
Abstract
Target identification and prioritisation are prominent first steps in modern drug discovery. Traditionally, individual scientists have used their expertise to manually interpret scientific literature and prioritise opportunities. However, increasing publication rates and the wider routine coverage of human genes by omic-scale research make it difficult to maintain meaningful overviews from which to identify promising new trends. Here we propose an automated yet flexible pipeline that identifies trends in the scientific corpus which align with the specific interests of a researcher and facilitate an initial prioritisation of opportunities. Using a procedure based on co-citation networks and machine learning, genes and diseases are first parsed from PubMed articles using a novel named entity recognition system together with publication date and supporting information. Then recurrent neural networks are trained to predict the publication dynamics of all human genes. For a user-defined therapeutic focus, genes generating more publications or citations are identified as high-interest targets. We also used topic detection routines to help understand why a gene is trendy and implement a system to propose the most prominent review articles for a potential target. This TrendyGenes pipeline detects emerging targets and pathways and provides a new way to explore the literature for individual researchers, pharmaceutical companies and funding agencies.
Similar content being viewed by others


Introduction
Pharmaceutical companies are actively looking for ways to reduce their attrition rates, the time taken for drug development, and the associated development costs1,2,3,4. One approach being explored to address this productivity challenge is the exploitation of big biomedical data sets through machine learning5,6. Evidence is emerging that machine learning can be used to speed-up and reduce the costs in all stages in drug discovery5,6: drug repurposing7,8, clinical trials9,10, de-novo drug design11,12,13,14,15,16,17,18,19,20, and target-disease associations21,22,23,24,25. However, target identification and prioritisation remain the first step for the majority of drug discovery programmes25,26,27,28. Only 10% of drug targets progress through clinical trials28,29,30 and this success rate appears lower for novel targets30,31,32. Historically, target identification has been broadly carried out on a case-by-case basis, based on the scientific interpretation of the available literature. However, thousands of peer-reviewed articles are published every day without taking into account pre-prints, patent data, and clinical trial reports33. PubMed alone contains more than 30 million publications as of 2020, and the scientific output doubles every nine years34, creating a corpus of "undiscovered public knowledge"35. Thus, there is a high demand for machine learning and other computational methods to exploit the current knowledge and facilitate the maintenance of an overview of this overwhelming literature volume. The development of (i) alert systems to identify and rank emerging targets at genomic-scale and (ii) recommendation systems to prioritise detailed reading of scientific reviews is of importance for both pharmaceutical companies and the whole scientific community25,27,36.
One of the most significant obstacles for the automatic analysis of biomedical literature is the use of non-redundant alternative gene synonyms, symbols, and acronyms from competing sources that can have other meanings in different areas of research37. Therefore, it is imperative to disambiguate biomedical entities in the scientific literature at the outset. There have been several attempts in this line of research21,22,23,24,37,38,39,40,41,42,43,44,45,46. However, these attempts do not unambiguously map gene and disease entities in scientific literature to controlled ontologies nor do they define an ambiguity measure for gene and disease synonyms. Although there have been multiple attempts about trend detection and burst term detection48,49 and more concretely about the biomedical literature of targets and small molecules50,51,52 to our knowledge this is the first attempt to analyse emerging trends about human protein coding genes.
Here we propose a new disambiguation algorithm based on co-citation networks and natural language processing to obtain accurate publication dynamics for every coding-gene in the human genome. This time-series data was used to train recurrent neural networks (RNN) in historical data and predict the state of the literature in recent years. We identify which genes are being mentioned in the literature more than expected in order to highlight and rank potential targets. This genome scale ranking is not alone sufficient for target assessment since this will not include assessment of tractability, commercial opportunity or clinical translatability, but identification of emerging biology is a key component of novel target identification. When the actual number of published articles exceeds predictions, there may have been a paradigm shift for that particular gene. Finally, we implemented topic detection algorithms along with recommendation systems to validate trendy targets. Therefore, the aims of this paper are fourfold: (i) to unambiguously detect genes and diseases within articles with a novel named entity recogniser (ii) to generate a ranking of genes and diseases based on a novel metric that defines its trendiness, (iii) to generate an automatic pipeline to analyse why these biological entities may be trendy, and (iv) to generate a recommendation system to suggest which articles to read which maximise the information coverage in subnetworks.
Results
Gene annotation
We gathered the human gene synonyms from different sources (Ensembl, UniProt, HGCN, Entrez and OpenTargets; Fig. 1B) to sample the potential publications mentioning human gene names. Human genes had around 10 synonyms on average and many of those synonyms are ambiguous (Table 1): More than 30% of gene symbols had at least one promiscuous synonym, around 10% of the gene symbols are unsafe and have at least one gene synonym in the English dictionary, and almost 50% of gene symbols had a nested synonym. Combining these problems, almost 60% of the 19,082 gene symbols have one or more of these four types of ambiguity. To determine which synonyms are potentially ambiguous (“unsafe gene synonyms”; Fig. 1C) we did feature engineering to obtain variables that characterise unsafe synonyms (e.g. longer gene names are less probable to be ambiguous; Table 2). Next, we used a positive-unlabelled bagging (PU) strategy following Mordelet et al. implementation55 with a random forest classifier with the engineered features to calculate the probability of a gene synonym to be “unsafe” (see Methods).

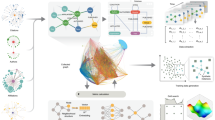
Workflow. Chart summarising the process from the downloading of the data to the detection and analysis of trends in the literature. (A) Creation of a graph database with the information contained in PubMed baseline 2020. (B) Acquisition of a comprehensive collection of human coding gene names and synonyms. (C) Automatic determination of potential ambiguous (unsafe) gene names. (D) Annotation of the graph database with unambiguous gene symbols by combining co-citation network topology and binary classifiers. (E) Prediction of per-gene publication trends using RNN. When a gene has significantly more publications or citations than expected by the model it is considered to be trendy. (F) Automatic topic detection of collections of publications. We used this algorithm to quantify the evolution of topics in trendy gene publications over time. (G) A review recommender system that uses information from the citation network and topic detection to recommend the most efficient set of reviews to explore the literature.
To link every human gene to a subset of publications we implemented a disambiguation pipeline based on co-citation networks and machine learning (Fig. 1D). We gathered the titles, abstracts and keywords of the publications that had a match for any of the synonyms using regex with ElasticSearch (Fig. 1D). Nevertheless, this original set of publications potentially contains false positives: publications that contain an ambiguous gene synonym in their titles or abstracts, that do not refer to the gene of interest.
We assumed that true and false positives synonyms will tend to belong to different communities of publications from different research fields. To detect these communities we used co-citation networks (Fig. 1D): a weighted graph where the weight of the edges represents the frequency of two publications being cited simultaneously (co-cited) by a third publication. When two publications are repeatedly co-cited it strongly suggests that both belong to the same field of study56. We used the fast greedy modulation algorithm from iGraph to determine communities in the co-citation network and distinguished communities of publications focusing on the gene of interest by detecting the presence of “safe gene synonyms” in their titles and abstracts (Fig. 1D). The process is summarised in Fig. 2.

Disambiguation pipeline. (A) Citation network for a subset of PubMed IDs mentioning any of the gene synonyms of the gene symbol LRWD1, including ORCA. (B) Co-citation network of the same subset of PubMed IDs as in (A). (C) Communities for the co-citation graph obtained after using iGraphs fast greedy algorithm: killer whale community, orca plant cluster, LRWD1 in drosophila and LRWD1 in heterochromatin. (D) Number of safe synonyms per PubMed ID in title or abstract in the same co-citation network. (E) Citation network with reviews citing any of the PubMed IDs. (E) Review information as defined by the recommender system scaled from 0 to 1.
Finally, because we only used citations from open-access publications contained in PubMed Central (PMC)57, 46% of the publications were disconnected in the PubMed co-citation graph. To tackle this problem, we used again the inductive bagging positive-unlabelled approach to train multiple classifiers to associate the disconnected publications with the previously computed co-citation network components (Fig. 1C) using the words, phrases and one to four n-grams, contained in titles and abstracts. All available machine classifiers in Scikit Learn were used but logistic regression was selected due to its speed to accuracy ratio (Table 3).
