
Abstract
Meat from wildlife species (bushmeat) represents a major source of dietary protein in low- and middle-income countries where humans and wildlife live in close proximity. Despite the occurrence of zoonotic pathogens in wildlife, their prevalence in bushmeat remains unknown. To assess the risk of exposure to major pathogens in bushmeat, a total of 3784 samples, both fresh and processed, were collected from three major regions in Tanzania during both rainy and dry seasons, and were screened by real-time PCR for the presence of DNA signatures of Bacillus anthracis (B. anthracis), Brucella spp. (Brucella) and Coxiella burnetii (Coxiella). The analysis identified DNA signatures of B. anthracis (0.48%), Brucella (0.9%), and Coxiella (0.66%) in a total of 77 samples. Highest prevalence rates of B. anthracis, Brucella, and Coxiella were observed in wildebeest (56%), dik-dik (50%), and impala (24%), respectively. Fresh samples, those collected during the rainy season, and samples from Selous or Serengeti had a greater relative risk of being positive. Microbiome characterization identified Firmicutes and Proteobacteria as the most abundant phyla. The results highlight and define potential risks of exposure to endemic wildlife diseases from bushmeat and the need for future investigations to address the public health and emerging infectious disease risks associated with bushmeat harvesting, trade, and consumption.
Similar content being viewed by others
Introduction
“Bushmeat”, the meat and organs derived from harvested wildlife species, is a common source of animal protein throughout the tropics of the Americas, Asia, Australia, and Africa. In Tanzania, although bushmeat hunting is unlawful without permits, illegal wildlife poaching is a common practice year-round1. Bushmeat is not only a source of food and income for the poachers, their families, and communities, but is also used in many cultural, social, and religious practices2. A recent survey in the regions surrounding the Greater Serengeti ecosystem revealed that between 2 and 5 meals per week in a household contained bushmeat3. While the majority of bushmeat is consumed locally, a considerable amount is smuggled illegally into the United States and Western Europe. Reports suggest that up to 5 tons of bushmeat is smuggled via a single airport in Western Europe each week4. Bushmeat smuggling from Africa and elsewhere could act as a conduit for pathogen spread as suggested by the recent discovery of retroviruses and herpesviruses in bushmeat illegally imported to the US5. This is of particular relevance in light of the current global pandemic of SARS-CoV-2, and the increasing risk of newly emerging infectious diseases of zoonotic origin6 . It is widely recognized that 60% of emerging infectious diseases are zoonotic in origin, and of these 70% originate in wildlife7. In this study, we shall examine the prevalence of endemic Bacillus anthracis (B. anthracis), Brucella spp. (Brucella), and Coxiella burnetii (Coxiella) in bushmeat samples collected within Tanzania.
Bacillus anthracis is a spore-forming, Gram-positive bacterium and the causative agent of anthrax, a zoonotic disease that mostly affects grazing livestock and wild ungulates and poses a threat to human health. Studies have estimated that 20,000 to 100,000 human cases of anthrax occur worldwide annually8,9. The disease is endemic in poor rural areas, especially where there are extensive interactions between human, livestock and wildlife8,9. Outbreaks are regularly reported with an early event in 1962, causing mortality in 1200 impalas in Tanzania10. More recently, an outbreak in 2016–2017 in Northern Tanzania recorded positive cases in 131 wildlife and 39 livestock carcasses that were also used for consumption8,10. The recurrence of outbreaks in Tanzania are mainly the result of interactions between humans, wildlife, and livestock at specific “hotspots”, where both domestic and wild animals are potential sources of infections to humans11. People become infected when handling and consuming infected livestock and also bushmeat. Periods of drought facilitate anthrax outbreaks since they lead to aggregations of animals close to water sources, heavy overgrazing that exposes soil, and atypical migration patterns12.
Brucella, a genus of Gram-negative bacteria, is the causative agent of a number of zoonotic infections including brucellosis. Infection passes to humans through direct contact with infected animals, by the consumption of unpasteurized dairy products (often raw milk), or via occupational contact with infected animals as well as by the inhalation of airborne agents13. In sub-Saharan Africa, including Tanzania, studies have indicated the presence of Brucella in wildlife and that livestock to humans is considered an endemic problem causing devastating economic losses and challenges for public health14,15,16,17. A recent study in Kenya on Brucella suggested that transmission to humans increased as a consequence of close contact between livestock and wildlife18. Another study from Tanzania identified geographical “hotspots” with high anti-Brucella antibodies in humans, cattle, goats, and buffalos14. Other studies have emphasized that transmission of Brucella to humans was caused by the preparation and consumption of bushmeat19,20,21.
Coxiella burnetii, a Gram-negative bacterial CDC select agent that causes Q fever is also transmitted to livestock, wildlife, and humans via byproducts and the direct contact with infected animals. There are few studies on the prevalence of Q fever in Tanzania. One study on humans, livestock and wildlife found that the incidence of Q fever in cattle and wildlife was associated with arthropod vectors22. Another cross sectional sero-epidemiological study conducted in South Africa concluded higher risk of zoonotic transmission and economic loss was associated with the livestock-wildlife interface23 and this has been supported by a number of studies across Africa21,24,25.
Metagenomics has transformed microbiology by leading the way for a cultivation-independent determination of microbial profiles in samples, especially in samples similar to bushmeat that are subjected to a supply chain before consumption. Recent microbiome analyses have provided preliminary evidence for the presence of select agents in bushmeat samples21, and suggest that characterization of the microbiomes of bushmeat samples with evidence of DNA signatures for the three select agents may help better assess the overall microbial burden and potential public health risks associated with bushmeat harvest, handling, and consumption.
