi.org/10.1097/ACI.0000000000000542 (2019).
Holland, K. T. & Bojar, R. A. Cosmetics: What is their influence on the skin microflora?. Am. J. Clin. Dermatol. 3, 445–449. https://doi.org/10.2165/00128071-200203070-00001 (2002).
Niemeyer-van der Kolk, T., van der Wall, H. E. C., Balmforth, C., Van Doorn, M. B. A. & Rissmann, R. A systematic literature review of the human skin microbiome as biomarker for dermatological drug development. Br. J. Clin. Pharmacol. 84, 2178–2193. https://doi.org/10.1111/bcp.13662 (2018).
Leyden, J. J., McGinley, K. J., Nordstrom, K. M. & Webster, G. F. Skin microflora. J. Invest. Dermatol. 88, 65s–72s. https://doi.org/10.1111/1523-1747.ep12468965 (1987).
Dethlefsen, L., McFall-Ngai, M. & Relman, D. A. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449, 811–818. https://doi.org/10.1038/nature06245 (2007).
Findley, K. et al. Topographic diversity of fungal and bacterial communities in human skin. Nature 498, 367–370. https://doi.org/10.1038/nature12171 (2013).
Bayal, N. et al. 16S rDNA based skin microbiome data of healthy individuals and leprosy patients from India. Sci. Data 6, 225. https://doi.org/10.1038/s41597-019-0232-1 (2019).
Fortenberry, J. D. The uses of race and ethnicity in human microbiome research. Trends Microbiol. 21, 165–166. https://doi.org/10.1016/j.tim.2013.01.001 (2013).
Perez Perez, G. I. et al. Body site is a more determinant factor than human population diversity in the healthy skin microbiome. PLoS ONE 11, e0151990. https://doi.org/10.1371/journal.pone.0151990 (2016).
Nagase, S. et al. Distinct skin microbiome and skin physiological functions between bedridden older patients and healthy people: A single-center study in Japan. Front. Med. (Lausanne) 7, 101. https://doi.org/10.3389/fmed.2020.00101 (2020).
Watanabe, H. et al. Minor taxa in human skin microbiome contribute to the personal identification. PLoS ONE 13, e0199947. https://doi.org/10.1371/journal.pone.0199947 (2018).
Shibagaki, N. et al. Aging-related changes in the diversity of women’s skin microbiomes associated with oral bacteria. Sci. Rep. 7, 10567. https://doi.org/10.1038/s41598-017-10834-9 (2017).
Ma, Z. S. & Li, W. How and why men and women differ in their microbiomes: Medical ecology and network analyses of the microgenderome. Adv. Sci. (Weinh) 6, 1902054, https://doi.org/10.1002/advs.201902054 (2019).
Gao, Z., Tseng, C. H., Strober, B. E., Pei, Z. & Blaser, M. J. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS ONE 3, e2719. https://doi.org/10.1371/journal.pone.0002719 (2008).
Blaser, M. J. et al. Distinct cutaneous bacterial assemblages in a sampling of South American Amerindians and US residents. ISME J. 7, 85–95. https://doi.org/10.1038/ismej.2012.81 (2013).
Leung, M. H. Y., Tong, X., Wilkins, D., Cheung, H. H. L. & Lee, P. K. H. Individual and household attributes influence the dynamics of the personal skin microbiota and its association network. Microbiome 6, 26. https://doi.org/10.1186/s40168-018-0412-9 (2018).
Kim, J. H. et al. Taxonomic profiling of skin microbiome and correlation with clinical skin parameters in healthy Koreans. Sci. Rep. 11, 16269. https://doi.org/10.1038/s41598-021-95734-9 (2021).
Nordstrom, N. K. & Noble, W. C. Colonization of the axilla by Propionibacterium avidum in relation to age. Appl. Environ. Microbiol. 47, 1360–1362. https://doi.org/10.1128/AEM.47.6.1360-1362.1984 (1984).
