
Abstract
Next-generation sequencing (NGS) technologies have been applied in bacterial flora analysis. However, there is no standardized protocol, and the optimal clustering threshold for estimating bacterial species in respiratory infection specimens is unknown. This study was conducted to investigate the optimal threshold for clustering 16S ribosomal RNA gene sequences into operational taxonomic units (OTUs) by comparing the results of NGS technology with those of the Sanger method, which has a higher accuracy of sequence per single read than NGS technology. This study included 45 patients with pneumonia with aspiration risks and 35 patients with lung abscess. Compared to Sanger method, the concordance rates of NGS technology (clustered at 100%, 99%, and 97% homology) with the predominant phylotype were 78.8%, 71.3%, and 65.0%, respectively. With respect to the specimens dominated by the Streptococcus mitis group, containing several important causative agents of pneumonia, Bray Curtis dissimilarity revealed that the OTUs obtained at 100% clustering threshold (versus those obtained at 99% and 97% thresholds; medians of 0.35, 0.69, and 0.71, respectively) were more similar to those obtained by the Sanger method, with statistical significance (p < 0.05). Clustering with 100% sequence identity is necessary when analyzing the microbiota of respiratory infections using NGS technology.
Similar content being viewed by others

Introduction
Bacterial pneumonia is a major infectious disease worldwide, and the number of patients with pneumonia is predicted to increase, particularly in Japan, where the population is rapidly aging. Therefore, it is important to properly evaluate the bacteriological etiology to provide optimal treatments for these patients in clinical practice1. Next-generation sequencing (NGS) technologies have been applied in bacterial flora analysis, and advances in these methods over the last decade have revealed the presence of microbiota in the lower respiratory tract, which was previously considered as sterile2,3,4. The use of NGS technology has become more common in microbiota analysis, including to assess respiratory infections5,6,7, but there are some limitations in interpreting the results.
In NGS analysis, technical methods, such as those used for the selection of target genes and gene region or length, as well as equipment are diverse. For example, untargeted metagenomic NGS of clinical samples may be the most promising approach for comprehensively detecting agents responsible for infectious diseases and evaluating normal flora, whereas the analysis of large datasets requires a combination of bioinformatics skills; therefore, few laboratories can use NGS diagnostically8,9. Targeted sequencing, including sequencing part of the 16S ribosomal RNA (rRNA) gene, is a superior approach for detecting and estimating bacterial species compared to metagenomic NGS methods because of their low complexity, low cost, and practical clinical application. However, the analysis pipelines, including assemblers and analytical parameters, can significantly influence the final results, and incorrect sequence results have been reported10,11. There is no standardization between amplicon studies with NGS technology12, leading to issues with research replication13. Clustering of 16S rRNA sequences into operational taxonomic units (OTUs) was generally analyzed in the gastrointestinal and urological areas with thresholds of 99% and 97%, respectively14,15,16,17, but it is unclear whether this approach is applicable to the microbiota in respiratory specimens. In recent years, analysis based on amplicon sequence variants (ASVs) has proven to be useful because it increases the accuracy of analysis per sequence compared to OTU analysis18,19,20. However, several problems have been highlighted regarding ASV analysis21,22,23, such as the possibility that species of the genus Streptococcus represented by Streptococcus pneumoniae, which are important in respiratory infections, are underestimated21. Therefore, further validation of the method is required before use in the assessment of pneumonia with aspiration risks in which Streptococcus species are frequently detected. OTU analysis has been utilized in various fields of research14,24,25, and verification of the accuracy of this method is required.
A disadvantage of performing clone library analysis using the Sanger method is the reduced analysis capacity per operation compared to NGS technology; however, the Sanger method can read comparatively long sequences (700–1000 bp) with high accuracy (99.999%)26 relative to NGS and can estimate the species level for some bacteria27. The combination of the clone library and Sanger methods can be used to estimate predominant bacterial species without the use of culture-based methods in clinical specimens. We have reported the usefulness and significance of this approach in evaluating respiratory infections28,29,30,31,32,33.
