
Abstract
Maternal and environmental factors influence brain networks and architecture via both physiological pathways and epigenetic modifications. In particular, prenatal maternal depression and anxiety symptoms appear to impact infant white matter (WM) microstructure, leading us to investigate whether epigenetic modifications (i.e., DNA methylation) contribute to these WM differences. To determine if infants of women with depression and anxiety symptoms exhibit epigenetic modifications linked to neurodevelopmental changes, 52 umbilical cord bloods (CBs) were profiled. We observed 219 differentially methylated genomic positions (DMPs; FDR p < 0.05) in CB that were associated with magnetic resonance imaging measures of WM microstructure at 1 month of age and in regions previously described to be related to maternal depression and anxiety symptoms. Genomic characterization of these associated DMPs revealed 143 unique genes with significant relationships to processes involved in neurodevelopment, GTPase activity, or the canonical Wnt signaling pathway. Separate regression models for female (n = 24) and male (n = 28) infants found 142 associated DMPs in females and 116 associated DMPs in males (nominal p value < 0.001, R > 0.5), which were annotated to 98 and 81 genes, respectively. Together, these findings suggest that umbilical CB DNA methylation levels at birth are associated with 1-month WM microstructure.
Similar content being viewed by others

Introduction
The first years of life are characterized by dynamic brain growth that is governed by genetic and environmental factors which help shape the structural and functional architecture of the brain and provide a foundation for subsequent cognition, behavior, and well-being1,2. In particular, recent neuroimaging studies indicate maternal depression and anxiety symptoms impact early brain development of the mother’s children, with higher depression and anxiety symptoms associated with reductions in cortical thickness and regional brain volumes3,4,5, differing patterns of functional connectivity6,7, and alterations in the maturing WM microstructure5,8,9,10,11,12,13,14. Despite evidence that maternal factors influence the brain prenatally, the molecular mechanisms underlying the effects on infant neurodevelopment associated with maternal depression and anxiety remain unclear.
Genome-wide DNA methylation levels vary dramatically throughout life and are responsive to early life adversity, including prenatal stress, separation from parents or variable maternal care15,16,17,18. DNA methylation (5-methylcytosine [5mC]) is an environmentally sensitive epigenetic modification serving important functions in chromatin remolding, gene silencing, embryonic development, cellular differentiation, and the maintenance of cellular identity19. Moreover, alterations in DNA methylation levels are emerging as important factors in the long-term biological trajectories leading to stress-related psychiatric disorders20,21. For instance, changes in 5mC levels in adults have been linked psychiatric disorders, including depression, anxiety, post-traumatic stress disorders and schizophrenia22,23,24,25. These studies suggest that DNA methylation is associated with neurobehavioral disorders in adults; however, the molecular mechanisms contributing to these disorders are likely to be initiated much earlier than adulthood26,27,28. In particular, placental and fetal DNA methylation levels provide tools to better understand the impact of prenatal depression and anxiety symptoms on molecular pathways in the infants. These indices may provide insights into the cellular and intracellular mediating pathways that contribute to the neurodevelopmental effects associated with prenatal anxiety and depression symptoms29,30.
Much remains to be learned about the role that adverse prenatal exposures have on DNA methylation levels and early brain development. A focused study on DNA methylation profiles from umbilical cord blood (CB) may potentially provide novel diagnostic tools to help identify at-risk infants and to improve assessment and clinical treatment strategies27,31. Previous work indicates prenatal exposure to maternal depression and anxiety symptoms can influence the developing brain, and in particular, the white matter (WM) microstructure3,4,5,6,7,8,9,10,11,12,13,14,32,33. In Dean et al. (2018), we observed associations between measures infant WM microstructure and prenatal maternal depression and anxiety symptoms8, including several WM regions, localized in regions spanning the corona radiata, superior longitudinal fasciculus, posterior thalamic radiations, among others, in which the association differed between male and female infants. To identify potential molecular mechanisms contributing to these observed sex-by-prenatal maternal depression and anxiety symptom interactions, here we examine the association between DNA methylation levels in cells from CB at delivery and measures of 1-month WM microstructure from regions identified in Dean et al. (2018). Specifically, we performed genome-wide DNA methylation profiling of infant leukocytes and examined how DNA methylation levels related to the WM microstructure in the preselected regions. Since the association between maternal depression and anxiety symptoms and 1-month WM microstructure was found to differ between males and females; we additionally performed sex-specific analyses. In sum, the aim was to identify genes and pathways involved in WM development that might be regulated by modifiable molecular substrates (e.g., DNA methylation), and that ultimately could be amenable to early intervention and treatment.
Methods
Study design and participants
Participants (N = 55 mother-infant dyads) were selected from a larger longitudinal study investigating the influence of early-life experience on child brain development8,34,35,36. Eligibility for the overall study included pregnant women aged 18–40 years, without major psychiatric illness (e.g., schizophrenia, bipolar or borderline personality disorder), or major autoimmune or infectious disorders during pregnancy. Infants were included if singleton, born between 37 and 42 weeks, and if discharged with the mother, without neurologic conditions, major birth head trauma, or a neonatal intensive care unit stay beyond observation8,34,35,36. Additional inclusion criteria for the current study included both an available CB sample collected at the time of delivery and a successful diffusion weighted MRI of the infant at 1 month of age. The University of Wisconsin–Madison Institutional Review Board approved all study procedures. Written informed consent was obtained from all mothers and written informed parental consent was obtained for all infants. All experiments were performed in accordance with relevant guidelines and regulations.