To test the performance of the disambiguation pipeline we compared the disambiguation results with the gene-publication annotations from GeneRif58 (manually curated annotations), DISEASES59 (computational annotations), and UniProt60 (computational and manually curated annotations) (Table 4). On average, the disambiguation recovers > 85% of all publications contained in these databases. Both GeneRif and Uniprot annotation do not necessarily contain a gene-synonym in the title or abstract, therefore those publications are out of our pipeline. Disambiguation results present on average a 70% precision with UniProt, the only collection of disambiguated publications of a similar magnitude. Finally, we included the disambiguated gene-publication annotations into the graph database.
Trend detection
To detect incoming trends in the literature we gathered the publication dynamics of a given human gene from the disambiguated graph database (Fig. 1E). These time series include the number of publications, clinical trials, reviews and publications from big and medium-sized pharmaceutical companies, as well as, citations of publications coming from the mentioned categories per calendar year. Specifically, if a manuscript with author affiliations to big pharma cites other publications these citations are categorized as big pharma citations. Conversely publications citing this manuscript whose authors are affiliated to big pharma are not categorized as big pharma citations.
Time-series data from 1980 to 2013 was used to predict the per gene publication dynamics in each category between 2014 and 2019 using a Recurrent Neural Network model with an encoder-decoder architecture preceded by an attention layer, where both the encoder and decoder are composed of five hidden layers of Gated Recurrent Units (GRU). The time-series were created in a cumulative fashion, where each year contains the new publications and citations in addition to the previous ones.
For most genes, the model produces accurate predictions of the publication dynamics (Table 5), but for a small subset of genes the real number of publications or citations is significantly higher than expected (Fig. 3A). When the number of publications or citations exceeds the predictions, we interpret that the publication dynamics changed substantially in a way that cannot be explained simply by the gene’s publication history, implying that a meaningful discovery in the field has recently occurred (Fig. 3A; orange). Trendiness is defined as the probability of the fold-change between predicted and real number of publications and citations for a given gene. We used this metric to identify the trendiest genes in the academic community-using all publications-, or in the pharmaceutical industry-using publications coming from pharmaceutical companies-(Table S1, supplementary material).

Trend detection and gene–gene-disease co-occurrence. (A) Logarithmic scatter plots showing the predicted number of publications, reviews, citations and citations from big pharma companies against real data in the year 2018. (B) Trendiness (log2(predicted/real)) for genes associated with groups of diseases (MeSH parent categories). Left; Average trendiness of publications, reviews, citations and citations from reviews. Right; Average trendiness of citations coming from big and medium sized pharmaceutical companies. (C) Gene–Gene–Disease co-occurrence network of the first neighbours of CD274. Orange nodes are diseases, grey nodes are genes and the size of gene nodes represents the trendiness The grey edges are gene-disease association, the blue edges are gene-diseases with the width of the edges reflecting the number of co-occurrences.
Finally, to identify trendy genes of pharmaceutical interest, we computed the normalised mutual information of genes and diseases in the titles and abstracts of publications (Fig. 3B). Disease names and their synonyms were obtained from the Medical Subject Headings (MeSH) ontology at the Bioportal61. MeSH ontology contains 4818 different disease nodes at different levels of the ontology. We created a dictionary for each disease with the preferred and alternative names (see Methods). The diseases were disambiguated in titles and abstract using the same disambiguation pipeline used with the genes.
We noticed that many trendy genes cluster forming trendy pathways when getting the gene–gene and gene-disease association networks (Fig. 3C). We used enrichment of gene ontology (GO) terms for biological processes to uncover common pathways among the top 100 trendiest genes (Table S1, supplementary material). Among the most enriched GO terms in both academia and pharma are T cell co-stimulation, execution phase of necroptosis and pyroptosis. These biological processes are enriched in trendy genes which presumably reflect these fields of study are generating the most innovation and expectations in current biomedical research.
Topic detection
After the detection of gene trends, the next step was to understand why those genes might be trendy and curate possible mistakes in the disambiguation. With this aim we implemented a topic detection pipeline as an automatic, fast discovery tool to study groups of publications that mention the gene of interests (Fig. 1F). In this context, we used topic modelling algorithms. A topic is a collection of similar words, specific to a group of documents62. We used non-Negative Matrix Factorisation to generate a set of latent topics for each query (Fig. 4A; word clouds).

Topic time-lines. Topic time lines. Topic timelines for publications mentioning any of the genes for the immune checkpoint inhibitor (A), necroptosis (B) and pyroptosis (C) pathways. The x-axis represents the time in years and the y-axis represents the likelihood of a given topic. Colors represent different topics defined by the keywords contained in the correspondent word clouds. The latent four topics were obtained using Non-Negative-Factorization all publications annotated with the genes after disambiguation. Word clouds were created using the phrases with highest TFIDF for groups of publications belonging to each topic. All timelines show at least one rising topic after 2013 that represents the reason why these genes became trendy, their implications in human disease: immune checkpoint inhibitors and monoclonal antibodies (yellow and orange in A), activation of necroptosis (orange in B), agonists of STING1 in cancer (black in C).
We explored the evolution of the topics associated with some trendiest genes. For the immune checkpoint inhibitors (CD274, PDCD1, TGIT and CTLA4) the topic timeline suggests that there was a rapid decrease in the likelihood of publications discussing the biological role of these immune checkpoint inhibitors since 2010 (Fig. 4A in grey), which coincides with a notable increase in topics that discuss cancer therapies (Fig. 4A in orange) and monoclonal antibodies that target these four different transmembrane immunoglobulins (Fig. 4A in yellow).This way, the topic-detection pipeline is able to capture the evolution of the research from its biological description to the clinical application.
The topic timeline of the members of the necroptosis pathway (RIPK1, RIPK3 and MLKL; Fig. 4B) suggests that in the last decade there has been a decrease in the likelihood of publications discussing these genes in the context of apoptosis (Fig. 4B in grey), in favour of publications that discuss the newly discovered form of cell death, the necroptotic pathway (Fig. 4B in orange), as well as, the translational medicine perspective of this pathway as is suggested by words like mouse, treatment and activity or cancer (Fig. 4B, in blue).
Finally, the topic timeline the members of the pyroptosis pathway (CGAS, TMEM173, GSDMA and GSDMAD; Fig. 4C) shows a fast increase from 2013 of publications discussing the therapeutic opportunity in cancer immunotherapy with agonists for TMEM173 (Fig. 4B in grey), while again, the remaining topics seemed to contain information on the biochemistry and biological role of the genes.
Recommender system
In addition to the automatic topic detection, we designed a review recommender system to accelerate the screening of the publications that cover most of the information in a network (Fig. 1G). There are an average of 2.9 reviews citing any publication that mentions at least one gene name. The aim was to minimise the time reading and maximising the information within a gene subnetwork. The algorithm aggregates both topic and network information from the citation subgraph of the publications that mention the gene of interest to obtain the most query-centric reviews. The topic information comes from the latent topics obtained from the topic detection algorithm. The network information was captured by the PageRank scores of the subgraph (see Methods). This approach ensures that reviews citing publications with highest PageRank scores are prioritised. To further minimize the number of reviews for initial human analysis we avoid repetition of information by simultaneously maximising the cumulative PageRank score whilst minimising the overlap of combined citations. This way, we expect to obtain a small set of reviews that will cover the main topics and publications in the field. We used this recommender system to select the optimal subset of reviews to assess why genes might be trendy (see Discussion). An example of the output can be found for the genes in the discussion in the supplementary data file.
Discussion and conclusions
We present TrendyGenes as a first attempt to (i) establish a systematic analysis of contemporary topics associated to human genes and diseases, (ii) develop an alert system for emerging targets and trends in the scientific literature across the human, protein-coding genome, (iii) to use topic modelling to rapidly generate timelines of phrases that facilitate the understanding of why these genes are trendy.