The genus Streptococcus, including Streptococcus pneumoniae or Streptococcus intermedius, is a major causative agent of bacterial pneumonia in elderly people30,34 and in patients with lung abscess31. Streptococcus pneumoniae is generally the major causative agent in respiratory infections, but species such as, oral streptococci, Streptococcus pseudopneumoniae35,36, and S. intermedius37,38 are important in estimating antibiotic resistance and the clinical course of the disease. The nucleotide sequences of 16S rRNA regions of these organisms, including S. pneumoniae and Streptococcus mitis, are very similar27, and thus correct nucleotide sequence identification is crucial; however, it is unclear whether these bacteria can be distinguished by targeted NGS analysis.
Respiratory infections exhibit diverse bacterial flora even in each sample directly obtained from the lung, which are difficult to characterize using commercially available bacterial cultures. Therefore, we retrospectively evaluated bronchoalveolar lavage fluid (BALF) samples from patients with pneumonia with aspiration risks and from patients with lung abscess, in which the genus Streptococcus plays an important role, to compare the results of the Sanger method combined with the clone library method and NGS methods. We also investigated the optimal threshold for clustering 16S rRNA gene sequences into OTUs by NGS technology by comparing the results of NGS technology with those of the Sanger method.
Methods
Study population
This was a retrospective study targeting 177 patients with community-onset pneumonia (community-acquired pneumonia and healthcare-associated pneumonia) and 61 patients with lung abscess on whom a bronchoscopy examination was performed at the University of Occupational and Environmental Health, Japan, and affiliated community hospitals between April 2010 and March 2016. Most patients were included in previous studies30,31. Patients less than 65 years of age and those who did not meet the definition of pneumonia with aspiration risks and lung abscess were excluded30,31. Two cases were also excluded during quality verification of the BALF samples. Finally, 45 of 177 patients with pneumonia with aspiration risks and 35 of 61 patients with lung abscess were included in this study (Fig. 1). This study was approved by the Ethics Committee of Medical Research, University of Occupational and Environmental Health, Japan (UOEHCRB18-016). All experiments were performed in accordance with relevant guidelines and regulations. The need for written informed consent was waived by the Ethics Committee of Medical Research, University of Occupational and Environmental Health, Japan because of the retrospective study design.

Patient inclusion and exclusion flow diagram.
Definitions
Community-onset pneumonia was defined according to the guidelines of the Infectious Diseases Society of America/American Thoracic Society39. Aspiration risks were defined according to the criteria of Marik et al.40 as in our previous reports30,31 and included neurologic dysphagia, disruption of the gastroesophageal junction, or anatomical abnormalities of the upper aerodigestive tract.
Lung abscess was diagnosed when the following three criteria were fulfilled: (a) the presence of new areas of infiltrations with a cavity or low-density area within the infiltrates on chest radiographs and/or computed tomography; (b) new clinical findings including at least two of the following: fever, sputum production, coughing, chest pain, and leukocytosis (peripheral white blood cell count ≥ 10,000/μL), and (c) exclusion of other causes31.
Sample collection
Fiberoptic bronchoscopy was performed as previously described29. Briefly, BALF samples were obtained using 40 mL of sterile saline from the affected lesions of patients with pneumonia or lung abscess.
Cell lysis efficiency analysis and DNA extraction
DNA samples were extracted from the BALF samples by adding sodium dodecyl sulfate (final concentration: 3.0%) and glass beads followed by vigorous shaking, as previously described29.
Microbiota analysis using the Sanger method
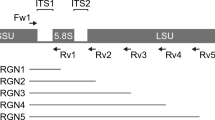
The partial 16S rRNA gene fragments (approximately 550 bp) were amplified by polymerase chain reaction (PCR) with universal primers (E341F and E907R), as previously described29. The amplified products were cloned into Escherichia coli using a TOPO TA cloning kit (Invitrogen, Carlsbad, CA, USA). Nucleic acid sequences of 96 randomly selected clones in each clone library were determined using a 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Highly accurate sequences selected by Phred quality values were trimmed from the primer and vector regions, and only sequences with good quality were used for analysis29.
NGS library preparation and Illumina MiSeq sequencing
Extracted DNA samples were purified using Agencourt AMPure XP (Beckman Coulter, Brea, CA, USA). Two-step PCRs were performed on the purified DNA samples to obtain sequence libraries. The first PCR was an amplification step performed using a 16S (V3‒V4) Metagenomic Library Construction Kit for NGS (Takara Bio, Shiga, Japan) with primer pairs, 341F (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′) and 806R (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGACTACHVGGGTWTCTAAT-3′), corresponding to the V3‒V4 region of the 16S rRNA gene. Thermal cycling was performed in a TaKaRa PCR Thermal Cycler Dice Gradient under the following conditions: initial denaturation at 94 °C for 1 min, followed by 28 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 15 s, and extension at 68 °C for 15 s. PCR amplicons were purified using AMPure XP magnetic purification beads.