Thus, to better assess the prevalence of select agents and risks associated with the handling and consumption of bushmeat, we screened a total of 3784 fresh and processed bushmeat samples representing 42 animal species from the Serengeti, Ruaha, and Selous regions in Tanzania collected during rainy and dry seasons over a 2-year period for the presence of DNA signatures of B. anthracis, Brucella spp., and Coxiella. Taken together, the results highlight the presence of select agent signatures and help define potential risks of exposure to endemic wildlife diseases from bushmeat in the country.
Methods
Study area
The sampling was carried out at three study savannah dominant sites in Tanzania: the Serengeti National Park (Serengeti), the Ruaha National Park (Ruaha), and the Selous Game Reserve (Selous). The ~ 25,000 km2 Serengeti ecosystem is located in northern Tanzania, bordering southern parts of Kenya26. It consists of several protected areas including the Serengeti National Park (14,760 km2, between 1° and 3° 30S and 34° and 36° E) and adjacent Ngorongoro Conservation Area, Game Reserves and Game Controlled Areas. Serengeti typically has a dry season (June-October) and a wet season (November–May). The Ruaha ecosystem (7° 30′ S 35° 00′ E), located in the southern region of Tanzania and at 20,226 km2, is comprised of Ruaha National Park and surrounding Game Reserves and Wildlife Management Areas. Ruaha typically has a dry season (May–October) and a wet season (November–April). The Selous Game Reserve (9° 0′ S 37° 24′ E) 50,000 km2 (partitioned to form Nyerere National Park, the largest Park in the country) is another protected area in Southern Tanzania. It is contiguous with the Mikumi National Park and Udzungwa Mountains National Park and contains one third of the abundant wildlife of Tanzania. Selous typically has a dry season (June–October) and a wet season (November–May). Typically, Tanzania has a short dry season (January–February) and a long dry season (June-October) and a short rainy season (November–December) and a long rainy season (March–May), although variations are observed in different regions27,28,29,30. For the purpose of this study, we considered the dry season from June-October while the rainy season was considered from the months of November–May for all the regions in the study.
Bushmeat sample collection
Sample collection occurred over a two-year period between July 2016 and July 2018, from both dry and rainy seasons. Samples were identified as either “fresh” or “processed”. Fresh samples were those that appeared to be recently harvested while the processed samples included those that were either heavily salted, sun or air-dried, semi-boiled, or a combination of methods that resulted in dehydrated meat samples31. The samples were collected by teams of veterinarians, anti-poaching units, and local enumerator networks from villages surrounding the protected areas, where bushmeat is sold32. All samples were collected through incidental sampling after obtaining requisite permits from national authorities and in consultation with village leaders. No wildlife species were harmed or directly surveyed as part of this investigation.
Bushmeat samples (~ 250–500 g) were collected in double zip lock freezer bags containing sterile non-toxic Silica Gel desiccant moisture absorbers/dehumidifiers. The samples were stored in − 20 °C vehicle freezers prior to transferring to − 20 °C solar freezers in the field. The samples were then transported to the Nelson Mandela African Institution of Science and Technology (NM-AIST) in Arusha via vehicle freezers, where they were stored at − 80 °C until processing. Cold chain maintenance was ensured through temperature trackers included with the samples throughout the entire process. Sample identification sheets were completed for all collected samples and the metadata, including information regarding sample condition, wildlife species, the collection site (using GPS coordinates), and seasonality.
Nucleic acid extraction
To avoid potential issues associated with surface contamination, three replicates of ~ 120 mg each, were obtained from the muscle tissue using sterile, disposable Rapid Core punch (World Precision Instruments, Sarasota, FL) and sterile disposable safety scalpels (VWR, Bridgeport, NJ). The samples were placed in the MagMAX™ Lysis/Binding Solution Concentrate buffer (Thermo Fisher Scientific, Waltham, Massachusetts). The homogenization protocol was optimized and the settings with the best DNA yield for both fresh and processed samples can be found in the Supplemental Appendix 1.
Nucleic acid extractions were performed using MagMAX™ 96 DNA Multi-Sample Kit (Thermo Fisher Scientific, Grand Island, NY) and KingFisher Flex automated DNA purification system (Thermo Fisher Scientific, Grand Island, NY) per manufacturer’s instructions with minor modifications. If the yield and purity of extractions were not adequate for a specific sample, such as the DNA concentration was less than 4 ng/µl and/or if the quality of the amplicons analyzed by the Bioanalyzer did not pass the quality control, DNA was extracted manually using DNeasy PowerSoil Kit (Qiagen, Hilden, Germany) per manufacturer’s instructions. Extracted DNA was quantified using Qubit™ 3.0 Fluorometer (Thermo Fisher scientific, Grand Island, NY). To ensure purified DNA was of high-quality, DNA was also visualized through agarose gel electrophoresis.