Prenatal maternal depression and anxiety symptoms
Mothers completed the Edinburgh Postnatal Depression Scale (EPDS)37 and State-Trait Anxiety Inventory (STAI)38 at 28 and 35 weeks of gestation. The EPDS is a widely used 10-item screening tool for perinatal depression, with scores ranging from 0 to 30 and a cut-off value of 10 or higher typically used to identify women who endorse depressive symptoms over the last month39. The 20-item STAI measures both state (14-items) and trait (6-items) level anxiety. Here, we focused on state anxiety, which captures maternal anxiety symptoms over the last month, with scores ranging from 0 to 42. As previously described, EPDS and STAI scores were strongly correlated, and a principal component analysis was used to construct a psychometrically valid depression and anxiety composite score from the prenatal EPDS and STAI measures8. This composite score was positively correlated with 28- and 35-week EPDS and STAI scores, with higher scores indicating more maternal symptoms during pregnancy. This composite score was used in subsequent analyses.
Cord blood collection and DNA extraction
After delivery, umbilical CB for whole blood genomic DNA was collected in sodium heparin vacutainer tubes. Whole cord blood samples were stored at 4 °C and processed at a median of 7.85 h after delivery (range 1.75–135 h, with two-thirds processed within 12 h and all but one before 48 h). gDNA was isolated by QIAamp DNA Mini Kit spin column protocol (Qiagen), quantitated by PicoGreen DNA dye (Turner BioSystems), using human gDNA standards (Promega E3401). DNA samples were stored frozen at − 70 °C.
Diffusion imaging data acquisition and processing
MRI was performed on a 3T scanner (MR750 Discovery scanner; General Electric) with a 32-channel head RF array coil (Nova Medical). Scanning was performed when the infant was 1-month of age during natural, nonsedated sleep34,35,40. Measures taken for successful image acquisition included acoustic noise reduction via fitting a foam insert inside the MRI scanner bore, utilizing ear plugs and MiniMuff (Natus Medical Incorporated) neonatal noise-attenuating ear covers, playing white noise through electrodynamic headphones (MR Confon, Germany) throughout the image acquisition, and limiting the MRI scanner slew rates. To help reduce motion throughout the scan, we swaddled each child using an infant MedVac vacuum immobilization bag and placed foam cushions around the head. Additional details about MRI acquisition have been previously described8,34,35,36,40.
A 10-min multi-shell diffusion imaging protocol was used to acquire 69 diffusion weighted images (DWIs), with 9/18/36 diffusion-encoding gradient directions at b = 350/800/1500 s/mm2, respectively, and 6 with no (b = 0 s/mm2) diffusion weighting. Additional imaging criteria consisted of the following: image resolution 2 × 2 × 2 mm; repetition time, 8400 ms; and echo time, 94 ms. DWI data were manually inspected for image artifacts, eddy current and motion corrected41, and skull-stripped (http://afni.nimh.nih.gov/pub/dist/doc/program_help/3dSkullStrip.html). Diffusion tensor imaging (DTI42) and Neurite Orientation Dispersion and Density (NODDI43) parameters were estimated and registered to a common, population specific template. Additional details regarding the diffusion image acquisition and processing are described elsewhere8,34.
Three WM regions previously identified to have differing associations between prenatal maternal depression and anxiety symptoms and DTI fractional anisotropy (FA), NODDI intracellular volume fraction (νIC), and NODDI orientation dispersion index (ODI) in males and females were examined. These regions spanned different areas of WM (Fig. 1A), including regions of the corona radiata and superior longitudinal fasciculus, among others (FA); the sagittal stratum and white matter adjacent to the hippocampus (νIC); and regions of posterior thalamic radiations, splenium of the corpus callosum, among others (ODI)8. For each subject, mean FA, νIC, and ODI values were extracted from the corresponding WM regions and used in subsequent DNA methylation analyses. Figure 1B provides a schematic of how average WM microstructures measures were computed for each subject.

(A) Representative sagittal, coronal, and axial sections of WM regions previously found to be differentially associated with maternal depression and anxiety symptoms during mid-late pregnancy. DTI FA associations were observed in regions of the corona radiata and superior longitudinal fasciculus, among others; NODDI νIC associations were found in the sagittal stratum and white matter adjacent to the hippocampus; and NODDI ODI associations were observed in areas of posterior thalamic radiations, splenium of the corpus callosum, among others8. (B) Schematic illustrating extraction of WM microstructural measures from WM regions. Mean FA, νIC, and ODI were extracted from the respective WM regions for each subject and used in subsequent analyses.
Preprocessing of human methylation EPIC data
Genome-wide DNA methylation levels were determined using the Infinium HumanMethylationEPIC array (Illumina, San Diego, CA) and raw intensity data files were imported into the R environment44. The R package minfi was used to assess sample quality, calculate the signal detection p value of each tested probe, estimate cell proportions in the CB specimens, predict sex, and normalize data45,46. Two samples were discarded because they did not pass quality control, as their mean signal detection p value exceeded 0.05. Probes were normal-exponential out-of-band (noob) normalized with dye correction, followed by quantile normalization. One additional sample was discarded as it failed sex prediction, suggesting unreliable DNA methylation levels for this sample. Probes were filtered if any one sample exhibited a signal detection p value > 0.01, contained a SNP, reported methylation at a SNP, were derived from sex chromosomes, measured methylation at a CH dinucleotide site, or are known cross-reactive probes (≥ 47 bp homology with an off-target site)47,48. These filtration criteria resulted in 763,917 probes used for further analysis.
Identification of differentially methylated loci
Beta-values were obtained through minfi and were further converted to logit-transformed M-values for differential analyses. Linear regression for each tested CpG using a multivariate model was employed using the R package limma49. For regression, separate models for key WM microstructure measures of FA, νIC, or ODI were constructed, controlling for CB cell proportions (i.e., CD8+T, CD4+T, Natural Killer, Monocyte, B cell, and Granulocyte), sex, infant gestational age-corrected postpartum age, Hollingshead socioeconomic status50, and an index of total motion during the diffusion acquisition51; gene length was not considered in the analysis. Surrogate variables were assessed by the R package sva52 and identified 6 surrogate variables that were adjusted for during model fitting. To assess systematic bias of the linear regression model, the genomic inflation factor was calculated for the obtained p values, yielding a genomic inflation factor of ~ 1, suggesting no bias. Pearson’s correlation coefficients (r) were calculated for continuous variables of interest with beta-values. Differential methylation was deemed significant if the Benjamini–Hochberg FDR adjusted p value < 0.05 (full data) or the unadjusted p value < 0.001 and the correlation (r) was > 0.5 (sex-specific data). Notably, minimizing family-wise error rates with conventional false discovery rates is overly conservative for methylation data, because DNA methylation levels are continuous variables when measured across a large number of cells and neighboring probes on the array are known to be correlated and many sites on the array are non-variable53,54. Thus, the DMPs identified in the full dataset are likely a conservative set. The smaller sample size of the sex-specific analysis likely resulted in the absence of FDR adjusted DMPs.