The second PCR was performed to add the index sequences for Illumina sequencing using a Nextera XT Index kit v2 (Illumina, San Diego, CA, USA) and Tks Gflex DNA polymerase S (Takara Bio). Thermal cycling was performed in a TaKaRa PCR Thermal Cycler Dice Gradient (Takara Bio) with the following conditions: initial denaturation at 94 °C for 1 min, followed by 8 cycles of denaturation at 98 °C for 10 s, annealing at 60 °C for 15 s, and extension at 68 °C for 15 s. PCR amplicons were evaluated for quality verification of the sequence library using an Agilent 4200 TapeStation (Agilent Technologies, Santa Clara, CA, USA).
After quality verification, mixed samples were prepared by pooling approximately equal amounts of each amplified DNA and sequenced using MiSeq Reagent Kit V3 (250 × 2 cycles) and a MiSeq sequencer (Illumina), according to the manufacturer’s instructions. The obtained nucleotide sequences were subjected to assembly and clustering processes using the CD-HIT-OTU algorithm41. First, low-quality reads (reads containing > 10 bases with an error rate ≥ 10%, more than 4 mismatches in the overlap region, or non-overlapping paired reads) were removed and assembled. The assembled sequences with 100% homology were clustered, and chimera sequences were removed. In addition, clusters with a low frequency (39 contigs or less) of occurrence estimated using simulations from the maximum cluster size were removed. The nucleotide sequences obtained were clustered based on homologies of 100%, 99%, and 97%. A representative sequence was selected; those of OTUs showed the longest lengths.
Homology searching
In the Sanger method, the resulting high-accuracy reads were directly homology searched, whereas for NGS technology, homology searching was performed for representative sequences of each OTU. The determined sequences were compared with an in-house database containing the 16S rRNA gene sequences of 5,878 type strains using the basic local alignment search tool (BLAST; NCBI, Bethesda, MD, USA) algorithm42. Bacterial type strains that were top hits with ≥ 97% homology identified using BLAST were classified as presumptive species43, and those with < 97% homology were classified as unidentified organisms29,30,31,44. The 16S rRNA gene sequences of type strains were obtained from the DNA Data Bank of Japan (http://www.ddbj.nig.ac.jp/) and the Ribosomal Database Project (http://rdp.cme.msu.edu/).
Interpretation of the results
The bacterial phylotype exhibiting the highest proportion of the total microbiota in each BALF sample was defined as the “predominant phylotype.” In addition, phylotypes that occupied less than 5% of each microbiota were classified as “Others,” as previously described29.
Bray Curtis dissimilarity
The Bray Curtis dissimilarity was calculated for each specimen to assess the degree of difference between the results of the Sanger method and NGS technology. This analysis is a standardized index that has a value of 0 when the species composition is similar between two microbiota and a maximum value of 1 when they are completely different45. Species included in “Others” and bacteria considered as unclassified organisms were excluded, and major populations were compared.
Statistical analysis
The statistical software PRISM 8 (GraphPad Software, Inc., San Diego, CA, USA) was used for the Kruskal–Wallis test and post hoc Dunn's test with Bonferroni adjustment for multiple comparisons, as appropriate.
Results
Patient characteristics
The characteristics of 80 patients (45 with pneumonia with aspiration risks and 35 with lung abscess) are shown in Table 1. The median age of the patients was 73 [interquartile range (IQR) 70–79] years and the inpatient mortality rate was 6.3%. In many cases (68 of 80 patients), the severity grade according to the Pneumonia Severity Index score46 was mild to moderate.
Comparison of the results of microbiota analysis between the Sanger method and NGS
There were 6,530 total clones, and the mean nucleotide sequence length using the Sanger method was 544.3 bases (per read). NGS generated 7,637,916 pairs of reads, and the total number of contigs after filtering and assembling was 4,103,775 with a mean nucleotide sequence length of 459.5 (see Supplementary Table S1 online). OTUs were created with 100%, 99%, and 97% clustering thresholds for subsequent analysis. The total numbers of OTUs clustered at 100%, 99%, and 97% identity were 3,663, 493, and 255, respectively. The rarefaction curves of the number of OTUs identified per sample almost plateaued with ~ 5000 sequence reads (see Supplementary Fig. S1 online).