Real time PCR
Amplification reactions for the detection of all pathogens (B. anthracis, Brucella and Coxiella) were carried out in 20 μl reactions with both positive and negative controls (Supplemental Table 1)33. Each PCR reaction included 10 μl of 2 × Taqman Fast Advanced master mix (Applied Biosystems, Woodward, Austin, USA), 2 μl of 5 uM of each forward and reverse primer (Supplemental Table 1), 1 μl of 5 μM probe (Supplemental Table 1), 1 μl of UltraPure™ DNase/RNase-Free distilled water (Thermo Fisher scientific, Grand Island, NY) and 4 μl of DNA template. Thermal cycling reactions were performed using 7500 Fast Real Time PCR System (Applied Biosystems, Waltham, MA). Targeted genes for B. anthracis included Protective antigen (PA) encoded by plasmid pX01 and Capsule B (CAPB2) encoded by plasmid pX02 (Supplemental Table 1). Samples were considered positive for B. anthracis when both plasmids, pXO1 and pXO2, were detected through RT-PCR. Outer Membrane Protein 2b (OMP2b) was the targeted gene to detect Brucella species and to detect Coxiella burnetti, Insertion element IS1111 was used as the target gene. Supplemental Table 1 contains the name of the target genes, sequences for forward and reverse primers and probes, and the PCR run condition. All positive samples were verified through repeating the nucleic acid extraction from the original sample and RT-PCRs were performed in technical triplicates.
Microbiome sequencing
The purified DNA was transported to the Biosciences eastern and central Africa-International Livestock Research Institute (BecA-ILRI) Hub in Nairobi, Kenya for the microbiome sequencing. The V3-V4 hypervariable region of the 16S rRNA gene was sequenced on the Illumina MiSeq System generating 2 × 300 bp paired-end reads (see methods in Supplemental Appendix 1). Sequence datasets are deposited in NCBI’s Sequence Read Archive (SRA) repository (Bioproject PRJNA477349).
Bioinformatics for microbiota composition
To guarantee a high quality data set, microbiome analyses were performed using an open source platform, Empowering the Development of Genomics Expertise (EDGE)34. The EDGE Bioinformatics platform (v2.3.0) implementation is based on QIIME v1.9.1 and includes demultiplexing and quality filtering, operational taxonomic unit (OTU) clustering, taxonomic assignment, phylogenetic reconstruction, diversity analyses, and visualizations of each of these analyses34. QIIME started the pre-processing step with 43,095,486 paired-end reads (in FASTQ format) by removing reads with a PHRED quality score lower than Q20. Data were further filtered to remove reads with more than one ambiguous base. If less than 50% of a read contained consecutive high-quality bases, the read was discarded. The forward and reverse reads were merged before analysis. A 94% nucleotide identity sequence clustering threshold (approximately genus level) was used to generate OTUs. The representative sequence for each OTU was then queried against the Greengenes database35 for taxonomic classification assignment. The depth filter was set to 1000 reads—any barcoded sample that had less than 1000 assembled reads did not undergo taxonomic classification or further alpha- and beta-diversity analyses. Next, the integrated phylotype command generated the consensus taxonomy using phylotype-based approach, where taxonomy-linked sequences were assigned to OTUs based on similarity.
Statistical analysis
All the statistical analyses were performed using the software R (v1.2.1335, RStudio, Inc), if not otherwise stated. The proportion of positive samples by season (dry, rainy), sampling region (Serengeti, Ruaha, Selous) and meat condition (fresh, processed) was estimated using prop.test with continuity correction function in the ‘stats’ package36. The relative risk of a sample being positive for one or more of the select pathogens was calculated using the risk-ratio function of the ‘epitools’ package36. To investigate the additive effects of all three categories on the outcome of a sample being positive for any of the pathogens, a multivariate analysis was performed using logistic Generalized Linear Model (GLM with logit link) with region, season and meat condition included as categorical variables37. To examine the potential for sample size biases in the microbiota sequencing, a rarefaction analysis was conducted where the fraction of taxa was captured for each sample38. Results show that we were able to consistently identify all the detectable OTUs present in every sample with a minimum reading depth of 14,050 sequences per sample. This suggests that the variation of the microbiota among samples is not likely caused by low sequence coverage and that further sequencing would not detect additional genera (Supplemental Figure 1). The microbiome alpha diversity (species richness) was calculated using the Shannon Diversity Index in the package ‘Vegan’39. Statistical analysis for the microbial diversity between the groups was performed using Welch’s t-test37. Hierarchical clustering of the samples based on relative abundance of phyla was performed using the package ‘pvclust’, with 1000 bootstrapped replications of the clusters40. To examine the beta diversity of the samples, Principal Coordinate Analysis (PCoA) was performed using the Bray–Curtis Matrix at the phylum level using the package ‘Vegan’39. Groups were clustered graphing ellipses estimating the 95% confidence interval (CI) around the average value, for each variable.

Source Geospatial Foundation Project. Version 3.4.9-Madeira. http://qgis.osgeo.org".