Statistical analyses
The distributions of demographic data were evaluated. Comparisons between female and male infants were conducted with t tests for continuous and χ2 for categorical variables, with p value < 0.05 considered significant. Infants were included if mothers’ prenatal depression and anxiety scores were obtained, CB volumes were sufficient for obtaining DNA for epigenetic analysis, and full NODDI MRI imaging data were obtained.
Genes exhibiting differential methylation were subjected to gene ontological analysis using the R package clusterProfiler4 to identify significant disruptions in biological processes, and to define gene concepts and map enrichment terms. The database used for the gene pathway analysis was the “Genome wide annotation for Human” (org.Hs.eg.db) in the following Bioconductor R package: https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html. The total number of genes subjected to the differential analysis was used as the background gene universe. Pathways were included in the analysis if they had a at least 10 and no more than 500 genes in the background gene set. Gene ontological terms were deemed significant if p value < 0.05.
Results
Cohort demographics
From a total 149 possible mother-infant dyads, 73 had insufficient CB volumes for DNA extraction or did not pass quality control procedures (see “Methods” section), and an additional 24 infants did not complete the entire MRI scan, resulting in a final sample of 52 (24 female). Demographic data are shown in Table 1. Infants were healthy at birth, without differences in demographic characteristics between females and males, except that more males were born in middle-income families and thus fewer males were born into the high-income families. Mothers with male infants reported higher state trait anxiety index (STAI) at 35 weeks of gestation (p value = 0.04) and tended to have a higher depression and anxiety composite score (p value = 0.07). Twenty-four mothers (46.2%) reported EPDS scores between 0 and 6, 17 mothers (32.7%) reported EPDS scores between 7 and 10, and 11 mothers (21.2%) reported EPDS scores ≥ 11. Six mothers reported EPDS scores higher than 12, which is generally consistent with a diagnosis of major depressive disorder. Six women reported antidepressant medication use for depression or anxiety during pregnancy. The EPDS scores and STAI scores conveyed relatively mild to moderate levels of depression and/or anxiety symptomology.
Sex-differing associations between prenatal maternal depression and anxiety symptoms and infant WM microstructure
To determine the extent to which CB DNA methylation levels were correlated with sex-differentiated relationships with prenatal maternal depression and anxiety symptoms, genomic DNA was analyzed from CB using the HumanMethylationEPIC beadchip array (Illumina). After filtering out unreliable probes (see “Methods” section), this analysis provided a reliable quantitative measure of DNA methylation levels at 763,917 CpG dinucleotides across the human genome at single-nucleotide resolution, including enhancers and all coding regions. A regression model using late pregnancy depression and anxiety composite scores during late pregnancy as the explanatory variable did not find correlations with CB DNA methylation levels after FDR correction; however, 130 DMPs corresponding to 91 genes were correlated at a lower stringency (p value < 0.001, R > 0.4).
In Dean et al. (2018), associations between prenatal maternal depression and anxiety symptoms and infant WM microstructure were previously observed to differ between males and females in the larger sample (N = 101)8. CB samples were available from only a subset of these infants (N = 52). Therefore, to confirm the previous findings, linear regressions using mean FA, νIC, and ODI measures were repeated for the smaller subset with an available CB specimen. The sex-by-prenatal maternal depression and anxiety symptom interactions remained significant for FA, νIC and ODI in the smaller sample (p value = 0.002, < 0.001, < 0.001, respectively; Supplementary Fig. 1), suggesting lower FA, νIC and ODI in females and higher FA, νIC and ODI in males exposed to higher levels of maternal depression and anxiety. These WM measures at 1 month of age were used to determine if there were significant correlations with CB DNA methylation levels.
Differential methylation related to infant WM microstructure
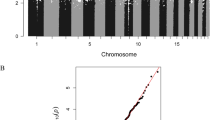
Individual regression models for 1-month infant FA, νIC, and ODI at 1 month were performed, with the WM microstructure indices considered as the explanatory variable. No correlation between DNA methylation levels in CB and FA and ODI that survived FDR correction; however, 319 (FA) and 122 (ODI) DMPs corresponding to 217 (FA) and 99 (ODI) genes were found at lower a stringency (p value < 0.001, R > 0.4). In contrast, we found 219 νIC-associated DMPs in CB (FDR p value < 0.05; Fig. 2A; Dataset 1). These νIC-associated DMPs were distributed across all autosomes with a total of 154 hyper- and 65 hypo-DMPs (Dataset 1), indicating the majority of sites contributing to WM microstructure had higher DNA methylation levels.

Cord blood DNA methylation levels associated with infant WM microstructure-related changes (νIC) showing a sex-by-prenatal maternal depression and anxiety symptom interaction. (A) Circos plot depicts genomic distribution of differentially methylated positions (DMPs) across the human genome. (Outer ring) Each chromosome is shown as a different color and the relative chromosome size is represented by the bar length. (Inner rings) Represent the relative location of DMPs across all chromosomes in the full dataset (black), females only (orange), and males only (blue). Sex chromosomes were omitted from the analysis. B) Pie chart showing the percent distribution of DMPs to standard genomic features. 5′UTR = 5′ untranslated region’ 3′UTR = 3′ untranslated region; TSS = transcription start site; TSS200 = 0–200 bp upstream of TSS; TSS1500 = 200–1500 bp upstream of TSS. Circos plot and pie chart were generated using the R environment44.