In patients with pneumonia with aspiration risks, the most frequently detected bacterial phylotype by the Sanger method was S. pneumoniae (17.8%), followed by Streptococcus oralis (15.6%) and Haemophilus influenzae (13.3%) (Fig. 2). In contrast, the most frequently detected bacterial phylotype was S. oralis (20.0%), followed by S. pseudopneumoniae (17.8%), and H. influenzae (13.3%) by NGS technology (100% identity thresholds for clustering). Streptococcus pseudopneumoniae (37.8%) was the most abundant phylotype, followed by H. influenzae (13.3%) in NGS technology (99% and 97% identity thresholds for clustering).

Predominant phylotype by the Sanger method and NGS technology (pneumonia with aspiration risks). This pie chart shows the percentage of predominant bacterial phylotypes in the bronchoalveolar lavage fluid for the Sanger method (a), NGS (100%) (b), NGS (99%) (c), and NGS (97%) (d) for patients with pneumonia with aspiration risks. The same color represents the same bacterial phylotypes. NGS (100%) = NGS results obtained with a clustering threshold of 100%; NGS (99%) = NGS results obtained with a clustering threshold of 99%; NGS (97%) = NGS results obtained with a clustering threshold of 97%.
In lung abscess, the most frequently detected bacterial phylotype by the Sanger method was S. intermedius (22.9%), followed by Fusobacterium nucleatum (17.1%) and Streptococcus salivarius (8.6%) (Fig. 3). Alternately, the most frequently detected bacterial phylotype was S. intermedius (22.9%), followed by F. nucleatum (11.4%) and S. salivarius (8.6%) in NGS technology (100% identity thresholds for clustering). At 99% and 97% identity thresholds for clustering in NGS technology, S. intermedius (22.9%) was the most frequently detected phylotype, followed by F. nucleatum (14.3%) and S. pseudopneumoniae (11.4% at 99% identity thresholds for clustering, 14.3% at 97% identity thresholds for clustering).

Predominant phylotype by the Sanger method and NGS technology (lung abscess). This pie chart shows the percentage of predominant bacterial phylotypes in the bronchoalveolar lavage fluid for the Sanger method (a), NGS (100%) (b), NGS (99%) (c), and NGS (97%) (d) for patients with lung abscess. The same color represents the same bacterial phylotypes. NGS (100%) = NGS results obtained with a clustering threshold of 100%; NGS (99%) = NGS results obtained with a clustering threshold of 99%; NGS (97%) = NGS results obtained with a clustering threshold of 97%.
The concordance rates in the results of the predominant phylotype determined by NGS technology, using 100%, 99%, and 97% identity thresholds for clustering, with those of the Sanger method were 78.8% (63 of 80), 71.3% (57 of 80), and 65.0% (52 of 80), respectively (see Supplementary Table S2 online).
Bray Curtis dissimilarity of Sanger method versus NGS technology with NGS clustering threshold changes
The results of Bray Curtis dissimilarity between the Sanger method and NGS technology showed no differences in microbiota similarity among the 100%, 99%, and 97% clustering thresholds in pneumonia with aspiration risks (NGS (100%): median 0.26 [(IQR) 0.07–0.44] vs NGS (99%): 0.31 [0.05–0.70] vs NGS (97%): 0.36 [0.05–0.75], p = 0.59) and lung abscess (NGS (100%): median 0.26 [IQR 0.09–0.48] vs NGS (99%): 0.26 [0.07–0.45] vs NGS (97%): 0.27 [0.05–0.51], p = 0.88) (Fig. 4).

Bray Curtis dissimilarity between the Sanger method and NGS technology. The results of the Bray Curtis dissimilarity to assess the similarity of microbiota analysis between the Sanger method and NGS technology in patients with pneumonia with aspiration risks (a) and patients with lung abscess (b) showed no difference in flora similarity between clustering thresholds (pneumonia with aspiration risks: p = 0.59, lung abscess: p = 0.88). P-values were determined by Kruskal–Wallis test. NGS (100%) = NGS results obtained with a clustering threshold of 100%; NGS (99%) = NGS results obtained with a clustering threshold of 99%; NGS (97%) = NGS results obtained with a clustering threshold of 97%.