Map and distribution of bushmeat samples collected from the three ecosystems in Tanzania. (A) Map of the sample collection sites. In the map, the three regions are shown in green. The points show the distribution of samples collected (black circles) and samples positive for one of the three pathogens (red circles). (B) Distribution of the collected bushmeat samples. The pie charts show the stratification of the collected bushmeat samples. For the species category (n = 3784), the top 5 most abundant species collected from the particular region are shown: Serengeti–Wildebeest (green), Buffalo (blue), Zebra (red), Impala (yellow), Gazelle (light blue); Ruaha–Dik-dik (pink), Gazelle (light blue), Kudu (light green), Impala (yellow), Buffalo (blue); Selous–Gazelle (light blue), Buffalo (blue), Dik-dik (pink), Kudu (light green), Hartebeest (grey). For the condition category (n = 3784), the red represents fresh samples and blue represents processed samples. For the season category (n = 3784), dry and rainy seasons are represented by the red and blue colors, respectively. The pathogen column (n = 77) shows the proportion of each pathogen from each region with B. anthracis shown in red, Brucella in blue, and Coxiella in green. The samples were collected from both dry and rainy seasons, Ruaha (dry; I = 1297; rainy = 391), Selous (dry = 813; rainy = 374), Serengeti (dry = 461; rainy = 448), and from two sample conditions, Ruaha (fresh = 1571; processed = 117), Selous (fresh = 962; processed = 223), Serengeti (fresh = 393; processed = 516). The map was generated using the QGIS Geographic Information System software. "QGIS Development Team (2019). Open-
Results
Bushmeat samples
A total of 3784 samples were collected in and around the three geographical study locations: Ruaha (n = 1688; 33 villages), Selous (n = 1187; 27 villages), and Serengeti (n = 909; 54 villages) as shown in Fig. 1A. The samples represent wildlife from both high and low densities and include the predominant herbivores, such as wildebeest (Connochaetes taurinus), zebra (Equus burchellii quagga), buffalo (Syncerus caffer), impala (Aepyceros melampus), dik-dik (Madoqua spp.), and other less common species, including eland (Tragelaphus oryx), topi (Damaliscus lunatus), and the African savannah hare (Lepus microtis) (Table 1).
Signatures of select agents in bushmeat samples
Of the 3784 collected bushmeat samples, 2.03% (n = 77, CI 1.6–2.5%) samples were identified as positive for at least one of the three tested pathogens, B. anthracis, Brucella, and Coxiella. There was an overall prevalence of 0.48% (n = 18, CI 0.3–0.8%) of B. anthracis in the collected bushmeat samples, 0.9% (n = 34, CI 0.6–1.3%) prevalence of Brucella, and 0.7% (n = 25, CI 0.4–1.0%) prevalence of Coxiella (Fig. 2). The proportion of the positive samples from each location indicates that the highest prevalence of B. anthracis was found in the Serengeti at 78% (n = 14, CI 0.6–1.0%), the highest percentage of Brucella in Ruaha at 53% (n = 18, CI 0.4–0.7%), and the highest percentage of Coxiella was found in Selous at 76% (n = 19, CI 0.6–1.0%) (Fig. 1B, Supplemental Table 2). Overall, from each location, there was a 2.3% (n = 21, CI 1.3–3.3%) prevalence of the three select agents in the Serengeti, 1.4% (n = 24, CI 0.9–2.0%) prevalence in Ruaha, and 2.7% (n = 32, CI 1.8–3.6%) prevalence in Selous (Table 1). All Confidence Intervals (CI) were calculated at 95% threshold.

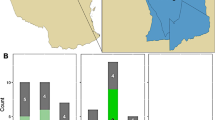
Counts of Bushmeat Samples Positive for Select Agents. The count of positives for each species are included in the bar charts (with the total count in the bar with white font), separated by B. anthracis-positive in the left panel, Brucella-positive in the middle panel, and Coxiella-positive in the right panel. The coloring above the bar graphs show the proportion of samples within the species that are in each category (Region, Season, and Condition). The blue color represents samples collected from Ruaha, collected during dry season, and fresh samples. The red color represents samples collected from Selous, collected during Rainy season, and processed samples. The green color represents samples collected from the Serengeti. For instance, in the B. anthracis panel, one positive fresh (blue) buffalo sample was collected during rainy season (red), from Serengeti region (green).
The samples positive for B. anthracis were primarily wildebeest (n = 10) and the samples positive for Brucella were mostly from dik-dik (n = 17) (Fig. 2). The samples positive for Coxiella had a larger range of species, with impala and dik-dik being the most prevalent (Fig. 2). Remarkably, all of the wildebeest samples that were positive for B. anthracis were fresh samples from the Serengeti collected during the rainy season (Fig. 2). Also, all of the dik-dik samples positive for Brucella were fresh samples from Ruaha collected during the dry season (Fig. 2). Only one of the Coxiella-positive samples was from the Serengeti and this was a fresh sample from an eland collected during rainy season (Fig. 2).
Relative risk and multivariate analyses
The relative risk (RR) associated with a sample being positive for one or more of the select pathogens was determined (Fig. 3). The relative risk for pathogens collected during rainy season was 1.4 (CI 1.1–1.7) and significantly higher compared to the RR of 0.64 (CI 0.5–0.8) for samples collected during dry season; in other words, there was a 71% higher risk of samples collected during rainy season to be positive. The relative risk of getting a pathogen from fresh samples was 1.96 (CI 1.1–3.6) and significantly higher than the relative risk of 0.87 (CI 0.08–0.95) for processed samples, indicating a 109% higher risk for a fresh sample being found to be positive than those that were processed. The RR of samples from Ruaha was 0.8 (CI 0.7–0.9) and it was significantly lower than samples collected from the other locations, in particular, Selous (RR = 1.2; CI 0.97–1.4) (Fig. 3).

Relative risk (RR) of the positive bushmeat samples associated with different variables. (A) Counts (positive and total samples), prevalence and RR associated with each variable. The 95% confidence intervals are reported in parethesis. (B) Graph of the RR for each variable. The relative risk (pointS) and 95% confidence interval (lines) for each variable are reported for the seasonal data (red), sample condition (blue) and regions (green). Results indicate that there is a 71% higher risk of a sample collected from rainy season being positive than samples collected during dry season. There is a 109% higher risk of a fresh sample being positive than processed sample. There is also a 64% lower risk of a sample collected from Ruaha being positive that the other regions (the RR of samples collected from both Selous and Serengeti is 1.44). Significant differences in RR analysis are reported (*p < 0.05).