Annotation of the νIC-associated DMPs to standard genomic structures revealed that nearly half (47%) resided in promoter regions of annotated genes (i.e., within 1,500 base pairs of the transcription start site or the first exon; Fig. 2B). Annotation of the 219 νIC-associated DMPs to genes revealed 143 unique genes, including genes known to contribute to the development of the nervous system (e.g., LDLR and KDM4A).
Differential methylation regions are linked to nervous system and Wnt signaling pathways
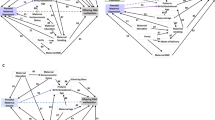
To characterize genes and pathways linked to infant WM microstructure at 1 month of age, we next examined the gene ontologies (GO) of the νIC-associated genes that had DMPs. These analyses revealed significant relationships between genes linked to processes involved in astrocyte differentiation, GTPase activity, and Wnt signaling (Fig. 3A,B; Supplementary Table 1). Moreover, gene network mapping of the enrichment results identified two major gene network hubs: nervous system development, including negative regulators of nervous system development, and regulation of the canonical Wnt signaling pathway (Fig. 3C).

Gene pathways analysis of differentially methylation position (DMP) in CB that were associated infant WM microstructure-related changes (νIC) showing a Sex x Symptom interaction. (A) Bar Graph of the top 20 gene ontological (GO) biological processes associated with the differentially methylated genes, ordered by statistical significance. X-axis indicates the number of DMP-associated genes contributing to each GO term. The shade of the bars shows the p value based on the legend, as determined by a Fischer test. Neg. = Negative; Pos. = Positive. (B) Gene-concept network plot shows the top five gene ontology terms (beige), the genes (gray) associated with each term, and the interconnectivity between genes and processes (lines). The size of the beige dot relates to the number of DMP-associated genes contributing to that term. (C) An enrichment map plot depicts the connectivity of associated terms, with hubs of similar processes clustering further apart. Node (spheres) size represent the relative number of DMP-associated genes contributing to each term, while the color represents the FDR p-value, shown in the legend, as determined by a Fischer test. The size of the edges (gray lines) depicts the strength of relatedness between terms. Bar graph, gene-concept network plot, and enrichment map were constructed using the R environment44.
Sex-specific differential methylation related to infant WM microstructure
Associations between νIC and prenatal maternal depression and anxiety symptoms that differed by sex8 led us to investigate sex-specific DNA methylation levels in relation to infant WM microstructure at 1 month. Using separate regression models for females (N = 24) and males (N = 28), we observed 142 νIC-associated DMPs in females and 116 νIC-associated DMPs in males (p value < 0.001, R > 0.5; Fig. 2A). These νIC-associated DMPs were annotated to 98 and 81 genes in females and males, respectively, with an overlap of 3 genes (Chondroitin Sulfate N-Acetylgalactosaminyltransferase 1 [CSGALNACT1], Phosphodiesterase 6B [PDE6B], and Transcription Factor 7 Like 2 [TCF7L2]; Dataset 1). There was only one female-specific DMP-associated gene (ACVR1B) and one male-specific DMP-associated gene (TLL2) that overlapped with the full dataset.
Discussion
This study extends our prior findings linking WM microstructure at 1 month of age to maternal depression and anxiety symptoms during the prenatal period8 and now identifies some associations with epigenetic modifications. Using CB leukocytes collected at delivery, we show that DNA methylation levels associated with the microstructure of several selected WM regions were enriched in gene pathways that negatively regulate neurodevelopmental processes.
Previous research examining the effect of maternal depression and anxiety on CB DNA methylation levels has yielded mixed results. A meta-analysis of two large birth cohorts with CB showed no epigenome-wide associations with prenatal stress55. Another study found 23 DMPs associated with depression in late pregnancy30. Although we did not find a robust, simple relationship between DMPs and prenatal depression and anxiety scores, we did find many DMPs associated with WM microstructure that were related to maternal depression and anxiety during the third trimester. These findings support the value of employing a targeted approach utilizing microstructural brain imaging to identify anatomical regions associated with CB DNA methylation levels.
DMPs were associated with WM neurite density in the left-hemisphere sagittal stratum8, a major WM bundle containing the inferior fronto-occipital fasciculus, inferior longitudinal fasciculus, and posterior thalamic radiations56,57,58. Several underlying processes contribute to the development of these WM tracts, including astrocyte, glial, neuronal and cellular proliferation and differentiation59. νIC-associated DMPs were detected on genes that negatively regulate astrocyte differentiation, including the low-density lipoprotein R (LDLR), Neurofibromin 1 (NF1), Wingless-Type MMTV Integration Site Family, Member 3A (WNT3A) and patched-1 (PTCH1). LDLR functions as a carrier of cholesterol in blood, which is important in neuronal differentiation and synaptogenesis60. NF1 encodes a protein that regulates cell growth through the Ras pathway, with NF1 dysregulation affecting brain development61,62,63, including WM microstructure64,65 and myelin66. NF1 is also linked to the regulation of the GTPase pathway, which plays a role in ectoderm differentiation and is linked to microcephaly, structural brain abnormalities, and intellectual disability67,68,69. Finally, WNT3A and PTCH1 provide proteins that trigger signals essential for cell fate, specialization, and patterning during embryogenesis70,71. These data suggest that νIC-associated DNA methylation levels may play a role in the negative regulation of neuronal differentiation, growth, and patterning of WM microstructure.
We found unique sets of νIC-associated alterations in DNA methylation levels for male and female infants, with an overlap of only three genes. Although the mechanisms underlying this difference are unknown, sex differences in DNA methylation levels may be related to variation in the timing of WM development between females and males, with WM usually maturing earlier in females than in males72,73,74. Moreover, environmental exposures during different gestational periods have unique effects on epigenetic programming of the developing embryo/fetus and influence males and females differently75. For example, stress-induced epigenetic changes during early gestation predominately affect male development in rats76, while stress during late gestation tends to have larger effects on development in females76. Thus, sex-specific effects may depend on the gestational timing of the adverse event. Whether these sex differences in DNA methylation are related to long-term neurodevelopmental and behavioral outcomes requires further study.