As shown in Fig. 2, the ability to detect bacterial phylotypes in the S. mitis group, such as S. oralis and S. pseudopneumoniae, differed according to the clustering threshold. Based on these results, we compared the results of the Sanger method and NGS technology in eight cases in which the S. mitis group (S. oralis and S. pseudopneumoniae), excluding S. pneumoniae, was detected as the predominant bacterial species using the Sanger method in pneumonia with aspiration risks (Fig. 5). Of note, Bray Curtis dissimilarity revealed that the OTUs obtained with the 100% threshold (median 0.35 [IQR 0.26–0.45]) were more similar to those obtained by the Sanger method than to those obtained with the 99% (0.69 [0.54–0.78]) and 97% (0.71 [0.58–0.77]) thresholds (p < 0.05) (Fig. 6).

Detected phylotypes using the Sanger method and NGS technology in the eight pneumonia cases in which Streptococcus mitis group (other than pneumococcus) was detected predominantly by the Sanger method. NGS (100%) = NGS results obtained with a clustering threshold of 100%; NGS (99%) = NGS results obtained with a clustering threshold of 99%; NGS (97%) = NGS results obtained with a clustering threshold of 97%.

Similarity of the Sanger method and NGS technology using Bray Curtis dissimilarity in the eight pneumonia cases in which Streptococcus mitis group (other than pneumococcus) was detected predominantly by the Sanger method. Bray Curtis dissimilarity revealed that OTUs obtained at 100% clustering threshold were more similar to those obtained by the Sanger method than did those obtained at 99% and 97% thresholds (p < 0.05). P-values were determined by Kruskal–Wallis test and a post hoc Dunn’s test with Bonferroni adjustment for multiple comparisons. NGS (100%) = NGS results obtained with a clustering threshold of 100%; NGS (99%) = NGS results obtained with a clustering threshold of 99%; NGS (97%) = NGS results obtained with a clustering threshold of 97%.
Total number of OTUs detected as unidentified organisms in each sample
The total number of unidentified organisms increased when the clustering threshold was set to 100% (median 180 [IQR 128–244]) compared to 99% (48 [36–63]) and 97% (39 [27–46]) (p < 0.001) (Fig. 7).

Total number of OTUs detected as unidentified organisms in each sample. The total number of unidentified organisms increased when the clustering threshold was set to 100% (p < 0.001). P-values were determined by Kruskal–Wallis test and a post hoc Dunn’s test with Bonferroni adjustment for multiple comparisons. NGS (100%) = NGS results obtained with a clustering threshold of 100%; NGS (99%) = NGS results obtained with a clustering threshold of 99%; NGS (97%) = NGS results obtained with a clustering threshold of 97%.
Discussion
In this study, we investigated the optimal threshold for clustering 16S rRNA gene sequences into OTUs using NGS technology (MiSeq sequencer; Illumina) by comparing the results with those obtained using the Sanger method. We demonstrated the difficulty in distinguishing the bacterial phylotype of the S. mitis group, which is the most common bacteria in respiratory infections, when the clustering threshold was set to low (< 99% OTUs). When analyzing the microbiota of respiratory infections by NGS technology, particularly in bacterial pneumonia, clustering at 100% sequence identity is necessary.
Generally, S. pneumoniae is the main causative agent of community-onset pneumonia47,48, but we previously reported that oral streptococci may also play an important role in elderly people with pneumonia with aspiration risks30,31. We detected S. pseudopneumoniae by NGS technology as the predominant phylotype in all nine cases in which S. pneumoniae phylotypes were detected by the Sanger method (Figs. 2 and 3). It was not possible to distinguish between S. pneumoniae and S. pseudopneumoniae because these two bacterial phylotypes were identical in the NGS amplification region (V3‒V4) used in this study (see Supplementary Fig. S2 online). Evaluating longer regions, such as the V3‒V5 region, using the MiSeq sequencer has been reported to reduce the accuracy of the second half of the read49. The Sanger method revealed that S. pseudopneumoniae dominated in one case (1.3%). Thus, the Sanger method is a better method for discriminating between these two species when examined in respiratory infections in the clinical setting.