We constructed a graph database containing PubMed data where publications are connected by citations and authors and are annotated with disambiguated human gene-names and diseases. We expect this new resource to provide new ways to navigate the scientific literature, detect and visualise networks of discussion and analyse networks of influence from key opinion leaders. Disambiguating author names from PubMed, MedRxiv, or BioRxiv would further improve the quality of the database. Machine or deep learning algorithms could be trained on already labelled data to improve on previously published approaches63,64,65 and address this issue.
Similarly further improvements in gene-name disambiguation would assist precision and recall metrics on our validation set suffer for different reasons GeneRIF and DISEASES include fewer publications in comparison to the genome wide metrics identified in our pipeline and there will be a lot of potential “false positives”. This makes the precision of our approach appear lower than what it may actually be. On the other hand, GeneRIF and Uniprot contain publications which either are not gene specific or do not mention the gene in the title or abstract.
However, the disambiguated genes and diseases can serve as labelled data for more sophisticated deep learning approaches to annotate biomedical entities. Gene and disease entities could be better annotated using both representation learning to capture the network topology and contextual information with transformer layers. Topic detection could be improved by using the state-of-the-art text summarisers with deep learning.
The number of publications per gene in aggregate is very predictable66. However, occasionally genes present significantly more publications than expected, meaning that a recent breakthrough occurred which cannot be accounted for from the publication dynamics. In this study, we show that trendiness can identify emerging targets from the literature for rapid profiling at genome-scale. We combined trendiness with gene-disease associations to prioritise potential drug targets: emergent genes associated with diseases but yet included in pharmaceutical publications are worthy of being investigated as potential targets. We observe that TrendyGenes usually cluster into the same biological pathways (Fig. 3C for CD274, PDCD1, CTLA4 and TIGIT). Here, using topic modelling and the recommendation system, we identify the trendiest genes and pathways and discuss some case studies to exemplify our pipeline. We selected genes pharmacological relevance by choosing genes with high trendiness both in the academia and the pharmaceutical industry with high association with disease and more than 100 publications. Reviews suggested by the recommender system for these genes are included in (whatReview2read.zip, supplementary material).
Immune checkpoint inhibitors: CTLA4, CD274, PDCD1, TIGIT
CTLA4, PDCD1 (PD-1), CD274 (PD-L1) and TIGIT are among the trendiest genes in academia and pharma in 2019 (Fig. 3A). CTLA4, PDCD1, CD274 and TIGIT genes encode four different transmembrane immunoglobulins that act as co-inhibitory receptors: checkpoints or ‘breaks’ for the adaptive immune response that prevent T cells from exerting their functions67,68. CTLA4 competes with its analogous CD28 for CD80 and CD86 to prevent a premature activation of T cells68. PDCD1-CD274 interaction counters the positive signals that may have already activated T effector cells68. TIGIT interacts with CD155 to down-regulate natural killer cells and T lymphocytes69. Cancer cells attempt to impair these checkpoints and currently there are 7 FDA approved monoclonal antibodies that target three of proteins (CTLA4: Ipilimumab70; PDCD1: Nivolumab71, Pembrolizumab72, Cemiplimab73; CD274: Atezolizumab74, Avelumab75) and multiple candidates targeting TIGIT (BGB-A121776, OMP-313M3277, MTIG7192A78, AB15479). Moreover, James Allison and Tasuku Honjo received the Nobel Prize in Medicine in 2018 for its research on immune checkpoint inhibitors47.
Neurodegeneration: TREM2 and C9orf72
C9orf72 encodes a guanine nucleotide exchange factor involved in endosomal trafficking and autophagy80,81. Hexanucleotide repeat expansions in promoter or intronic regions of C9orf72 are some of the major causes of sporadic and familial forms of both amyotrophic lateral sclerosis and frontotemporal dementia80. Antisense oligonucleotides are being used to impede the transcription of C9orf7282,83,84 or CRISPR–Cas9 system to target the GGGGCC repeat in the DNA85 or RNA85,86.
TREM2 gene encodes a transmembrane immunoglobulin receptor expressed in macrophages, osteoclasts, dendritic cells, and brain microglia87,88. TREM2 variants have been associated with Nasu-Hakola disease89,90, late-onset Alzheimer’s disease91,92,93,94, frontotemporal dementia95,96,97,98,99,100, amyotrophic lateral sclerosis101,102 and Parkinson’s disease101,103. TREM2 activates a pathway—through TYROBP/DAP12—that promotes inflammation87,88 and promotes phagocytosis of cellular waste, remains of apoptotic cells, and pathogens87,88. Currently, two independent groups have generated anti-TREM2 antibodies to stimulate microglia to remove amyloid plaques104. Furthermore, the mAb generated by one of these groups, Alenco, in collaboration with Abbvie, has entered Phase I clinical trials105,106.
DNA sensing by cGAS–STING: cGAS, TMEM173, GSDMD, GSDMA
The cytosolic nucleic acid-sensing pathway leads to pyroptosis, a lytic pro-inflammatory type of cell death involved in antiviral, antibacterial, and anticancer response107. cGAS is a nucleotidyl-transferase that catalyzes production of cyclic GMP-AMP (cGAMP) upon the recognition of double-stranded DNA107. TMEM173 (STING) binds to cGAMP and promotes the activation of both TBK1 and IRF3, increasing the transcription of genes encoding type I interferons107. GSDMA and GSDMD are pore-forming effector proteins in the plasma membrane to release proinflammatory interleukins like IL-1β and IL-18108. The cGAS-STING pathway has been associated to multiple autoimmune and chronic inflammatory diseases like non-alcoholic fatty liver disease109, systemic lupus erythematosus110, vascular and pulmonary syndrome111, macular degeneration112, Bloom syndrome113, Aicardi-Goutières syndrome114, cancer115, DNA damage116, neurodegeneration117 and beyond. Currently, there are ongoing clinical trials for TMEM173118,119,120 and GSDMD121 although there are no reported trials for GSDMA nor cGAS.
Necroptosis: RIPK1, RIPK3, and MLKL
RIPK1, RIPK3 and MLKL form part of the tumour necrosis factor-induced necroptosis pathway122,123,124. This pathway has been associated with multiple pathologies: systemic inflammatory response syndrome125,126, ulcerative colitis127,128, psoriasis128, rheumatoid arthritis128, neurodegenerative diseases129 and even cancer130,131,132. TNFR1, FasL, TRAIL, and TLR can all activate RIPK1 to decide the cell’s fate: inflammation, apoptosis or necrosis133. If caspase-8 is inhibited, RIPK1 and RIPK3 form the necrosome that subsequently phosphorylates MLKL134. MLKL forms homo-trimers135,136, migrates to the plasma membrane135,136, binds to highly phosphorylated inositol phosphates137, creates pores in the membrane138 and disrupts the cell integrity. The discovery of RIPK1 dates back to 1995139. Since then, four inhibitor programs have progressed through human phase II safety trials140,141,142,143. The first publication mentioning MLKL is more recent144 and, despite the lack of kinase activity, pharmaceutical companies have cited its publications by 60 times more since 2013. Although there are no clinical trials yet, there are at least three known different chemical inhibitors145.
Mechanobiology: YAP1/WWTR1, PIEZO1 and PIEZO2
Cells use mechanical cues from their environment to guide behaviours such as proliferation and migration. Forces act as signals which are transduced to the nucleus where they control gene expression146. Mechanical forces are critical regulators of organ and tissue homeostasis, morphogenesis and regeneration, and are important aspects of diseases like cancer, metastasis, fibrosis and cardiac hypertrophy. YAP1/WWTR1 (TAZ) are transcriptional co-activators and mechanotransducers147. YAP/TAZ is hyperactivated in cancers148, its inhibition reduces atherogenesis149 and fibrosis150, it triggers pulmonary hypertension151 , and it is necessary for epithelial regeneration in the intestine152. PIEZO1 and PIEZO2 are two mechano-sensitive cation channels that play a key role in cell number regulation153,154 and migration155, hearing156, neural157 and vascular158 development, somatosensory functions159, proprioception160 and beyond. Piezo channels have been recently associated with multiple pathologies like arthrogryposis161, apnea162, congenital lymphatic dysplasia163, hyperalgesia164,165, malaria166, pancreatitis167, xerocytosis168, Gordon syndrome, Marden-Walker Syndrome, and Distal Arthrogryposis Type 5169. The discovery of mechanotransduction signalling pathways has received notable attention in the last years and may open the door to new therapeutic strategies to treat these diseases147.