A descriptive analysis of the CB methylation events related to gene structure revealed that more than 50% of the νIC-associated DMPs were located in the body of the gene, of which nearly 90% (70/79) were νIC-associated decreases in DNA methylation levels. Because active transcription is generally associated with increased gene body DNA methylation levels77, the findings suggest that these 70 DMP-associated genes moderate gene expression in CB. Future studies are needed to confirm the function and expression levels of these candidate genes.
A strength of this study is the multi-method birth cohort design, which combined rich phenotypic data, CB samples collected at delivery for whole genome epigenetic analysis, and quantitative diffusion MRI for characterizing early brain development. Blood has the highest proportion of CpGs that are nominally correlated to brain, when compared to other available tissues, including buccal swabs and saliva78. Although the need for data across three domains limited our sample size, it allowed us to detect a novel association between the prenatal environment, CB molecular indices, and WM microstructure at 1 month of age. Importantly, the cord blood DNA methylation levels presented here could conceivably provide bioindicators that reflect how the prenatal environment shapes early brain development, independent of DNA methylation levels in brain. Another strength is the finding of DMPs on genes relevant to neurodevelopment in circulating nucleated blood cells; however, these DNA methylation changes may not be functional (i.e., alter gene expression) and thus still require further analysis. Further, despite the neuroimaging being performed in close proximity to birth, the influence of postnatal parenting during the first month of life could contribute to the findings. Our data are from a short developmental period; longitudinal studies are now needed to examine the impact of DNA methylation on subsequent neurodevelopmental trajectories at older ages.
Here, we identify a potential mechanism by which prenatal maternal depression and anxiety symptoms could negatively regulate the development of WM microstructure through epigenetic alterations. Although we had a relatively small cohort, several other studies have also found significant associations with processes or pathways in smaller cohorts79,80,81,82. The majority of mothers reported depression and anxiety scores in the subclinical range, limiting our ability to draw inferences about effects of overt maternal psychopathology, but presumably the alterations might be larger in clinically depressed mothers. In addition, only a few mothers reported using antidepressants during pregnancy, a potential limiting confounder, precluding our ability to examine the influence of medication use on DNA methylation and early brain development. Of note, previous analyses indicated no differences with the exclusion of these women8; however, future work would benefit from examining prenatal medication use in larger, longitudinal samples. Additional environmental or experiential factors not taken into account in the current study may also have some contributing impact on the processes investigated and should be considered in future studies of larger cohorts. Though not investigated here, the maturation of cortical and subcortical gray matter and functional connectivity may also be influenced by epigenetic modifications and are of interest for future study. Nonetheless, our findings provide evidence for an association between epigenetic modifications and WM microstructure even after the mild symptoms commonly experienced by a significant number of pregnant women83. Our findings suggest that these environmentally sensitive epigenetic modifications detected in umbilical CB are associated with altered WM microstructure at 1 month of age. The sex-specific differences in DNA methylation levels were present on genes that negatively regulate neurodevelopment, regulate GTPase, and modulate canonical Wnt signaling, and thus implicate candidate genes for further investigation of sex-related differences in risk for neurodevelopmental disorders. Understanding these early microstructural developmental patterns are essential for appreciating the changes that occur at later stages of growth and informs a deeper understanding of the role of epigenetic modifications on the development of early white matter and brain development.
References
Bick, J. & Nelson, C. A. Early adverse experiences and the developing brain. Neuropsychopharmacology 41, 177–196. https://doi.org/10.1038/npp.2015.252 (2016).
Davidson, R. J. & McEwen, B. S. Social influences on neuroplasticity: stress and interventions to promote well-being. Nat. Neurosci. 15, 689–695. https://doi.org/10.1038/nn.3093 (2012).
Buss, C., Davis, E. P., Muftuler, L. T., Head, K. & Sandman, C. A. High pregnancy anxiety during mid-gestation is associated with decreased gray matter density in 6–9-year-old children. Psychoneuroendocrinology 35, 141–153. https://doi.org/10.1016/j.psyneuen.2009.07.010 (2010).
Sandman, C. A., Buss, C., Head, K. & Davis, E. P. Fetal exposure to maternal depressive symptoms is associated with cortical thickness in late childhood. Biol. Psychiatry 77, 324–334. https://doi.org/10.1016/j.biopsych.2014.06.025 (2015).
Lebel, C. et al. Prepartum and postpartum maternal depressive symptoms are related to children’s brain structure in preschool. Biol. Psychiatry 80, 859–868. https://doi.org/10.1016/j.biopsych.2015.12.004 (2016).
Posner, J. et al. Alterations in amygdala–prefrontal circuits in infants exposed to prenatal maternal depression. Transl. Psychiatry 6, e935–e935. https://doi.org/10.1038/tp.2016.146 (2016).
Qiu, A. et al. Prenatal maternal depression alters amygdala functional connectivity in 6-month-old infants. Transl. Psychiatry 5, e508–e508. https://doi.org/10.1038/tp.2015.3 (2015).
Dean, D. C. 3rd. et al. Association of prenatal maternal depression and anxiety symptoms with infant white matter microstructure. JAMA Pediatr. 172, 973–981. https://doi.org/10.1001/jamapediatrics.2018.2132 (2018).
Lautarescu, A. et al. Maternal prenatal stress is associated with altered uncinate fasciculus microstructure in premature neonates. Biol. Psychiatry 87, 559–569. https://doi.org/10.1016/j.biopsych.2019.08.010 (2020).
Rifkin-Graboi, A. et al. Prenatal maternal depression associates with microstructure of right amygdala in neonates at birth. Biol. Psychiatry 74, 837–844. https://doi.org/10.1016/j.biopsych.2013.06.019 (2013).
Coe, C. L., Lulbach, G. R. & Schneider, M. L. Prenatal disturbance alters the size of the corpus callosum in young monkeys. Dev. Psychobiol. 41, 178–185. https://doi.org/10.1002/dev.10063 (2002).