NGS analysis with 99% and 97% homology revealed S. pseudopneumoniae as the predominant phylotype in 21 cases (26.3%) and 22 cases (27.5%), respectively (Figs. 2 and 3). In addition, the nine cases in which S. oralis was detected as the predominant phylotype by the Sanger method showed a concordance rate of 88.9% (8/9) with the results of NGS when the clustering threshold was set to 100%. However, at clustering thresholds under 100%, S. pseudopneumoniae was identified in all nine cases. Streptococcus oralis was identified as a minor population when the clustering threshold was above 99%. These findings indicate that the phylotype distinction depends on the clustering threshold; therefore, it is necessary to set the clustering threshold at 100% to estimate bacterial phylotypes more accurately.
Clustering of 16S rRNA sequences into OTUs removes minor artifactual sequence variants due to PCR amplification and sequencing errors when collapsing sequences into groups16. In this study, when the clustering threshold was set to 100%, the number of bacterial species defined as unidentified organisms increased. It is not possible to evaluate whether the bacterial species included in the unidentified organism group are an actual minor population or artifact. Using a high threshold, such as 100%, will result in the exclusion of samples because in several instances the number of reads falls considerably below the threshold in downstream processing; however, this can be compensated by re-sequencing the samples.
When the clustering threshold was set to 97%, some bacterial phylotypes, such as Corynebacterium spp. differed in the results of the Sanger method and NGS technology at the species level. Generally, the genus Corynebacterium is often regarded as an oral contaminant. In recent years, pneumonia due to Corynebacterium striatum has been reported50,51, and we also reported the importance of the genus Corynebacterium in pneumonia29,44, particularly when hospital-acquired52. The genus Corynebacterium has been reported to have different drug sensitivities depending on the bacterial species; therefore, species-level identification is desirable when Corynebacterium is suspected as the causative agent of pneumonia53. When the clustering threshold was set to 97%, Corynebacterium simulans and C. striatum were classified into the same OTUs. Comparison of the nucleotide sequences of the type strain (C. simulans, accession No. AJ012837; C. striatum, accession No. X84442) in the V3‒V4 amplification region for these two strains showed mismatches of three base pairs and homology of more than 99%. Therefore, when the clustering threshold was set to 97%, C. simulans and C. striatum were classified into the same OTUs by NGS technology.
There were several limitations to this study. First, this study was retrospective and included only patients with sufficient specimens available for analysis. Second, there was a difference in the analysis primer region between NGS technology and the Sanger method. Third, bacterial species with abundances of 5% or less were treated as “Others” as previously reported29, but there is no clear definition of the makeup of this group. In this study, we did not examine the minor population, but pathogenic bacteria, such as Mycobacterium tuberculosis may be included; thus, it is necessary to examine the minor population in further studies. Fourth, accurate identification of bacterial species is difficult via analysis of the 16S rRNA gene sequence alone. However, predicting the bacterial species is highly useful in deciding treatment strategies such as antibiotic selection. Therefore, it is important to utilize methods based on 16S rRNA gene sequencing that can help evaluate microbiome information as accurately as possible.
Microbiome analysis using NGS technology will continue to progress through technological innovation. When analyzing the microbiota of respiratory infections using NGS technology, it is necessary to cluster with a 100% sequence identity. Alternatively, it is necessary to use an analysis method that can maintain high accuracy for sequencing analysis, such as that for ASVs. If the analysis is performed using a low threshold on sequence clustering, important pathogens may not be identified, and/or incorrect phylotype information may be obtained.
Data availability
The sequence read data were submitted to a public database (DNA Data Bank of Japan Sequence Read Archive under the BioProject identifier PRJDB11117).
References
Budden, K. F. et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir. Med. 7, 907–920 (2019).
Hilty, M. et al. Disordered microbial communities in asthmatic airways. PLoS ONE 5, e8578 (2010).
Charlson, E. S. et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care Med. 184, 957–963 (2011).
Huang, Y. J. et al. The role of the lung microbiome in health and disease. A National Heart, Lung, and Blood Institute workshop report. Am. J. Respir. Crit. Care Med. 187, 1382–1387 (2013).
Chen, X. et al. Blood and bronchoalveolar lavage fluid metagenomic next-generation sequencing in pneumonia. Can. J. Infect. Dis. Med. Microbiol. 2020, 6839103 (2020).
Xu, A. et al. Diagnosis of severe community-acquired pneumonia caused by Acinetobacter baumannii through next-generation sequencing: A case report. BMC Infect. Dis. 20, 45 (2020).