Trends in scientific literature are useful for pharmaceutical and biomedical companies. Moreover, this approach can offer crucial information to funding agencies to prioritise projects and a new way to study the research impact. Finally, individual researchers may benefit from a new methodology to explore the literature and from algorithms to maximise the efficiency of navigating over an increasingly vast biomedical literature.
Material and methods
Terminology
Here we use the term gene symbol to mean the approved symbol for any of the 19,084 human, protein-coding genes accepted by the HUGO Gene Nomenclature Committee. We refer to gene synonyms as any of the possible gene name variations by which the scientific community has ever referred to a given gene. Approved gene symbols are also included in the gene synonyms. For example: ‘EGFR’ is the approved gene symbol whereas ‘EGFR’, ‘Epidermal Growth Factor Receptor’, ‘ERBB1’, ‘ErbB-1’, ‘c-erbB1’, ‘HER1’, ‘ERBB’ are gene synonyms. We define promiscuous gene names as any gene synonym that is a synonym of more than one gene. This can include previous official gene symbols since these will not have been expunged from the literature. An example of this could be ‘ARP1’ which is a promiscuous gene synonym for the gene symbols ‘NR2F2’, ‘ACTR1A’, ‘ACTR1B’, ‘ANGPTL1’, ‘APOBEC2’, ‘ARFRP1’, ‘PITX2’47. Unsafe gene synonyms are gene synonyms that may have a different meaning in other areas of research or in another context, for instance in standard English. The ‘STAR’ gene symbol is unsafe as opposed to its gene synonym ‘Steroidogenic Acute Regulatory Protein’ or CCP4 is both a gene synonym and the name for crystallography software. The final type of synonym we distinguish are Nested gene synonyms. These are gene synonyms that are part of another gene synonym. For instance ‘insulin’ is a nested gene synonym of ‘insulin receptor’, ‘TNF’ is nested gene synonym of ‘TNF Receptor Superfamily Member 1A’ (gene symbol ‘TNFRSF1A’) and ‘TNF Receptor Associated Factor 2’ (gene symbol ‘TRAF2’).
Pubmed as a graph database
PubMed baseline 202053 comprises 30,419,056 publications for biomedical literature from MEDLINE and life science journals and 173,572,773 citations from the full-text archive of open-source publications PubMed Central (PMC). PubMed was imported into a graph database (Fig. 1A) for a fast performance in the retrieval of highly relational data like authorship and citation networks. In a graph database information is represented as nodes and edges, allowing the fast retrieval of queries about relationships. We loaded PubMed 2020 base-line into Neo4J54, an open source graph-database management system. We introduced four node types (publications, authors, human protein-coding genes, human diseases, medical subheadings), and four edge types (published, from authors to publications; cited by between publications, gene annotation from genes to publications; and disease annotation from diseases to publications). Furthermore, PUBLICATION nodes have the following attributes: PubMed identifier, title, abstract, affiliations, is_review, is_clinical_trial, big_pharma, med_pharma and date of publication. Profiling of the graph is included in Table 6, Database Profiling. Neo4J offers an interactive approach to navigate through PubMed (i) easily accessing references of publications, (ii) with the ability to query for specific genes and diseases already disambiguated, and (iii) with the aim of creating a knowledge graph for further exploration of gene-disease associations. The database is accessible to download at: https://zenodo.org/record/8362679.
Gold standard sets
GeneRif58, UniProt, and DISEASES59 were used as a golden-standard for validation.
Pharmaceutical companies
A list of organisation names was generated from Cortellis170. Organisations with more than 100 patents in Cortellis were considered ‘big pharma’ and ‘mid pharma’ otherwise.
Gene annotation
Search for publications
A ElasticSearch API search engine was used to retrieve PubMed IDs of publications containing a gene or disease synonym in their title, abstract or keywords (Fig. 1D; GET PMIDs and GET corpus). These PubMed IDs were later used to retrieve the publications’ attributes from Neo4J using Cypher language through its python driver171 (Fig. 1E; GET trends). Regular expressions were used avoid nested name ambiguity with lookarounds and fuzzy matching to account for case and punctuation and letter case variations (e.g. ‘ErbB-1’, ‘erbB1’, ‘ERBB1’, ‘ErbB 1’).
Unsafe synonym detection
We used 19,082 protein-coding human genes annotated by HUGO Gene Nomenclature Committee (HGNC). Gene synonyms were obtained from Ensembl, HUGO, Entrez, UniProt and Open Targets (Fig. 1C). Gene synonyms which were identical to disease names contained in the Medical Subject Headings (MeSH) database were eliminated. This mainly occurs when genes are named after diseases that are associated with e.g. ‘Li Fraumeni syndrome’ as a gene synonym for gene TP53172 or ‘Marfan syndrome’ in ‘FBN1’173.
Gene synonyms were classified into “safe” or “unsafe” categories using a modified version of the positive-unlabelled learning with bootstrap-aggregating as implemented by Mordelet et Vert (PU-learning; Fig. 1C)55. PU learning is a form of semi-supervised learning which iteratively finds positive examples within a-priori unlabeled data. To build a binary classifier able to distinguish the unlabelled class (U) into unsafe (P, positive) and safe (N, Negative) we engineered a series of features (Table 2, Unsafe features) such as the combined frequency of the characters in a gene synonym (example: ‘ZNF’ will be safer than ‘EDA’ because ‘Z’ and ‘F’ characters are less frequent in PubMed corpus than ‘E’, ‘D’, and ‘A’) or the probability of a gene synonym given that other gene synonym appeared in the text (the probability of ‘STAR’ given ‘Steroidogenic Acute Regulatory Protein’ is high but the probability of ‘Steroidogenic Acute Regulatory Protein’ given ‘STAR’ is low because ‘STAR’ is more ambiguous).
The PU learning was run for five iterations with a random forest classifier. The pure positive class (unsafe) was constructed combining gene synonyms present in the Enchant English dictionary174, gene synonyms with less than three characters, and promiscuous gene synonyms. In an active learning fashion, after each iteration, the top 1000 examples with the highest probability of being unsafe were manually relabelled if they were wrongly classified. For example, true positive unsafe synonyms like gene families (e.g. ‘G protein coupled receptor’), phenotypes (e.g. ‘Williams Beuren Syndrome’) and other biological entities (e.g. ‘Cell surface antigen’) were included in the true positive set for the next iteration. False positives like ‘thymopoietin’ or ‘tubulin alpha-1C chain’ were included into a new true negative class for the remaining iterations.
After the five iterations, a gene synonym was considered unsafe if: (i) it is included in the English dictionary from Enchant, (ii) it is a word with less than three characters, (iii) if the predicted score for the random forest classifier was higher than 0.5, and (iv) it is a promiscuous gene synonym .
Community detection
We produced weighted, undirected co-citation networks from unweighted, directed citation networks (Fig. 1D, GET communities). Subsequently, connected components were broken into communities using the fast greedy modulation algorithm implementation in iGraph175.
Gene annotation
Communities in co-citation networks represent different areas in the scientific literature. We used this feature to disambiguate large groups of publications (Fig. 1D, LABEL communities). We labelled all the publications in a community with the gene symbol of interest if the ratio of publications mentioning at least one safe synonym with respect of publications that only mention unsafe synonyms was higher than 0.1%.
Nevertheless, the co-citation graph is incomplete because PMC only contains citations of open-source publications. Because of that, 46% of the publications were disconnected from the co-citation graph. Disconnected publications that mention a safe-synonym were automatically linked to the gene symbol of interest. The rest of disconnected publications were linked to gene of interest using PU bagging strategy55 with a binary logistic regression classifier based on the words in the text corpus (keywords, titles and abstracts) of communities already linked to the gene of interest and discarded communities.