Multivariate analysis was undertaken to investigate the combined effects of all three categories on the outcome of a sample being positive for any of the pathogens. Results showed that samples collected during the rainy seasons increase the log odds of being positive by 0.70, compared to the dry season. On the contrary, processed samples decrease the log odds of being positive by 1.1, compared to fresh samples. Among the regions, analysis indicated that compared to Ruaha, samples collected from Selous increase the log odds of being positive by 0.70, and samples collected from Serengeti increase the log odds of being positive by 0.70 (Table 2).
The microbiome associated with select pathogen-positive bushmeat samples
Microbiome characterization of the 77 positive samples showed that the two most abundant phyla across all samples were Firmicutes and Proteobacteria (Fig. 4) (Supplemental Table 2). Rarefaction analyses showed consistent identification of the detectable OTUs present in each sample with a minimum reading depth of 14,050 sequences per sample38. This suggests that the variation of the microbiota among samples is not likely caused by low sequence coverage (Supplemental Figure 1). The alpha and beta diversity for the different variables associated with each sample was assessed. The first two principal coordinates of the Principal Coordinate Analysis (PCoA) accounted for 95% of the total variation. The first principal coordinate was driven by the effect of the relative abundance of Proteobacteria (coefficient = 0.86) and Firmicutes (− 0.90) and the second principal coordinate by the relative abundance of Bacteroidetes (0.99) (Supplemental Figure 2).

Hierarchical clustering of the samples positive for one of the three pathogens based on the relative abundance of phyla in each sample. (A) Hierarchical clustering of samples. The samples grouped into 5 clusters: I—blue, II—tan, III—purple, IV—grey, V—red. The samples are labeled with the first letter representing the pathogen (A—B. anthracis, B—Brucella, C—Coxiella) and the ending representing the ecosystem (R—Ruaha, Sel—Selous, Ser—Serengeti). The AU/BP values for the clustering are shown at the base of each node. (B) Sample metadata. These columns show the other metadata associated with each sample. The open circles represent processed samples, and the closed are fresh samples. The open squares are from rainy season and the closed squares are from dry season. The shapes are colored according to species of wildlife from which the bushmeat originated: dik-dik—red, wildebeest—green, all other species—blue. (C) Bar graph of the relative abundance of phyla. The relative abundance of phyla > 1% is shown in the stacked bar graph. The colors corresponding to each respective phylum is in the legend at the bottom.
In addition, lower alpha diversity and significant differences in beta diversity were observed in B. anthracis-positive samples, and samples collected from the Serengeti, which were primarily wildebeest. The results showed that B. anthracis-positive samples had significantly lower alpha diversity than either of the Brucella- or Coxiella-positive samples (p-value < 0.001; Supplemental Figure 3) and tended to cluster separately in the Principal Coordinate Analysis (PCoA) (Supplemental Figure 3). There was a similar pattern for wildebeest samples, which showed lower overall diversity and clustering separately from dik-dik or other species (p-value < 0.001; Supplemental Figures 2 and 3).
Hierarchical clustering based on the relative abundance of phyla grouped the samples into five distinct clusters (Fig. 4A). The associated Approximately Unbiased (AU) and Bootstrap Probability (BP) are represented at the base node of each cluster (AU > 95 are statistically significant). Cluster I (n = 6) contained six samples, of which five were Brucella-positive with the majority of the samples being freshly collected (Fig. 4). Cluster II (n = 18) included 12 B. anthracis-positive samples collected from Serengeti, of which eight were from fresh wildebeest samples collected during the rainy season. Cluster III (n = 24) included 16 Coxiella-positive samples, of which 15 were fresh, with thirteen of these collected from the Selous ecosystem. Samples in cluster IV (n = 24) included 17 Brucella-positive samples collected from Ruaha and Selous, four Coxiella-positive samples collected from Selous, and three B. anthracis-positive samples collected from the Serengeti ecosystem. The final cluster, cluster V (n = 5) was the most diverse and included three Brucella-positive and two Coxiella-positive samples (Fig. 4).
The relative abundance was also analyzed at the family level (Supplemental Figure 4). At this level, the top three most abundant taxa in these samples include, Streptococcaceae (26%), Enterobacteriaceae (22%), and Moraxellaceae (14%). The alpha diversity revealed differences according to pathogen, host species, and region (Supplemental Figure 5) and the beta diversity clustered the wildebeest samples separately from the other species (Supplemental Figure 6).

Heatmap of the genus-level relative abundance in the samples positive for one of the three pathogens. The heatmap shows the relative abundance for any genus present at > 1%. The color ranges from pale yellow (low abundance) to dark orange (high abundance). Hierarchical clustering was performed for both the samples and the genera. The samples are labeled with the first letter representing the pathogen (A—B. anthracis, B—Brucella, C—Coxiella) and the ending representing the regions (R—Ruaha, Sel—Selous, Ser—Serengeti).
Interestingly, when the data were investigated at the genus-level, hierarchical clustering tended to group the samples by pathogen. The B. anthracis-positive samples were high in Bacillus, which was not found in the Brucella- or Coxiella-positive samples (Fig. 5). On the other hand, the Brucella- and Coxiella-positive samples showed a higher abundance of species represented by the Lactococcus and Macrococcus genera (Fig. 5). These differences were further confirmed through examining the alpha and beta diversity associated with the samples. Within each variable, samples that had lower alpha diversity were B. anthracis-positive, wildebeest samples, and those collected from Serengeti (Supplemental Figure 7), which were also shown to be the most significant in the PCoA analysis (Supplemental Figure 8).