O'Sullivan, J. N., Rea, M. C., O'Connor, P. M., Hill, C. & Ross, R. P. Human skin microbiota is a rich source of bacteriocin-producing staphylococci that kill human pathogens. FEMS Microbiol. Ecol. 95, fiy241, https://doi.org/10.1093/femsec/fiy241 (2019).
Sims, G. K., Sommers, L. E. & Konopka, A. Degradation of pyridine by micrococcus luteus isolated from soil. Appl. Environ. Microbiol. 51, 963–968. https://doi.org/10.1128/AEM.51.5.963-968.1986 (1986).
Azua-Bustos, A. & Gonzalez-Silva, C. Biotechnological applications derived from microorganisms of the Atacama Desert. Biomed. Res. Int. 2014, 909312. https://doi.org/10.1155/2014/909312 (2014).
Lennon, J. T. & Jones, S. E. Microbial seed banks: The ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 9, 119–130. https://doi.org/10.1038/nrmicro2504 (2011).
Lennon, J. T., Aanderud, Z. T., Lehmkuhl, B. K. & Schoolmaster, D. R. Jr. Mapping the niche space of soil microorganisms using taxonomy and traits. Ecology 93, 1867–1879. https://doi.org/10.1890/11-1745.1 (2012).
Grice, E. A. et al. A diversity profile of the human skin microbiota. Genome Res. 18, 1043–1050. https://doi.org/10.1101/gr.075549.107 (2008).
Park, E. J., Kim, M. S., Roh, S. W., Jung, M. J. & Bae, J. W. Kocuria atrinae sp. nov., isolated from traditional Korean fermented seafood. Int. J. Syst. Evol. Microbiol. 60, 914–918, https://doi.org/10.1099/ijs.0.014506-0 (2010).
Kandi, V. et al. Emerging bacterial infection: Identification and clinical significance of Kocuria species. Cureus 8, e731. https://doi.org/10.7759/cureus.731 (2016).
Hospodsky, D. et al. Hand bacterial communities vary across two different human populations. Microbiology (Reading) 160, 1144–1152. https://doi.org/10.1099/mic.0.075390-0 (2014).
Ying, S. et al. The influence of age and gender on skin-associated microbial communities in urban and rural human populations. PLoS ONE 10, e0141842. https://doi.org/10.1371/journal.pone.0141842 (2015).
Gupta, V. K., Paul, S. & Dutta, C. Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front. Microbiol. 8, 1162. https://doi.org/10.3389/fmicb.2017.01162 (2017).
Boxberger, M., Cenizo, V., Cassir, N. & La Scola, B. Challenges in exploring and manipulating the human skin microbiome. Microbiome 9, 125. https://doi.org/10.1186/s40168-021-01062-5 (2021).
Lehtimaki, J. et al. Patterns in the skin microbiota differ in children and teenagers between rural and urban environments. Sci. Rep. 7, 45651. https://doi.org/10.1038/srep45651 (2017).
Khmaladze, I., Leonardi, M., Fabre, S., Messaraa, C. & Mavon, A. The skin interactome: A holistic, “genome-microbiome-exposome” approach to understand and modulate skin health and aging. Clin. Cosmet. Investig. Dermatol. 13, 1021–1040. https://doi.org/10.2147/CCID.S239367 (2020).
Wilantho, A., Deekaew, P., Srisuttiyakorn, C., Tongsima, S. & Somboonna, N. Diversity of bacterial communities on the facial skin of different age-group Thai males. PeerJ 5, e4084. https://doi.org/10.7717/peerj.4084 (2017).
Harker, M. et al. Functional characterisation of a SNP in the ABCC11 allele—effects on axillary skin metabolism, odour generation and associated behaviours. J. Dermatol. Sci. 73, 23–30. https://doi.org/10.1016/j.jdermsci.2013.08.016 (2014).
Yoshiura, K. et al. A SNP in the ABCC11 gene is the determinant of human earwax type. Nat. Genet. 38, 324–330. https://doi.org/10.1038/ng1733 (2006).
Dimitriu, P. A. et al. New insights into the intrinsic and extrinsic factors that shape the human skin microbiome. mBio 10, https://doi.org/10.1128/mBio.00839-19 (2019).