Each corpus was pre-processed by (i) removal of non-alphanumeric characters, (ii) tokenization or split by whitespace, (iii) deletion of stop words from NLTK176, (iv) lower case conversion, (v) deletion of tokens whose length is less than three characters, (vi) deletion of token representing integers and (vii) stemming (‘disambiguated’, ‘disambiguations’, ‘disambiguating’ is converted to ‘disambiguat’). List of tokens (uni-, bi-, tri-, tetra-grams) with at least 2 counts and a frequency lower than 0.6 in the complete corpus were vectorised using TF IDF177. When there were less than 1000 unlabeled publications in the training set for the gene of interest, we generated an auxiliary negative class to augment the negative examples in the training data. This auxiliary negative class comprised a random sample of 1000 publications that mentioned genes different from the gene of interest.
These vectors were fed to all available machine learning classifiers from the Python library sklearn: Extra Tree Classifier, Gaussian Process Classifier, K-Nearest Neighbour, Logistic Regression, Ridge Classifier, Random Forest Classifier, and Support Vector Machine. All classifiers were trained with hyper-parameter tuning and threefold cross-validation to avoid over-fitting in each of the 50 PU-bagging iterations (Table 7). The implementation of this PU learning algorithm is the same as the inductive bagging positive-unlabelled learning with bootstrap-aggregating approach described by Mordelet55 (PU-learning; Fig. 1D) and also the same as in the section of “Unsafe Synonym Detection” of the Methods (PU-learning; Fig. 1C). To maximise specificity and sensitivity simultaneously we selected the models with highest weighted F1 score (Scikit Learn) to maximise precision and recall at the same time. We selected the logistic regression (LOG, Table 3) classifier for the disambiguation pipeline given its accuracy-speed balance (Table 3).
Disease annotation
The same procedure used for gene entity recognition was used to detect disease entities, co-citation networks and machine learning. The Medical Subject Headings (MeSH) ontology was downloaded by querying their Rest-API available at BioOntology178. We constructed a key-value dictionary. Each disease was a node in the ontology. The disease synonyms were obtained from the 'Concept List Terms' field in the ontology to gather the preferred and the alternative ways of denoting the disease. We generated more synonyms of the diseases by reversing the order of synonyms with commas: 'Insipidus, Diabetes' to 'Diabetes Insipidus'.
Gene–gene-disease co-occurrence
Co-occurrence
Co-occurrence of genes and diseases was computed using the simultaneous occurrence of gene/disease tags in publications after disambiguation, normalized by the total number of publications presenting those tags. We also computed mutual information metrics for gene–gene and gene-disease associations.
Gene mesh-parent association
Every disease MeSH term was associated with its lowest ancestor in the MeSH ontology under the node Disease179. After computing the gene-disease co-occurrence, each gene is linked with the most frequent ancestor disease term (Fig. 3B).
TrendyGenes
Trendiness
In this paper the trendiness for a gene is defined as the probability of observing the magnitude of fold-change between the predicted and the real number of publications for that given gene. The error in the predictions is inevitably higher with genes associated with small numbers of publications. To correct for this, we generated five bins based on the initial number of publications (percentiles 20, 40, 60, 80 and 100). We computed the distribution of the fold-changes between the predictions and observed reality in each of the five bins using a gaussian kernel density estimator available at Scikit Learn (bandwidth = 0.1, remaining parameters with default values). The area under the obtained probability density function is equal to 1. Trendiness is the area of the right tail of the probability density function bounded to the left by the observed fold change. This gives us an estimate of how extreme the fold change was for that gene in a specific bin.
RNN model
The model consists of an encoder-decoder architecture preceded by an attention layer. Both the encoder and decoder were composed of five hidden layers of Gated Recurrent Units (GRU). The model was implemented in Keras using the Tensorflow-GPU backend. Min–max normalization was used to rescale the time series before training. The optimizer was RMSprop and the loss was computed as the log error. 30 percent of the time series was reserved for validation during the training.
Model optimisation
Input data was in both forms: cumulative and differential. In the cumulative model each year contains all the publication and citations published until then, while the differential model only contains the publications published in that year. Multiple normalisations were used ('none', ‘minmax’, ‘log’, ‘standard’, and combinations of them). Similar results were obtained with different normalisations and minmax was finally selected. Multiple Recurrent Neural Networks (RNNs) architectures were used (GRU, LSTM) in the form of encoder-decoder, with different numbers of neurons (1, 5, 10, 20, 50; Table 8). Models were compared with the Mean Accuracy Scaled Error (MASE), an unbiased method to compare time-series prediction models by comparing how much each model out-performs a naive model that repeats the last value. The 5-neuron-GRU with cumulative time-series was selected because it was the most parsimonious model with the smallest MASE.
Gene ontology terms enrichment
For the enrichment of gene ontology terms (Biological Process) associated with the 100 trendiest genes in academia (all publications) and the pharmaceutical industry we used the online tool GeneCodis 4.0180 with default parameters.
Recommender system
Topic detection
We implemented algorithms to detect topics in collections of publications. This is useful to determine in which areas trendy genes are relevant. Furthermore, topic detection allows the fast identification of errors during the disambiguation. We used two different topic detection algorithms: Latent Dirichlet Allocation62 (LDA), and Non-Negative Matrix Factorisation181 (NMF). Both algorithms factor a nonnegative matrix ‘A’ with size NxM, where N is the number of publications and M is the dimension of the TF IDF vector obtained for Named Entity Recognition (see above), into non-negative factors matrix W of size NxK and matrix H with size KxM where WxH is an approximation of matrix A. The matrix W contains the strength of the association of a given publication to belong to a latent topic while H contains the strength of the association between a latent topic and a given n-gram. Scikit Learn implementations for both algorithms were used to generate 'K' number of topics defined by the user with the default parameters until convergence (tolerance of 1e−12). We previously used perplexity182 to select the optimal number of topics but we disagreed with the output: the number of topics that model the corpus better was not necessarily the most human interpretable. Topic timelines were obtained by calculating the mean and standard deviations of the topic probabilities for all publications mentioning the gene of interest per calendar year (Fig. 4).
Recommendation algorithm
We implemented an automatic pipeline to guide users about which reviews to read for a specific query in PubMed. To do that, we used the topic probability of the publications and an aggregated PageRank score of the citation networks. The user can select an interval number of reviews (R) that is willing to read: between 2–3 or 3–50. Then, three matrices are defined for each group of publications: (i) a binary, sparse matrix of size NxR with N publications and R reviews that comprised the citation adjacency network; (ii) a Nx1 weight matrix that comprise a PageRank scores; and (iii) a NxK matrix with the topic probabilities for N publications and K user-defined topics.
The score for each review was defined as the sum of the PageRank scores of its references while the score for a combination of reviews is defined as the row sum of the indexed NxR matrix multiplied by the Nx1 PageRank vector and the sum of the obtained vector. Results were later normalized by the total maximum score, defined as a hypothetical review citing all gene publications. Finally, every combination of R reviews is presented to the user with the score, the average of the cited publication dates and the probability to belong to one of the K previously defined topics.
Change history
17 November 2023
The original online version of this Article was revised: In the original version of this Article, the database URL was incorrectly indexed. The original Article has been corrected.
References
Kola, I. & Landis, J. Can the pharmaceutical industry reduce attrition rates?. Nat. Rev. Drug Discov. 3, 711–716 (2004).
Paul, S. M. et al. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 9, 203–214 (2010).
Wong, C. H., Siah, K. W. & Lo, A. W. Estimation of clinical trial success rates and related parameters. Biostatistics 20, 273–286 (2019).
Waring, M. J. et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 14, 475–486 (2015).
Ekins, S. et al. Exploiting machine learning for end-to-end drug discovery and development. Nat. Mater. 18, 435–441 (2019).
Vamathevan, J. et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 18, 463–477 (2019).
Napolitano, F. et al. Drug repositioning: A machine-learning approach through data integration. J. Cheminform. 5, 1–9 (2013).
Dudley, J. T. et al. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med. 3, 96ra76–96ra76 (2011).
Shah, P. et al. Artificial intelligence and machine learning in clinical development: a translational perspective. npj Digit. Med. 2, 1–5 (2019).