Discussion
A goal of this study was to assess the potential for risk of human exposure to three select agents, Bacillus anthracis, Brucella spp., and Coxiella burnetii associated with bushmeat handling and consumption in Tanzania. Our initial focus was on three major locations in Tanzania: Ruaha and Selous in the south and the Serengeti in the north. Sites were selected to represent the distinct environments (habitat and climate) with diverse wildlife composition and density, and proximity to major urban centers (including Arusha, Dodoma and Dar es Salaam) where the bushmeat is often sold.
While the presence of DNA signatures does not imply the presence of a viable organism, given that bushmeat consumption is as high as 5 meals per week in some households, and with an overall pathogen prevalence of 2.03% (1.6–2.5%), this risk of exposure remains a concern and worthy of further investigation. This is especially problematic considering that the risks associated with endemic infections such as Brucella and Coxiella often go unrecognized and remain under-reported despite the considerable risks of causing disease41.
Relative risk associated with positive DNA signatures was assessed for variables including season, sample condition, animal species, and regions (ecosystems) and revealed notable associations. There was a higher risk of a sample being positive if it was collected during a rainy season, or if the sample was fresh, in contrast there was a lower risk if the sample was collected from Ruaha, than Serengeti or Selous (Fig. 3). Studies have shown that climate-related factors, such as prolonged rain, can contribute to disease outbreaks42. Changes in weather conditions force the movement and interactions of animals where rainy seasons and/or droughts affect animals spatial grazing patterns43,44. Also, exposure to anthrax may be higher for migratory ungulates like wildebeest that are found in different parts of the regions throughout the year3. This may be one reason why there is a higher likelihood of positive samples being collected during the rainy season. Also, during the rainy season there may be a higher consumption of fresh samples, which is consistent with the higher risk of positives in fresh samples, probably associated with methods of processing being unavailable or unsuitable during this season, or simply a higher availability of fresh meat. High salt, UV exposure, and boiling of processed samples may reduce pathogen loads associated with the fresh meat samples45,46; however, UV light does not necessarily decrease the microbial content of meat47,48. Results also indicate that there is a lower risk of a sample being positive if it was collected from Ruaha. There are some substantial differences among the three ecosystems, both biotic and abiotic, and we have no evidence of a dominant factor affecting the samples, and suggest that multiple components might be contributing to the relative risks associated with each ecosystem49,50.
Findings indicated that most of the differences in the microbiome are driven by the relative abundance of Firmicutes and Proteobacteria, as demonstrated by the PCoA (Supplemental Figure 2). This is expected, since B. anthracis belongs to the Firmicutes phylum and Brucella and Coxiella both belong to Proteobacteria. The microbiome analysis revealed significant lower alpha diversity and PCoA clustering associated with the B. anthracis-positive samples, wildebeest samples, and samples collected from the Serengeti (Supplemental Figures 2, 3, 5, 6, 7, and 8). These results are also supported by the hierarchical clustering at both the phylum- and the genus-levels, where the B. anthracis-positive samples tended to cluster together (Fig. 4–Cluster II and Fig. 5). This is expected since the highest abundance of B. anthracis-positive samples were in the Serengeti, where most of the wildebeest samples were collected (Fig. 1). These results are consistent with previous studies that documented many Anthrax outbreaks in Northern Tanzania, and particularly in the Serengeti8,11,21,51.
Samples that were collected in the same condition were generally available to be sold to the public, hence they represent microbial communities that are endogenous to the samples, as well as the microbiota that is acquired during harvesting, processing, and transporting. While the analysis suggests that fresh samples, samples collected during rainy season, and samples collected from the Serengeti and Selous had a higher likelihood of being positive for one of the three pathogens, positive samples carrying DNA signatures of the tested select agents were also collected during dry season, from other animal species, and ecosystems. Interestingly, the results also revealed that a subset (n = 39) of purported “bushmeat” samples from Selous that were instead speciated as being of cattle in origin had a combined prevalence of 28.7% of the three select agents, highlighting a potential public health risk associated with the endemic diseases in livestock production. This suggests that B. anthracis, Brucella, and Coxiella circulate throughout the year in all the ecosystems and confirms their endemicity and the recurrent risk for the nearby communities, including the larger markets where the meat is regularly sold.
While this study represents the first comprehensive assessments of the prevalence of DNA signatures of select agents in bushmeat in Tanzania, there are several unanswered questions that remain. For instance, it remains unclear how these DNA signatures represent / correlate with presence of viable pathogens and, with this, the risk of transmission to humans. It is also unclear whether there are any differences in the health risks associated with exposure during harvest, butchering, handling or consuming of bushmeat. Future studies investigating the role of other select agents (bacterial and viral) in the ecosystems, and the role of endemic and emerging strains in the microbial community, caused by sedentary and migrating animal species in the region are also warranted since the extent of an outbreak varies from year to year and place to place. In conclusion, the current investigations provide important insights and a rational basis for the formulation of future studies to assess and address the public health and emerging infectious disease risks associated with bushmeat harvesting, trade, and consumption in Tanzania and elsewhere.
Data availability
The datasets generated and analyzed during the current study are available in the NCBI Sequence Read Archive (SRA) repository, https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP151593.
References
Nielsen, M. R., Meilby, H. & Smith-Hall, C. How could the bushmeat trade in the Kilombero Valley of Tanzania be regulated? Insights from the rural value chain. Oryx 50(1), 84–93 (2016).