Fernández-Barat, L., López-Aladid, R. & Torres, A. Reconsidering ventilator-associated pneumonia from a new dimension of the lung microbiome. EBioMedicine 60, 102995 (2020).
Chiu, C. Y. & Miller, S. A. Clinical metagenomics. Nat. Rev. Genet. 20, 341–355 (2019).
Sabat, A. J. et al. Targeted next-generation sequencing of the 16S–23S rRNA region for culture-independent bacterial identification: Increased discrimination of closely related species. Sci. Rep. 7, 3434 (2017).
Jünemann, S. et al. GABenchToB: A genome assembly benchmark tuned on bacteria and benchtop sequencers. PLoS ONE 9, e107014 (2014).
Mee, E. T., Preston, M. D., Minor, P. D., Schepelmann, S. & Participants, C. S. S. Development of a candidate reference material for adventitious virus detection in vaccine and biologicals manufacturing by deep sequencing. Vaccine 34, 2035–2043 (2016).
Sinha, R. et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat. Biotechnol. 35, 1077–1086 (2017).
Sinha, R., Abnet, C. C., White, O., Knight, R. & Huttenhower, C. The microbiome quality control project: Baseline study design and future directions. Genome Biol. 16, 276 (2015).
Wang, J. et al. Uncovering the microbiota in renal cell carcinoma tissue using 16S rRNA gene sequencing. J. Cancer Res. Clin. Oncol. 147, 481–491 (2021).
Wirth, U. et al. Microbiome analysis from paired mucosal and fecal samples of a colorectal cancer biobank. Cancers 12, 3702 (2020).
Johnson, J. S. et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 10, 5029 (2019).
Park, J. I. et al. Comparative analysis of the tonsillar microbiota in IgA nephropathy and other glomerular diseases. Sci. Rep. 10, 16206 (2020).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 13, 581–583 (2016).
García-López, R. et al. OTUs and ASVs produce comparable taxonomic and diversity from shrimp microbiota 16S profiles using tailored abundance filters. Genes 12, 564 (2021).
Callahan, B. J. et al. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 47, e103 (2019).
Prodan, A. et al. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 15, e0227434 (2020).
Schloss, P. D. Amplicon sequence variants artificially split bacterial genomes into separate clusters. mSphere 6, e0019121 (2021).
Brandt, M. I. et al. Bioinformatic pipelines combining denoising and clustering tools allow for more comprehensive prokaryotic and eukaryotic metabarcoding. Mol. Ecol. Resour. 21, 1904–1921 (2021).
Sodhi, K. K., Kumar, M. & Singh, D. K. Assessing the bacterial diversity and functional profiles of the River Yamuna using Illumina MiSeq sequencing. Arch. Microbiol. 203, 367–375 (2021).
Zhang, Y., Zhu, C., Feng, X. & Chen, X. Microbiome variations in preschool children with halitosis. Oral Dis. 27, 1059–1068 (2021).
Shendure, J. & Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 26, 1135–1145 (2008).
Yatera, K., Noguchi, S. & Mukae, H. The microbiome in the lower respiratory tract. Respir. Investig. 56, 432–439 (2018).
Kawanami, T. et al. A higher significance of anaerobes: The clone library analysis of bacterial pleurisy. Chest 139, 600–608 (2011).
Yamasaki, K. et al. Significance of anaerobes and oral bacteria in community-acquired pneumonia. PLoS ONE 8, e63103 (2013).
Akata, K. et al. The significance of oral streptococci in patients with pneumonia with risk factors for aspiration: The bacterial floral analysis of 16S ribosomal RNA gene using bronchoalveolar lavage fluid. BMC Pulm. Med. 16, 79 (2016).
Mukae, H. et al. The importance of obligate anaerobes and the Streptococcus anginosus group in pulmonary abscess: A clone library analysis using bronchoalveolar lavage fluid. Respiration 92, 80–89 (2016).
Naito, K. et al. Bacteriological incidence in pneumonia patients with pulmonary emphysema: A bacterial floral analysis using the 16S ribosomal RNA gene in bronchoalveolar lavage fluid. Int. J. Chron. Obstruct. Pulm. Dis. 12, 2111–2120 (2017).
Hata, R. et al. Poor oral hygiene is associated with the detection of obligate anaerobes in pneumonia. J. Periodontol. 91, 65–73 (2020).