Coplan, J. D. et al. The role of early life stress in development of the anterior limb of the internal capsule in nonhuman primates. Neurosci. Lett. 480, 93–96. https://doi.org/10.1016/j.neulet.2010.06.012 (2010).
Howell, B. R. et al. Brain white matter microstructure alterations in adolescent rhesus monkeys exposed to early life stress: associations with high cortisol during infancy. Biol. Mood Anxiety Disord. 3, 21. https://doi.org/10.1186/2045-5380-3-21 (2013).
Rifkin-Graboi, A. et al. Antenatal maternal anxiety predicts variations in neural structures implicated in anxiety disorders in newborns. J. Am. Acad. Child. Adolesc. Psychiatry 54, 313-321.e312. https://doi.org/10.1016/j.jaac.2015.01.013 (2015).
Braithwaite, E. C., Kundakovic, M., Ramchandani, P. G., Murphy, S. E. & Champagne, F. A. Maternal prenatal depressive symptoms predict infant NR3C1 1F and BDNF IV DNA methylation. Epigenetics 10, 408–417. https://doi.org/10.1080/15592294.2015.1039221 (2015).
Murgatroyd, C., Quinn, J. P., Sharp, H. M., Pickles, A. & Hill, J. Effects of prenatal and postnatal depression, and maternal stroking, at the glucocorticoid receptor gene. Transl. Psychiatry 5, e560. https://doi.org/10.1038/tp.2014.140 (2015).
Oberlander, T. F. et al. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 3, 97–106. https://doi.org/10.4161/epi.3.2.6034 (2008).
Radtke, K. M. et al. Epigenetic modifications of the glucocorticoid receptor gene are associated with the vulnerability to psychopathology in childhood maltreatment. Transl. Psychiatry 5, e571. https://doi.org/10.1038/tp.2015.63 (2015).
Robertson, K. D. DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610. https://doi.org/10.1038/nrg1655 (2005).
Papale, L. A., Seltzer, L. J., Madrid, A., Pollak, S. D. & Alisch, R. S. Differentially methylated genes in saliva are linked to childhood stress. Sci. Rep. 8, 10785. https://doi.org/10.1038/s41598-018-29107-0 (2018).
Roth, T. L., Lubin, F. D., Funk, A. J. & Sweatt, J. D. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol. Psychiatry 65, 760–769. https://doi.org/10.1016/j.biopsych.2008.11.028 (2009).
Abdolmaleky, H. M. et al. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum. Mol. Genet. 15, 3132–3145. https://doi.org/10.1093/hmg/ddl253 (2006).
Kuratomi, G. et al. Aberrant DNA methylation associated with bipolar disorder identified from discordant monozygotic twins. Mol. Psychiatry 13, 429–441. https://doi.org/10.1038/sj.mp.4002001 (2008).
Pidsley, R. et al. Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome Biol. 15, 483. https://doi.org/10.1186/s13059-014-0483-2 (2014).
Poulter, M. O. et al. GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biol. Psychiatry 64, 645–652. https://doi.org/10.1016/j.biopsych.2008.05.028 (2008).
Dubovický, M. Neurobehavioral manifestations of developmental impairment of the brain. Interdiscip. Toxicol. 3, 59–67. https://doi.org/10.2478/v10102-010-0012-4 (2010).
Hodyl, N. A., Roberts, C. T. & Bianco-Miotto, T. Cord blood DNA methylation biomarkers for predicting neurodevelopmental outcomes. Genes (Basel) 7, 117. https://doi.org/10.3390/genes7120117 (2016).
Mitchell, C., Schneper, L. M. & Notterman, D. A. DNA methylation, early life environment, and health outcomes. Pediatr. Res. 79, 212–219. https://doi.org/10.1038/pr.2015.193 (2016).
Nemoda, Z. & Szyf, M. Epigenetic alterations and prenatal maternal depression. Birth Defects Res. 109, 888–897. https://doi.org/10.1002/bdr2.1081 (2017).
Viuff, A. C. et al. Maternal depression during pregnancy and cord blood DNA methylation: findings from the Avon Longitudinal Study of Parents and Children. Transl. Psychiatry 8, 244–244. https://doi.org/10.1038/s41398-018-0286-4 (2018).
Herbstman, J. B. et al. Predictors and consequences of global DNA methylation in cord blood and at 3 years. PLoS ONE 8, e72824. https://doi.org/10.1371/journal.pone.0072824 (2013).
Graham, A. M. et al. Maternal cortisol concentrations during pregnancy and sex-specific associations with neonatal amygdala connectivity and emerging internalizing behaviors. Biol. Psychiatry 85, 172–181. https://doi.org/10.1016/j.biopsych.2018.06.023 (2019).
Qiu, A. et al. Effects of antenatal maternal depressive symptoms and socio-economic status on neonatal brain development are modulated by genetic risk. Cereb. Cortex 27, 3080–3092. https://doi.org/10.1093/cercor/bhx065 (2017).
Dean, D. C. 3rd. et al. Mapping white matter microstructure in the 1 month human brain. Sci. Rep. 7, 9759. https://doi.org/10.1038/s41598-017-09915-6 (2017).
Dean, D. C. 3rd. et al. Investigation of brain structure in the 1-month infant. Brain Struct. Funct. 223, 1953–1970. https://doi.org/10.1007/s00429-017-1600-2 (2018).
Dowe, K. N. et al. Early microstructure of white matter associated with infant attention. Dev. Cogn. Neurosci. 45, 100815–100815. https://doi.org/10.1016/j.dcn.2020.100815 (2020).
Cox, J. L., Holden, J. M. & Sagovsky, R. Detection of postnatal depression: development of the 10-item Edinburgh Postnatal Depression Scale. Br. J. Psychiatry 150, 782–786 (1987).
Spielberger, C. D. The Corsini Encyclopedia of Psychology (Wiley, 2010).