Yauney, G. & Shah, P. Reinforcement learning with action-derived rewards for chemotherapy and clinical trial dosing regimen selection. in Machine Learning for Healthcare Conference 161–226 (2018).
Besnard, J. et al. Automated design of ligands to polypharmacological profiles. Nature 492, 215–220 (2012).
Bjerrum, E. J. SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules. (2017).
Segler, M. H. S., Kogej, T., Tyrchan, C. & Waller, M. P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. (2017).
Popova, M., Isayev, O. & Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 4, eaap7885 (2018).
Merk, D., Grisoni, F., Friedrich, L. & Schneider, G. Tuning artificial intelligence on the de novo design of natural-product-inspired retinoid X receptor modulators. Commun. Chem. 1, 1–9 (2018).
Müller, A. T., Hiss, J. A. & Schneider, G. Recurrent neural network model for constructive peptide design. J. Chem. Inf. Model. 58, 472–479 (2018).
Gómez-Bombarelli, R. et al. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent Sci. https://doi.org/10.1021/acscentsci.7b00572 (2016).
Blaschke, T., Olivecrona, M., Engkvist, O., Bajorath, J. & Chen, H. Application of generative autoencoder in de novo molecular design. (2017).
Jin, W., Barzilay, R. & Jaakkola, T. Junction Tree Variational Autoencoder for Molecular Graph Generation. (2018).
Simonovsky, M. & Komodakis, N. GraphVAE: Towards Generation of Small Graphs Using Variational Autoencoders. (2018).
Kim, J., Kim, J.-J. & Lee, H. An analysis of disease-gene relationship from Medline abstracts by DigSee. Sci. Rep. 7, 1–13 (2017).
Jie Zhou, B.-Q. F. The research on gene-disease association based on text-mining of PubMed. BMC Bioinform. 19 (2018).
Bhasuran, B. & Natarajan, J. Automatic extraction of gene-disease associations from literature using joint ensemble learning. PLoS One 13, e0200699 (2018).
Bravo, À., Piñero, J., Queralt-Rosinach, N., Rautschka, M. & Furlong, L. I. Extraction of relations between genes and diseases from text and large-scale data analysis: Implications for translational research. BMC Bioinform. 16, 1–17 (2015).
Ferrero, E., Dunham, I. & Sanseau, P. In silico prediction of novel therapeutic targets using gene-disease association data. J. Transl. Med. 15, 1–16 (2017).
Jeon, J. et al. A systematic approach to identify novel cancer drug targets using machine learning, inhibitor design and high-throughput screening. Genome Med. 6, 1–18 (2014).
Isik, Z., Baldow, C., Cannistraci, C. V. & Schroeder, M. Drug target prioritization by perturbed gene expression and network information. Sci. Rep. 5, 1–13 (2015).
Brown, K. K. et al. Approaches to target tractability assessment—A practical perspective. Med. Chem. Commun. 9, 606–613 (2018).
Plenge, R. M. Disciplined approach to drug discovery and early development. Sci. Transl. Med. 8, 349ps15–349ps15 (2016).
Nguyen, P. A., Born, D. A., Deaton, A. M., Nioi, P. & Ward, L. D. Phenotypes associated with genes encoding drug targets are predictive of clinical trial side effects. Nat. Commun. 10, 1–11 (2019).
Knowles, J. & Gromo, G. Target selection in drug discovery. Nat. Rev. Drug Discov. 2, 63–69 (2003).
Lindsay, M. A. Target discovery. Nat. Rev. Drug Discov. 2, 831–838 (2003).
Lee, J. et al. BioBERT: A pre-trained biomedical language representation model for biomedical text mining. Bioinformatics https://doi.org/10.1093/bioinformatics/btz682 (2019).
Bornmann, L. & Mutz, R. Growth rates of modern science: A bibliometric analysis based on the number of publications and cited references. (2014).
Swanson, D. R. & Smalheiser, N. R. Undiscovered Public Knowledge: A Ten-Year Update. (1996).
Cook, D. et al. Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 13, 419–431 (2014).
Becker, K. G., Barnes, K. C., Bright, T. J. & Alex Wang, S. The genetic association database. Nat. Genet. 36, 431–432 (2004).
Chen, Y.-A., Tripathi, L. P. & Mizuguchi, K. TargetMine, an integrated data warehouse for candidate gene prioritisation and target discovery. PLoS One 6, e17844 (2011).
Mallory, E. K., Zhang, C., Ré, C. & Altman, R. B. Large-scale extraction of gene interactions from full-text literature using DeepDive. Bioinformatics 32, 106 (2016).
Piñero, J. et al. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 45, D833–D839 (2017).
Liu, Y., Liang, Y. & Wishart, D. PolySearch2: A significantly improved text-mining system for discovering associations between human diseases, genes, drugs, metabolites, toxins and more. Nucleic Acids Res. 43, W535–W542 (2015).
Li, J. et al. BioCreative V CDR task corpus: A resource for chemical disease relation extraction. Database 2016 (2016).
Bauer-Mehren, A., Rautschka, M., Sanz, F. & Furlong, L. I. DisGeNET: A Cytoscape plugin to visualize, integrate, search and analyze gene–disease networks. Bioinformatics 26, 2924–2926 (2010).
Bundschus, M., Dejori, M., Stetter, M., Tresp, V. & Kriegel, H.-P. Extraction of semantic biomedical relations from text using conditional random fields. BMC Bioinform. 9, 1–14 (2008).
Kafkas, Ş. & Hoehndorf, R. Ontology based text mining of gene-phenotype associations: Application to candidate gene prediction. Database 2019 (2019).
Smith, L. et al. Overview of BioCreative II gene mention recognition. Genome Biol. 9, S2 (2008).
Website. HGNC Database, HUGO Gene Nomenclature Committee (HGNC), European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI). http://www.genenames.org.
Tattershall, E., Nenadic, G. & Stevens, R. D. Detecting bursty terms in computer science research. Scientometrics 122, 681–699 (2019).
Zdrazil, B., Richter, L., Brown, N. & Guha, R. Moving targets in drug discovery. Sci. Rep. 10, 1–15 (2020).
Rask-Andersen, M., Almén, M. S. & Schiöth, H. B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 10, 579–590 (2011).
ACS Publications. https://doi.org/10.1021/acs.jmedchem.7b00954.
Index of /pubmed/baseline. https://ftp.ncbi.nlm.nih.gov/pubmed/baseline/.
Neo4j Graph Platform—The Leader in Graph Databases. Neo4j Graph Database Platform. https://neo4j.com/.
Mordelet, F. & Vert, J.-P. A bagging SVM to learn from positive and unlabeled examples. (2010).
Small, H. Co-citation in the scientific literature: A new measure of the relationship between two documents. J. Am. Soc. Inf. Sci. 24, 265–269 (1973).
PubMed Help. (2005).
About Gene RIF—Gene—NCBI. https://www.ncbi.nlm.nih.gov/gene/about-generif.
DISEASES: Text mining and data integration of disease–gene associations. Methods 74, 83–89 (2015).
* in Literature citations. https://www.uniprot.org/citations/.
Martínez-Romero, M. et al. NCBO ontology recommender 2.0: An enhanced approach for biomedical ontology recommendation. J. Biomed. Semant. 8, 21 (2017).
Blei, D. M., Ng, A. Y. & Jordan, M. I. Latent Dirichlet allocation. in Advances in Neural Information Processing Systems 14 (Neural Information Processing Systems: Natural and Synthetic, NIPS 2001, December 3–8, 2001, Vancouver, British Columbia, Canada) Vol. 3 601–608 (Journal of Machine Learning Research, 2001).
Liu, W. et al. Author name disambiguation for PubMed. J. Am. Soc. Inf. Sci. 65, 765–781 (2014).
Lerchenmueller, M. J. & Sorenson, O. Author disambiguation in PubMed: Evidence on the precision and recall of authority among NIH-funded scientists. PLoS One 11, e0158731 (2016).