Goodrich, J. K., Davenport, E. R., Clark, A. G. & Ley, R. E. The relationship between the human genome and microbiome comes into view. Annu. Rev. Genet. 51, 413–433. https://doi.org/10.1146/annurev-genet-110711-155532 (2017).
Benderli, N. C. et al. Feasibility of microbial sample collection on the skin from people in Yaounde, Cameroon. Drug Discov. Ther. 13, 360–364. https://doi.org/10.5582/ddt.2019.01075 (2019).
Ogai, K. et al. A comparison of techniques for collecting skin microbiome samples: Swabbing versus tape-stripping. Front. Microbiol. 9, 2362. https://doi.org/10.3389/fmicb.2018.02362 (2018).
Castelino, M. et al. Optimisation of methods for bacterial skin microbiome investigation: Primer selection and comparison of the 454 versus MiSeq platform. BMC Microbiol. 17, 23. https://doi.org/10.1186/s12866-017-0927-4 (2017).
Zeeuwen, P. et al. Reply to Meisel et al. J. Invest. Dermatol. 137, 961–962, https://doi.org/10.1016/j.jid.2016.11.013 (2017).
Joshi, N. A. F. J. N. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files., <https://github.com/najoshi/sickle> (2011).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. https://doi.org/10.1093/bioinformatics/btq461 (2010).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. https://doi.org/10.1093/bioinformatics/btr381 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. https://doi.org/10.1038/nmeth.f.303 (2010).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 41, D590-596. https://doi.org/10.1093/nar/gks1219 (2013).
R Core Team. R: A language and environment for statistical computing. https://www.r-project.org/ (2020).
Acknowledgements
We thank the staff of the Efoulan district hospital for their collaboration and support, especially Dr Akame, the HIV clinic coordinator. We also thank the researchers and laboratory staff at the Biotechnology Centre, University of Yaoundé I, Cameroon, for processing and archiving the samples. We appreciate all the participants of this study for their time and contribution. This work was supported in part from International Research Training Program, National Institute on Minority Health and Health Disparities (T37MD008636); Global Infectious Disease Research Training, Fogarty International Center, National Institutes of Health (NIH) (D43TW009074); Centers of Biomedical Research Excellence, National Institute of General Medical Sciences, NIH (P30GM114737); National Institute of Allergy and Infectious Diseases, NIH (1R21 AI105286); Japan Society for the Promotion of Science Kakenhi (17H04428 and 19KK0243 to S.O.); AMED-PRIME, AMED (17gm6010011h0001 to T.K.); JST FOREST Program (Grant Number JPMJFR2063 to T.K.); and Kanazawa University Sakigake Project. Y.M.L.’s travels were supported by the Career Development Award from the American Association of University Women Hawaii Branch. K.O.’s travel and accommodation expenses were supported in part by the Overseas Research Support for Young Scientists in Kanazawa University.
Author information
Authors and Affiliations
Contributions
Conceptualization, G.L., S.O., and T.K.; methodology, K.O., Y.M.L., A.H., A.M., S.O. and T.K.; software, K.O.; validation, K.O., N.C., A.M., F.N. and T.K.; formal analysis, K.O., N.C., A.M.; investigation, K.O., N.C., Y.M.L, J.P.A., B.J., A.H.A., L.F.E., A.H., A.M., G.L. and T.K.; resources, N.C., A.H.A., L.F.E., R.M., R.G.F.L. and G.L.; data curation, K.O., A.M., F.N. and T.K.; writing—original draft preparation, K.O., F.N. and T.K.; writing—review and editing, K.O., F.N. and T.K.; visualization, K.O. and A.M.; supervision, R.M., R.G.F.L., G.L., S.O. and T.K.; project administration, G.L., S.O. and T.K.; funding acquisition, K.O., Y.M.L., G.L., S.O. and T.K. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ogai, K., Nana, B.C., Lloyd, Y.M. et al. Skin microbiome profile of healthy Cameroonians and Japanese. Sci Rep 12, 1364 (2022). https://doi.org/10.1038/s41598-022-05244-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-05244-5
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