Levis, B., Negeri, Z., Sun, Y., Benedetti, A. & Thombs, B. D. Accuracy of the Edinburgh Postnatal Depression Scale (EPDS) for screening to detect major depression among pregnant and postpartum women: systematic review and meta-analysis of individual participant data. BMJ 371, m4022. https://doi.org/10.1136/bmj.m4022 (2020).
Dean, D. C. 3rd. et al. Pediatric neuroimaging using magnetic resonance imaging during non-sedated sleep. Pediatr. Radiol. 44, 64–72. https://doi.org/10.1007/s00247-013-2752-8 (2014).
Jenkinson, M., Bannister, P., Brady, M. & Smith, S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage 17, 825–841 (2002).
Basser, P. J. & Pierpaoli, C. Microstructural and physiological features of tissues elucidated by quantitative–diffusion–tensor MRI. J. Magn. Reson. B 111, 209–219 (1996).
Zhang, H., Schneider, T., Wheeler-Kingshott, C. A. & Alexander, D. C. NODDI: practical in vivo neurite orientation dispersion and density imaging of the human brain. Neuroimage 61, 1000–1016. https://doi.org/10.1016/j.neuroimage.2012.03.072 (2012).
Team, R. C. R: A Language and Environment for Statistical Computing (Team, R. C., 2013).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. https://doi.org/10.1093/bioinformatics/btu049 (2014).
Gervin, K. et al. Systematic evaluation and validation of reference and library selection methods for deconvolution of cord blood DNA methylation data. Clin. Epigenet. 11, 125. https://doi.org/10.1186/s13148-019-0717-y (2019).
Pidsley, R. et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 17, 208. https://doi.org/10.1186/s13059-016-1066-1 (2016).
Chen, Y. A. et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 8, 203–209. https://doi.org/10.4161/epi.23470 (2013).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Hollingshead, A. B. Four factor index of social status. New Haven, CT: Yale University Department of Psychology, (1975).
Yendiki, A., Koldewyn, K., Kakunoori, S., Kanwisher, N. & Fischl, B. Spurious group differences due to head motion in a diffusion MRI study. Neuroimage 88, 79–90. https://doi.org/10.1016/j.neuroimage.2013.11.027 (2014).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics (Oxford, England) 28, 882–883. https://doi.org/10.1093/bioinformatics/bts034 (2012).
Michels, K. B. et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat. Methods 10, 949–955. https://doi.org/10.1038/nmeth.2632 (2013).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. 100, 9440–9445. https://doi.org/10.1073/pnas.1530509100 (2003).
Rijlaarsdam, J. et al. An epigenome-wide association meta-analysis of prenatal maternal stress in neonates: a model approach for replication. Epigenetics 11, 140–149. https://doi.org/10.1080/15592294.2016.1145329 (2016).
Di Carlo, D. T. et al. Microsurgical anatomy of the sagittal stratum. Acta Neurochir. (Wien) 161, 2319–2327. https://doi.org/10.1007/s00701-019-04019-8 (2019).
Oishi, K. et al. Human brain white matter atlas: identification and assignment of common anatomical structures in superficial white matter. Neuroimage 43, 447–457 (2008).
Mori, S. et al. Stereotaxic white matter atlas based on diffusion tensor imaging in an ICBM template. Neuroimage 40, 570–582. https://doi.org/10.1016/j.neuroimage.2007.12.035 (2008).
Dubois, J. et al. The early development of brain white matter: a review of imaging studies in fetuses, newborns and infants. Neuroscience 276, 48–71. https://doi.org/10.1016/j.neuroscience.2013.12.044 (2014).
Lane-Donovan, C., Philips, G. T. & Herz, J. More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron 83, 771–787. https://doi.org/10.1016/j.neuron.2014.08.005 (2014).
Moore, B. D. 3rd., Slopis, J. M., Jackson, E. F., De Winter, A. E. & Leeds, N. E. Brain volume in children with neurofibromatosis type 1: relation to neuropsychological status. Neurology 54, 914–920. https://doi.org/10.1212/wnl.54.4.914 (2000).
Greenwood, R. S. et al. Brain morphometry, T2-weighted hyperintensities, and IQ in children with neurofibromatosis type 1. Arch. Neurol. 62, 1904–1908. https://doi.org/10.1001/archneur.62.12.1904 (2005).
Dubovsky, E. C. et al. MR imaging of the corpus callosum in pediatric patients with neurofibromatosis type 1. AJNR Am. J. Neuroradiol. 22, 190–195 (2001).
Alkan, A. et al. Neurofibromatosis type 1: diffusion weighted imaging findings of brain. Eur. J. Radiol. 56, 229–234. https://doi.org/10.1016/j.ejrad.2005.05.008 (2005).
Karlsgodt, K. H. et al. Alterations in white matter microstructure in neurofibromatosis-1. PLoS ONE 7, e47854. https://doi.org/10.1371/journal.pone.0047854 (2012).
Viskochil, D. et al. The gene encoding the oligodendrocyte-myelin glycoprotein is embedded within the neurofibromatosis type 1 gene. Mol. Cell Biol. 11, 906–912. https://doi.org/10.1128/mcb.11.2.906 (1991).
North, K. et al. Specific learning disability in children with neurofibromatosis type 1, significance of MRI abnormalities. Neurology 44, 878–878. https://doi.org/10.1212/wnl.44.5.878 (1994).
Ozonoff, S. Cognitive impairment in neurofibromatosis type 1. Am. J. Med. Genet. 89, 45–52. https://doi.org/10.1002/(sici)1096-8628(19990326)89:1%3c45::Aid-ajmg9%3e3.0.Co;2-j (1999).
Payne, J. M., Moharir, M. D., Webster, R. & North, K. N. Brain structure and function in neurofibromatosis type 1: current concepts and future directions. J. Neurol. Neurosurg. Psychiatry 81, 304–309. https://doi.org/10.1136/jnnp.2009.179630 (2010).