Torvik, V. I. & Smalheiser, N. R. Author name disambiguation in MEDLINE. ACM Trans. Knowl. Discov. Data 3, 1–29 (2009).
Stoeger, T., Gerlach, M., Morimoto, R. I. & Nunes Amaral, L. A. Large-scale investigation of the reasons why potentially important genes are ignored. PLoS Biol. 16, e2006643 (2018).
Zerdes, I., Matikas, A., Bergh, J., Rassidakis, G. Z. & Foukakis, T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: Biology and clinical correlations. (2018).
Sharpe, A. H. & Pauken, K. E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18, 153–167 (2017).
Stamm, H. et al. Targeting the TIGIT-PVR immune checkpoint axis as novel therapeutic option in breast cancer. Oncoimmunology 8, e1674605 (2019).
Weber, J. S. Review: Anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. (2007).
Topalian, S. L., Stephen Hodi, F., Brahmer, J. R., Gettinger, S. N. & Sznol, M. Safety, Activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
McDermott, J. & Jimeno, A. Pembrolizumab: PD-1 inhibition as a therapeutic strategy in cancer. Drugs Today 51, 7–20 (2015).
Migden, M. R., Rischin, D., Schmults, C. D., Guminski, A. & Fury, M. G. PD-1 blockade with Cemiplimab in advanced cutaneous squamous-cell carcinoma. N. Engl. J. Med. 379, (2018).
Hamid, O., Robert, C., Daud, A., Stephen Hodi, F. & Ribas, A. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 369 (2013).
Bellmunt, J., Powles, T. & Vogelzang, N. J. A review on the evolution of PD-1/PD-L1 immunotherapy for bladder cancer: The future is now. Cancer Treat. Rev. 54 (2017).
Inc., K. N. & Kernel Networks Inc. Study of BGB-A1217 in combination with Tislelizumab in advanced solid tumors. Case Med. Res. 10.31525/ct1-nct04047862 (2019).
A Study of OMP-313M32 in Subjects with Locally Advanced or Metastatic Solid Tumors—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03119428.
A Study of MTIG7192A in Combination With Atezolizumab in Chemotherapy-Naïve Patients with Locally Advanced or Metastatic Non-Small Cell Lung Cancer—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03563716.
A Study to Evaluate the Safety and Tolerability of AB154 in Participants with Advanced Malignancies—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03628677.
Rostalski, H. et al. Astrocytes and microglia as potential contributors to the pathogenesis of C9orf72 repeat expansion-associated FTLD and ALS. Front. Neurosci. 13 (2019).
Balendra, R. & Isaacs, A. M. C9orf72 -mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558 (2018).
Jiang, J. & Cleveland, D. W. Bidirectional transcriptional inhibition as therapy for ALS/FTD caused by repeat expansion in C9orf72. Neuron 92, 1160–1163 (2016).
Donnelly, C. J. et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428 (2013).
Sareen, D. et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5, 208ra149–208ra149 (2013).
Pinto, B. S. et al. Impeding transcription of expanded microsatellite repeats by deactivated Cas9. Mol. Cell 68, 479-490.e5 (2017).
Batra, R. et al. Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell 170, 899-912.e10 (2017).
Colonna, M. & Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 17, 201–207 (2016).
Ulland, T. K. & Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 14, 667–675 (2018).
Klünemann, H. H. et al. The genetic causes of basal ganglia calcification, dementia, and bone cysts. Neurology 64, 1502–1507 (2005).
Paloneva, J. et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656 (2002).
Jonsson, T. et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107 (2013).
Rajagopalan, P., Hibar, D. P. & Thompson, P. M. TREM2 risk variant and loss of brain tissue. N. Engl. J. Med. 369, 1565 (2013).
Benitez, B. A., Cooper, B., Pastor, P., Jin, S.-C. & Cruchaga, C. TREM2 is associated with the risk of Alzheimer’s disease in Spanish population. Neurobiol. Aging 34 (2013).
Guerreiro, R. et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117 (2013).
Ruiz, A., Dols-Icardo, O., Bullido, M. J., Pastor, P. & Clarimón, J. Assessing the role of the TREM2 p.R47H variant as a risk factor for Alzheimer’s disease and frontotemporal dementia. Neurobiol. Aging 35, 444.e1–444.e4 (2014).
Guerreiro, R. J. et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 70, 78 (2013).
Lattante, S., Le Ber, I., Camuzat, A., Dayan, S. & The French Research Network on Ftd and FTD-ALS. TREM2 mutations are rare in a French cohort of patients with frontotemporal dementia. (2013).
Guerreiro, R. et al. A novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family☆. (2013).
Le Ber, I., De Septenville, A., Guerreiro, R., Bras, J. & Brice, A. Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol. Aging 35 (2014).
Cuyvers, E., Bettens, K., Philtjens, S., Van Langenhove, T. & Sleegers, K. Investigating the role of rare heterozygous TREM2 variants in Alzheimer’s disease and frontotemporal dementia. Neurobiol. Aging 35 (2013).
Lill, C. M. et al. The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimers Dement. 11, 1407 (2015).
Cady, J. et al. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 71, 449–453 (2014).
Rayaprolu, S. et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 8, 1–5 (2013).
Antibodies Against Microglial Receptors TREM2 and CD33 Head to Trials | ALZFORUM. https://www.alzforum.org/news/conference-coverage/antibodies-against-microglial-receptors-trem2-and-cd33-head-trials.
Alector Announces First Alzheimer’s Disease Patient Dosed in Phase 1b Study of AL002 | Alector. Alector. https://investors.alector.com/news-releases/news-release-details/alector-announces-first-alzheimers-disease-patient-dosed-phase.
A Phase I Study for Safety and Tolerability of AL002.—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03635047.
Motwani, M., Pesiridis, S. & Fitzgerald, K. A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 20, 657–674 (2019).
Broz, P., Pelegrín, P. & Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 20, 143–157 (2019).
Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology 155, 1971–1984.e4 (2018).
Sharma, S. et al. Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc. Natl. Acad. Sci. USA 112, E710–E717 (2015).
Liu, Y. et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 371, 507–518 (2014).
Kerur, N. et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med. 24, 50–61 (2017).
Gratia, M. et al. Bloom syndrome protein restrains innate immune sensing of micronuclei by cGAS. J. Exp. Med. 216, 1199 (2019).
Crow, Y. J. & Manel, N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol. 15, 429–440 (2015).
Woo, S.-R. et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830 (2014).
Bakhoum, S. F. et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018).
Activation of the STING-dependent type I interferon response reduces microglial reactivity and neuroinflammation. Neuron 96, 1290–1302.e6 (2017).
A Study of Stimulator of Interferon Genes (STING) Agonist E7766 in Non-muscle Invasive Bladder Cancer (NMIBC) Including Participants Unresponsive to Bacillus Calmette-Guerin (BCG) Therapy, INPUT-102—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04109092.
Phase 1 First Time in Humans (FTIH), Open Label Study of GSK3745417 Administered to Subjects with Advanced Solid Tumors—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03843359.
Study of Intratumorally Administered Stimulator of Interferon Genes (STING) Agonist E7766 in Participants With Advanced Solid Tumors or Lymphomas—INSTAL-101—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04144140.
New Signaling Pathway Targeting Systemic Lupus Erythematosus—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03984227.
Weber, K., Roelandt, R., Bruggeman, I., Estornes, Y. & Vandenabeele, P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun. Biol. 1, 1–13 (2018).
Chen, H. et al. RIPK3-MLKL-mediated necroinflammation contributes to AKI progression to CKD. Cell Death Dis. 9, 1–15 (2018).
Pasparakis, M. & Vandenabeele, P. Necroptosis and its role in inflammation. Nature 517, 311–320 (2015).
Duprez, L., Takahashi, N., Van Hauwermeiren, F., Vandendriessche, B. & Vandenabeele, P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35, 908–918 (2011).
Berger, S. B. et al. Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol. 192, 5476–5480 (2014).
Liu, Z.-Y. et al. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. Am. J. Cancer Res. 5, 3174 (2015).
Harris, P. A., Berger, S. B., Jeong, J. U., Nagilla, R. & Bertin, J. J. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J. Med. Chem. 60 (2017).