Sadowy, E. & Hryniewicz, W. Identification of Streptococcus pneumoniae and other Mitis streptococci: Importance of molecular methods. Eur. J. Clin. Microbiol. Infect. Dis. 39, 2247–2256 (2020).
Mohammadi, J. S. & Dhanashree, B. Streptococcus pseudopneumoniae: An emerging respiratory tract pathogen. Indian J. Med. Res. 136, 877–880 (2012).
Dupont, C. et al. Streptococcus pseudopneumoniae, an opportunistic pathogen in patients with cystic fibrosis. J. Cyst Fibros. 19, e28–e31 (2020).
Noguchi, S. et al. The clinical features of respiratory infections caused by the Streptococcus anginosus group. BMC Pulm. Med. 15, 133 (2015).
Dyrhovden, R., Nygaard, R. M., Patel, R., Ulvestad, E. & Kommedal, Ø. The bacterial aetiology of pleural empyema. A descriptive and comparative metagenomic study. Clin. Microbiol. Infect. 25, 981–986 (2019).
Mandell, L. A. et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin. Infect. Dis. 44(Suppl 2), S27-72 (2007).
Marik, P. E. Aspiration pneumonitis and aspiration pneumonia. N. Engl. J. Med. 344, 665–671 (2001).
Li, W., Fu, L., Niu, B., Wu, S. & Wooley, J. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief Bioinform. 13, 656–668 (2012).
Otsuji, K. et al. Dynamics of microbiota during mechanical ventilation in aspiration pneumonia. BMC Pulm. Med. 19, 260 (2019).
Drancourt, M. et al. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J. Clin. Microbiol. 38, 3623–3630 (2000).
Noguchi, S. et al. Bacteriological assessment of healthcare-associated pneumonia using a clone library analysis. PLoS ONE 10, e0124697 (2015).
Faith, D. P., Minchin, P. R. & Belbin, L. Compositional dissimilarity as a robust measure of ecological distance. Vegetatio 69, 57–68 (1987).
Fine, M. J. et al. A prediction rule to identify low-risk patients with community-acquired pneumonia. N. Engl. J. Med. 336, 243–250 (1997).
Metlay, J. P. et al. Diagnosis and treatment of adults with community-acquired pneumonia. An Official Clinical Practice Guideline of the American Thoracic Society and Infectious Diseases Society of America. Am. J. Respir. Crit. Care Med. 200, e45–e67 (2019).
Harju, I. et al. Improved differentiation of Streptococcus pneumoniae and other S. mitis group Streptococci by MALDI Biotyper using an improved MALDI Biotyper database content and a novel result interpretation algorithm. J. Clin. Microbiol. 55, 914–922 (2017).
McGovern, E., Waters, S. M., Blackshields, G. & McCabe, M. S. Evaluating established methods for rumen 16S rRNA amplicon sequencing with mock microbial populations. Front. Microbiol. 9, 1365 (2018).
Yang, K., Kruse, R. L., Lin, W. V. & Musher, D. M. Corynebacteria as a cause of pulmonary infection: A case series and literature review. Pneumonia 10, 10 (2018).
Shariff, M., Aditi, A. & Beri, K. Corynebacterium striatum: An emerging respiratory pathogen. J. Infect. Dev. Ctries. 12, 581–586 (2018).
Yatera, K. & Mukae, H. Corynebacterium species as one of the major causative pathogens of bacterial pneumonia. Respir. Investig. 58, 131–133 (2020).
Reddy, B. S. et al. Isolation, speciation, and antibiogram of clinically relevant non-diphtherial Corynebacteria (Diphtheroids). Indian J. Med. Microbiol. 30, 52–57 (2012).
Funding
This work was partly funded by KYORIN Pharmaceutical Co., Ltd.
Author information
Authors and Affiliations
Contributions
All key decisions were made by all authors. H. I. had full access to all of the data in the study and takes responsibility for the integrity of the data and accuracy of the data analysis. H. I., S. N., K. F., K. A., K. Yamasaki, T. K., H. M., and K. Yatera contributed substantially to the study design, data analysis and interpretation, and the writing of the manuscript. K. Yatera had the final responsibility for the decision to submit the article for publication. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ikegami, H., Noguchi, S., Fukuda, K. et al. Refinement of microbiota analysis of specimens from patients with respiratory infections using next-generation sequencing. Sci Rep 11, 19534 (2021). https://doi.org/10.1038/s41598-021-98985-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-98985-8
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