Sethi, J. K. & Vidal-Puig, A. Wnt signalling and the control of cellular metabolism. Biochem. J. 427, 1–17. https://doi.org/10.1042/bj20091866 (2010).
Komiya, Y. & Habas, R. Wnt signal transduction pathways. Organogenesis 4, 68–75. https://doi.org/10.4161/org.4.2.5851 (2008).
Dean, D. C. 3rd. et al. Characterizing longitudinal white matter development during early childhood. Brain Struct. Funct. 220, 1921–1933. https://doi.org/10.1007/s00429-014-0763-3 (2015).
Koolschijn, P. C. & Crone, E. A. Sex differences and structural brain maturation from childhood to early adulthood. Dev. Cogn. Neurosci. 5, 106–118. https://doi.org/10.1016/j.dcn.2013.02.003 (2013).
Schmithorst, V. J., Holland, S. K. & Dardzinski, B. J. Developmental differences in white matter architecture between boys and girls. Hum. Brain Mapp. 29, 696–710. https://doi.org/10.1002/hbm.20431 (2008).
McCarthy, M. M. et al. The epigenetics of sex differences in the brain. J. Neurosci. 29, 12815–12823. https://doi.org/10.1523/jneurosci.3331-09.2009 (2009).
Li, H. et al. NF-κB regulates prenatal stress-induced cognitive impairment in offspring rats. Behav. Neurosci. 122, 331–339. https://doi.org/10.1037/0735-7044.122.2.331 (2008).
Jones, P. A. The DNA methylation paradox. Trends Genet. 15, 34–37. https://doi.org/10.1016/s0168-9525(98)01636-9 (1999).
Braun, P. R. et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl. Psychiatry 9, 47. https://doi.org/10.1038/s41398-019-0376-y (2019).
Mordaunt, C. E. et al. Epigenomic signatures in liver and blood of Wilson disease patients include hypermethylation of liver-specific enhancers. Epigenet. Chromatin 12, 10. https://doi.org/10.1186/s13072-019-0255-z (2019).
Ham, S. et al. Epigenetic analysis in rheumatoid arthritis synoviocytes. Exp. Mol. Med. 51, 1–13. https://doi.org/10.1038/s12276-019-0215-5 (2019).
Grimm, S. A. et al. DNA methylation in mice is influenced by genetics as well as sex and life experience. Nat. Commun. 10, 305. https://doi.org/10.1038/s41467-018-08067-z (2019).
Rizzardi, L. F. et al. Neuronal brain-region-specific DNA methylation and chromatin accessibility are associated with neuropsychiatric trait heritability. Nat. Neurosci. 22, 307–316. https://doi.org/10.1038/s41593-018-0297-8 (2019).
Bennett, H. A., Einarson, A., Taddio, A., Koren, G. & Einarson, T. R. Prevalence of depression during pregnancy: systematic review. Obstet. Gynecol. 103, 698–709. https://doi.org/10.1097/01.AOG.0000116689.75396.5f (2004).
Acknowledgements
We sincerely thank the children and families who participated in this research. We wish to acknowledge and thank Dr. Andrew L. Alexander, Ronald Fisher, Michael Anderle, and Scott Mikkelson for their thoughtful discussions and contributions. This work was supported by grants P50 MH100031 (Dr. Davidson) and R01 MH101504 (Dr. Goldsmith), R00 MH11056 (Dr. Dean), and K01 MH113710 (Dr. Planalp) from the National Institute of Mental Health, National Institutes of Health. Infrastructure support was also provided, in part, by grant U54 HD090256 from the Eunice Kennedy Shriver NICHD, National Institutes of Health (Waisman Center), UnityPoint Meriter Foundation (Dr. Kling), University of Wisconsin-Madison Department of Neurosurgery (Dr. Alisch), NARSAD Young Investigator Grant from the Brain & Behavioral Research Foundation #22669 & 25212 (Dr. Papale), Ruth L. Kirschstein National Research Service Award MH113351-02 (Mr. Madrid).
Author information
Authors and Affiliations
Contributions
D.C.D. designed and developed the imaging strategy for parent study, imaged children, conceptualized data analysis, analyzed data, drafted sections of current manuscript, revised later versions and approved final version. J.F.M. helped design and develop imaging analyses for the current manuscript, co-wrote and approved final version. A.M. and R.S.A. conceptualized and designed epigenetic analyses, analyzed epigenetic samples, drafted epigenetic sections, revised versions, and approved final versions. E.M.P. enrolled participants, developed an approach to generate and analyze data relating to stress and anxiety assessments, drafted sections of manuscript, co-wrote, and approved final version. K.M.K. designed the sample collection and processing method for the parent study and L.A.P. designed the processing and analysis for the current study; while both analyzed samples, co-wrote sections, and approved the final version. C.L.C., E.K.W., R.M.M. all designed and developed the approach and analysis of the current manuscript, drafted sections and co-wrote the manuscript, and approved final version. H.H.G. and R.J.D. designed and received funding for the parent study, co-wrote the manuscript and approved final manuscript. P.J.K. designed and developed the strategy for the current study, received funding for the epigenetic analyses, supervised the team, drafted sections of current manuscript, revised and approved the final version.
Corresponding author
Ethics declarations
Competing interests
Dr. Richard J. Davidson is the founder, president, and serves on the board of directors for the non-profit organization, Healthy Minds Innovations, Inc. In addition, Dr. Davidson served on the board of directors for the Mind & Life Institute from 1992 to 2017. All other authors declare that they have no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dean, D.C., Madrid, A., Planalp, E.M. et al. Cord blood DNA methylation modifications in infants are associated with white matter microstructure in the context of prenatal maternal depression and anxiety. Sci Rep 11, 12181 (2021). https://doi.org/10.1038/s41598-021-91642-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-91642-0
This article is cited by
-
Epigenome-wide association studies of prenatal maternal mental health and infant epigenetic profiles: a systematic review
Translational Psychiatry (2023)
-
Applications of advanced diffusion MRI in early brain development: a comprehensive review
Brain Structure and Function (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