Yuan, J., Amin, P. & Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33 (2018).
Hou, J. et al. Discovery of potent necroptosis inhibitors targeting RIPK1 kinase activity for the treatment of inflammatory disorder and cancer metastasis. Cell Death Dis. 10, 1–13 (2019).
Strilic, B. et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 536, 215–218 (2016).
Wang, W., Marinis, J. M., Beal, A. M., Wong, K.-K. & Miller, G. RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell 34, 757–774 (2018).
Sheridan, C. Death by inflammation: drug makers chase the master controller. Nat. Biotechnol. 37, 111–113 (2019).
Degterev, A., Ofengeim, D. & Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. USA 116, 9714–9722 (2019).
Davies, K. A. et al. The brace helices of MLKL mediate interdomain communication and oligomerisation to regulate cell death by necroptosis. Cell Death Differ. 25, 1567–1580 (2018).
Cai, Z. et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 16, 55–65 (2013).
Dovey, C. M., Diep, J., Clarke, B. P., Hale, A. T. & Carette, J. E. MLKL requires the inositol phosphate code to execute necroptosis. Mol. Cell 70, 936-948.e7 (2018).
Xia, B. et al. MLKL forms cation channels. Cell Res. 26, 517–528 (2016).
RIP: A novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 81, 513–523 (1995).
First-Time-in-Human (FTIH) Study of GSK3145095 Alone and in Combination with Other Anticancer Agents in Adults with Advanced Solid Tumors—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03681951.
Safety and Tolerability, Pharmacokinetics (PK), Pharmacodynamics (PD) and Efficacy of Repeat Doses of GSK2982772 in Subjects with Moderate to Severe Rheumatoid Arthritis (RA)—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02858492.
Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Efficacy of Repeat Doses of GSK2982772 in Subjects with Psoriasis—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02776033.
GSK2982772 Study in Subjects with Ulcerative Colitis—Full Text View—ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02903966.
Zhao, J. et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 109, 5322 (2012).
Yan, B. et al. Discovery of a new class of highly potent necroptosis inhibitors targeting the mixed lineage kinase domain-like protein. Chem. Commun. 53, 3637–3640 (2017).
Ladoux, B. & Mège, R.-M. Mechanobiology of collective cell behaviours. Nat. Rev. Mol. Cell Biol. 18, 743–757 (2017).
Panciera, T., Azzolin, L., Cordenonsi, M. & Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 18, 758–770 (2017).
Moya, I. M. & Halder, G. Hippo–YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 20, 211–226 (2018).
Wang, L. et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540, 579–582 (2016).
The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 63, 679–688 (2015).
Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ-miR-130/301 circuit. Cell Rep. 13, 1016–1032 (2015).
Cai, J. et al. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 24, 2383–2388 (2010).
Gudipaty, S. A. et al. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature 543, 118–121 (2017).
Stewart, T. A. & Davis, F. M. Formation and function of mammalian epithelia: roles for mechanosensitive PIEZO1 ion channels. Front. Cell Dev. Biol. 7 (2019).
Pardo-Pastor, C. et al. Piezo2 channel regulates RhoA and actin cytoskeleton to promote cell mechanobiological responses. Proc. Natl. Acad. Sci. USA 115, 1925–1930 (2018).
Wu, Z. et al. Mechanosensory hair cells express two molecularly distinct mechanotransduction channels. Nat. Neurosci. 20, 24–33 (2016).
Pathak, M. M. et al. Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells. Proc. Natl. Acad. Sci. USA 111, 16148–16153 (2014).
Ranade, S. S. et al. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc. Natl. Acad. Sci. USA 111, 10347–10352 (2014).
Ranade, S. S. et al. Piezo2 is the major transducer of mechanical forces for touch sensation in mice. Nature 516, 121–125 (2014).
Woo, S.-H. et al. Piezo2 is the principal mechanotransduction channel for proprioception. Nat. Neurosci. 18, 1756–1762 (2015).
Haliloglu, G. et al. Recessive PIEZO2 stop mutation causes distal arthrogryposis with distal muscle weakness, scoliosis and proprioception defects. J. Hum. Genet. 62, 497–501 (2016).
Nonomura, K. et al. Piezo2 senses airway stretch and mediates lung inflation-induced apnoea. Nature 541, 176–181 (2016).
Lukacs, V. et al. Impaired PIEZO1 function in patients with a novel autosomal recessive congenital lymphatic dysplasia. Nat. Commun. 6, 1–7 (2015).
Bai, T. et al. Piezo2: A candidate biomarker for visceral hypersensitivity in irritable bowel syndrome?. J. Neurogastroenterol. Motil. 23, 453–463 (2017).
Functional and molecular characterization of mechanoinsensitive ‘silent’ nociceptors. Cell Rep. 21, 3102–3115 (2017).
Ma, S. et al. Common PIEZO1 allele in African populations causes RBC dehydration and attenuates plasmodium infection. Cell 173 (2018).
-J. Romac, J. M., Shahid, R. A., Swain, S. M., Vigna, S. R. & Liddle, R. A. Piezo1 is a mechanically activated ion channel and mediates pressure induced pancreatitis. Nat. Commun. 9, 1–10 (2018).
Cahalan, S. M. et al. Piezo1 links mechanical forces to red blood cell volume. Elife 4 (2015).
Mutations in PIEZO2 cause Gordon syndrome, Marden-Walker syndrome, and distal arthrogryposis type 5. Am. J. Hum. Genet. 94, 734–744 (2014).
Home-Cortellis. Cortellis https://clarivate.com/cortellis/.
The Py2neo v4 Handbook—The Py2neo v4 Handbook. https://py2neo.org/v4/.
Gene Symbol Report. https://www.genenames.org/data/gene-symbol-report/#!/hgnc_id/HGNC:11998.
Gene Symbol Report. https://www.genenames.org/data/gene-symbol-report/#!/hgnc_id/HGNC:3603.
pyenchant. PyPI. https://pypi.org/project/pyenchant/.
Clauset, A., Newman, M. E. J. & Moore, C. Finding community structure in very large networks. Phys. Rev. E https://doi.org/10.1103/PhysRevE.70.066111 (2004).
Natural Language Toolkit—NLTK 3.5 documentation. https://www.nltk.org/.
Roelleke, T. & Wang, J. TF-IDF uncovered: A study of theories and probabilities. in Proceedings of the 31st Annual International ACM SIGIR Conference on Research and Development in Information Retrieval, SIGIR 2008, Singapore, July 20–24, 2008 435–442 (2008).
MeSH Browser. https://meshb.nlm.nih.gov/search.
MeSH Browser. https://meshb.nlm.nih.gov/record/ui?ui=D004194.
Tabas-Madrid, D., Nogales-Cadenas, R. & Pascual-Montano, A. GeneCodis3: A non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 40, W478–W483 (2012).
Lee, D. D. & Sebastian Seung, H. Algorithms for non-negative matrix factorization. Adv. Neural Inf. Process. Syst. 13 (2001).
Chen, S., Beeferman, D. & Rosenfeld, R. Evaluation Metrics for Language Models. (2001).
Acknowledgements
Mark Swindells, Ewan Z. Pearson and Cornelis J Weijer for discussions.
Author information
Authors and Affiliations
Contributions
G.S.N. and D.N.C. designed and implemented the disambiguation algorithm, detection of trends and initial topic detection. D.N.C. generated the recommendation system. D.N.C. and D.J.C. interpreted the output. D.J.C. was responsible for the project conception and supervision. All authors contributed to the manuscript. G.S.N. and D.N.C. contributed equally to this project.
Corresponding author
Ethics declarations
Competing interests
D Crowther is an employee of Exscientia and is a member of the life sciences corporate advisory board for Elsevier. G Serrano-Nájera and D Narganes-Carlón declare no conflicts of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Serrano Nájera, G., Narganes Carlón, D. & Crowther, D.J. TrendyGenes, a computational pipeline for the detection of literature trends in academia and drug discovery. Sci Rep 11, 15747 (2021). https://doi.org/10.1038/s41598-021-94897-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-94897-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