Kaltenborn, B. P., Nyahongo, J. W. & Tingstad, K. M. The nature of hunting around the Western Corridor of Serengeti National Park. Tanzania. Eur. J. Wildl. Res. 51(4), 213–222 (2005).
Rentsch, D., The nature of bushmeat hunting in the serengeti ecosystem, Tanzania: socio-economic drivers of consumption of migratory wildlife. 2011.
McNaughton, S. J. Ecology of a grazing ecosystem: the Serengeti. Ecol. Monogr. 55(3), 260–294 (1985).
Shombe, H. A. A. R. Fire history and management as determinant of patch selection by foraging herbivores in western Serengeti, Tanzania. Int. J. Biodivers. Sci. Ecosyst. Serv. Manag. 7, 122–133 (2011).
Dobson, A. D. M. et al. A framework for assessing impacts of wild meat hunting practices in the tropics. Hum. Ecol. 47(3), 449–464 (2019).
World Organisation for Animal Health, OIE Terrestrial Manual. Chapter 2.1.5. Foot and Mouth Disease. https://www.oie.int/eng/A_FMD2012/docs/2.01.05_FMD.pdf. Accessed 02 May 2020 (2012).
Mwakapeje, E. R. et al. Anthrax outbreaks in the humans—livestock and wildlife interface areas of Northern Tanzania: a retrospective record review 2006–2016. BMC Public Health 18(1), 106 (2018).
Swartz, M. N. Recognition and management of anthrax—an update. N. Engl. J. Med. 345(22), 1621–1626 (2001).
Mlengeya, T. et al. Anthrax epizootics in Tanzania’s national parks with special interest in a recent anthrax outbreak in Serengeti National Park. Bull. Anim. Health Prod. Africa 46, 65–73 (1998).
Lembo, T. et al. Serologic surveillance of anthrax in the Serengeti ecosystem, Tanzania, 1996–2009. Emerg. Infect. Dis. 17(3), 387–394 (2011).
Mwakapeje, E. R., et al., Prevention, detection, and response to anthrax outbreak in Northern Tanzania using one health approach: a case study of Selela ward in Monduli district. 2017.
Njau, P.F., et al., Brucellosis outbreak in Kiteto district, Tanzania 2012. East African Community Integrated Disease Surveillance Network, 2013.
Assenga, J. A. et al. Epidemiology of Brucella infection in the human, livestock and wildlife interface in the Katavi-Rukwa ecosystem, Tanzania. BMC Vet. Res. 11(1), 189 (2015).
Cordes, D. O. & Carter, M. E. Persistence of Brucella abortus infection in six herds of cattle under brucellosis eradication. N. Z. Vet. J. 27(12), 255–259 (1979).
Kunda, J. et al. Quantifying risk factors for human brucellosis in rural northern Tanzania. PLoS ONE 5(4), e9968 (2010).
Shirima, G. M. The epidemiology of brucellosis in animals and humans in Arusha and Manyara regions of Tanzania, 234 (University of Glasgow, 2005).
Enstrom, S. et al. Brucella seroprevalence in cattle near a wildlife reserve in Kenya. BMC Res. Notes 10(1), 615 (2017).
Alexander, K. A. et al. Buffalo, bush meat, and the zoonotic threat of brucellosis in Botswana. PLoS ONE 7(3), e32842 (2012).
Ndengu, M. et al. Seroprevalence of brucellosis in cattle and selected wildlife species at selected livestock/wildlife interface areas of the Gonarezhou National Park, Zimbabwe. Prev. Vet. Med. 146, 158–165 (2017).
Katani, R. et al. Microbial diversity in bushmeat samples recovered from the serengeti ecosystem in Tanzania. Sci. Rep. 9(1), 18086 (2019).
Hummel, P. H. Incidence in Tanzania of CF antibody to Coxiella burneti in sera from man, cattle, sheep, goats and game. Vet. Rec. 98(25), 501–505 (1976).
Adesiyun, A. A. et al. Sero-epidemiological study of selected zoonotic and abortifacient pathogens in cattle at a wildlife-livestock interface in South Africa. Vector Borne Zoonotic Dis. 20(4), 258–267 (2020).
Njeru, J. et al. Q fever is an old and neglected zoonotic disease in Kenya: a systematic review. BMC Public Health 16(1), 297 (2016).
Simpson, G. J. G. et al. Prevalence of selected zoonotic diseases and risk factors at a human-wildlife-livestock interface in Mpumalanga province, South Africa. Vector Borne Zoonotic Dis. 18(6), 303–310 (2018).
Sinclair, A. R. E. & Arcese, P. Serengeti II: Dynamics, Management, and Conservation of an Ecosystem, Vol. 9, 517–518. Journal of Evolutionary Biology (University of Chicago Press, 1995).
Pennycuick, L. Movements of the migratory wildebeest population in the Serengeti area between 1960 and 1973. Afr. J. Ecol. 13(1), 65–87 (1975).
Sinclair, A. R. E. & Arcese, P. Serengeti II: dynamics, management, and conservation of an ecosystem. J. Evolut. Biol. 9(4), 517–518 (1996).
Zorita, E. & Tilya, F. F. Rainfall variability in Northern Tanzania in the March–May season (long rains) and its links to large-scale climate forcing. Clim. Res. 20(1), 31–40 (2002).
Barnes, R. F. W. The decline of the baobab tree in Ruaha National Park, Tanzania. Afr. J. Ecol. 18(4), 243–252 (1980).
Ceppi, S. L. & Nielsen, M. R. A comparative study on bushmeat consumption patterns in ten tribes in Tanzania. Trop. Conserv. Sci. 7(2), 272–287 (2014).
Rentsch, D. & Damon, A. Prices, poaching, and protein alternatives: an analysis of bushmeat consumption around Serengeti National Park, Tanzania. Ecol. Econ. 91, 1–9 (2013).
Christensen, D. R. et al. Detection of biological threat agents by real-time PCR: comparison of assay performance on the R.A.P.I.D., the LightCycler, and the Smart Cycler platforms. Clin. Chem. 52(1), 141–145 (2006).
Li, P.-E. et al. Enabling the democratization of the genomics revolution with a fully integrated web-based bioinformatics platform. Nucleic Acids Res. 45(1), 67–80 (2017).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72(7), 5069–5072 (2006).
Aragon, T. J. Epitools: Epidemiology Tools. R package version 0.5-10. https://CRAN.R-project.org/package=epitools (2017).
Development Core Team R. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austia. http://www.R-project.org (2010).
McMurdie, P. J. & Holmes, S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 10(4), e1003531 (2014).
Oksanen, J., et al., Vegan: Community Ecology Package. R Package Version. 2.0-10. 2013.
Suzuki, R. & Shimodaira, H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22(12), 1540–1542 (2006).
Brashares, J. S. et al. Economic and geographic drivers of wildlife consumption in rural Africa. Proc. Natl. Acad. Sci. U. S. A. 108(34), 13931–13936 (2011).
Hampson, K. et al. Predictability of anthrax infection in the Serengeti. Tanzania. J. Appl. Ecol. 48(6), 1333–1344 (2011).
Egondi, T. et al. Time-series analysis of weather and mortality patterns in Nairobi’s informal settlements. Glob. Health Action 5(1), 19065 (2012).
Mrema, S. et al. The influence of weather on mortality in rural Tanzania: a time-series analysis 1999–2010. Glob. Health Action 5, 33–43 (2012).
Baker, G. L. Food safety impacts from post-harvest processing procedures of molluscan shellfish. Foods (Basel, Switzerland) 5(2), 29 (2016).
Rajkovic, A. et al. The effect of pulsed UV light on Escherichia coli O157:H7, Listeria monocytogenes, Salmonella Typhimurium, Staphylococcus aureus and staphylococcal enterotoxin A on sliced fermented salami and its chemical quality. Food Control 73, 829–837 (2017).
Farquhar, W. B. et al. Dietary sodium and health: more than just blood pressure. J. Am. Coll. Cardiol. 65(10), 1042–1050 (2015).
Lee, E.-S., Park, S. Y. & Ha, S.-D. Effect of UV-C light on the microbial and sensory quality of seasoned dried seafood. Food Sci. Technol. Int. 22(3), 213–220 (2015).
Miller, P. The effect of weather on prevalence of disease. Am. Biol. Teach. 28(6), 469–472 (1966).
Fares, A. Factors influencing the seasonal patterns of infectious diseases. Int. J. Prev. Med. 4(2), 128–132 (2013).
Prins, H. H. T. & Weyerhaeuser, F. J. Epidemics in populations of wild ruminants: anthrax and impala, rinderpest and Buffalo in Lake Manyara National Park. Tanzania. Oikos 49(1), 28–38 (1987).
Acknowledgements
The authors would like to thank the following agencies for permission to conduct research in the Serengeti ecosystem in Tanzania: Tanzania Commission for Science and Technology, Tanzania Wildlife Research Institute (Permit Number TWRI/RS-331/VOL.II/2013/58 and TWRI/RS-331/VOL.II/2013/88), Tanzania National Parks (Permit Number TNP/HQ/C.10/1), Ngorongoro Conservation Area Authority (Permit Number NCAA/D/240/Vol.XXVIII/54), and Tanzania Wildlife Authority (Permit Number CB.517/519/01/14). Collection of samples would not have been possible without the cooperation of enumerators, rangers and management authorities. The authors would also like to thank Karen Davenport, Blake Hovde, and Mark Flynn from Los Alamos National Laboratory for their technical support with The Empowering the Development of Genomics Expertise- “EDGE” analysis. The authors would also like to thank other Bushmeat Biosecurity Research Consortium members for their continued hard work and dedication throughout the project.
Funding
This research was supported by grant from the U.S. Department of Defense, Defense Threat Reduction Agency, Biological Threat Reduction Program, Project # HDTRA1-16-1-0005 (to V.K. and J.J.B.).
Author information
Authors and Affiliations
Contributions
V.K. and J.J.B. developed the study concept and design. E.E., A.M., and A.B.E. collected the samples and the metadata associated with each sample. R.K., M.A.S., B.L., S.L., and L.M. performed sample processing. T.T., C.K.T., and F.S. performed sequencing. R.K. and M.A.S. performed the bioinformatic and statistical analysis. R.K. and M.A.S. analyzed data and drafted the manuscript. I.M.C and P.J.H. advised on the data analysis. B.L., E.E., A.M., T.T., I.M.C., S.C.F., A.B.E., D.R., S.S., S.L., L.M., C.K.T., F.S., P.G., F.M., P.J.H., J.J.B., and V.K. critically revised the content. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Katani, R., Schilling, M.A., Lyimo, B. et al. Identification of Bacillus anthracis, Brucella spp., and Coxiella burnetii DNA signatures from bushmeat. Sci Rep 11, 14876 (2021). https://doi.org/10.1038/s41598-021-94112-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-94112-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
