
Abstract
Although recent advances in the treatment of acute coronary heart disease have reduced mortality rates, few therapeutic strategies exist to mitigate the progressive loss of cardiac function that manifests as heart failure. Nuclear factor, erythroid 2 like 2 (Nfe2l2, Nrf2) is a transcriptional regulator that is known to confer transient myocardial cytoprotection following acute ischemic insult; however, its sustained activation paradoxically causes a reductive environment characterized by excessive antioxidant activity. We previously identified a subset of 16 microRNAs (miRNA) significantly diminished in Nrf2-ablated (Nrf2−/−) mouse hearts, leading to the hypothesis that increasing levels of Nrf2 activation augments miRNA induction and post-transcriptional dysregulation. Here, we report the identification of distinct miRNA signatures (i.e. “reductomiRs”) associated with Nrf2 overexpression in a cardiac-specific and constitutively active Nrf2 transgenic (caNrf2-Tg) mice expressing low (TgL) and high (TgH) levels. We also found several Nrf2 dose-responsive miRNAs harboring proximal antioxidant response elements (AREs), implicating these “reductomiRs” as putative meditators of Nrf2-dependent post-transcriptional regulation. Analysis of mRNA-sequencing identified a complex network of miRNAs and effector mRNAs encoding known pathological hallmarks of cardiac stress-response. Altogether, these data support Nrf2 as a putative regulator of cardiac miRNA expression and provide novel candidates for future mechanistic investigation to understand the relationship between myocardial reductive stress and cardiac pathophysiology.
Similar content being viewed by others


Introduction
During ischemia–reperfusion injury, generation of reactive oxygen and nitrogen species results in oxidative stress, which in turn, perturbs cardiac structure and function through calcium mishandling, inflammatory signaling and extracellular matrix degradation1,2,3. However, the findings from clinical studies have largely discredited the efficacy of antioxidant therapies4,5. Furthermore, our laboratory has identified the presence of a “reductive stress” wherein aberrant induction of antioxidant response element (ARE)-dependent antioxidant genes produces pathological cardiac hypertrophy and dysfunction6,7,8. While the deleterious consequences of reductive stress appear highly-conserved9,10, the transcriptional and post-transcriptional mechanisms of the myocardial redox milieu remain unknown.
Among the mechanisms known to regulate postnatal heart function, microRNAs (miRNAs) are a class of short (~ 22 nucleotide) RNAs that post-transcriptionally regulate mRNA stability and translational efficiency, often in a tissue-specific manner11. While miRNAs are necessary for physiologic cardiac function and development12,13, many have been found to be dysregulated in the failing heart14,15,16. Specifically, miRNAs have been directly linked to the structural and functional deficits in cardiac function17. Nevertheless, it remains unclear whether ARE-linked miRNAs contribute to cardiac pathogenesis.
As a transcriptional activator of cis-regulatory AREs18, nuclear factor erythroid 2-related factor 2 (Nfe2l2, a.k.a. Nrf2) plays a critical role in regulating cardiac redox status. Transient Nrf2 signaling is cardioprotective immediately following ischemic insult19, but chronic transactivation of AREs causes reductive stress and cardiac dysfunction20,21. We have recently shown that Nrf2 deficiency (Nrf2−/−) inhibits the expression of several miRNAs in the heart22, but the genome-wide impact of reductive stress on miRNA expression remains unknown.
In this investigation, we identify a miRNA signature for reductive stress to gain insight into potential biomarkers and/or effectors of this novel pathological phenomenon in the heart. The cardiomyocyte-specific and constitutively-active Nrf2 transgenic mouse model (CaNrf2-Tg) was used to conduct a multi-omics analysis of Nrf2-dependent and ARE-bearing miRNAs, which we term “reductomiRs”. Our use of both mRNA-seq and small RNA sequencing (miRNA-seq) in caNrf2 low (TgL) and high-expressing (TgH) mouse lines reveals a distinct signature of transgenic Nrf2 dose-responsive miRNAs linked to a number of suppressed cardiac genes under pro-reductive and reductive stress conditions23. Collectively, this analysis uncovers several novel miRNA candidates for which future mechanistic studies will investigate the interplay between post-transcriptional regulatory responses and redox state in the myocardium of Nrf2-Tg mice.
Methods
Animals
Our method for establishing the cardiac-specific constitutively active Nrf2 transgenic mouse model (caNrf2-Tg) has been described previously23. Briefly, cDNA encoding a truncated Nrf2 protein lacking the Neh2 domain was ligated into an α myosin heavy chain (αMHC) expression vector, the plasmid backbone was digested, and the αMHC-caNrf2 insert was used for pronuclear injection. Transgenic low (TgL) and transgenic high (TgH) founders were determined using caNrf2 primer sets in real-time qPCR which compared relative transgene expression to endogenous Nrf2 mRNA, and transgenic mice were back-crossed onto the C57BL/6J background for six generations. For expression analyses, sex-matched male and female TgL, TgH and non-transgenic (NTg) littermate heart tissue was harvested at 6 months of age. All the animal studies including non-invasive cardiac imaging were conducted in accordance with the Guide for Care and Use of Laboratory Animals developed by the National Research Council at the National Institutes of Health (NIH). The Institutional Animal Care and Use Committee (IACUC#14-10160) at the University of Alabama at Birmingham has approved the study, which was carried out in compliance with the Guidelines for Animal Research Reporting of In Vivo Experiments (ARRIVE).
GSH-NEM immunofluorescence assay
To measure the redox status of Tg & NTg mouse hearts, we performed immunofluorescence using anti-GSH staining24. The mouse was anesthetized (2% isoflurane) and the heart was perfused in situ with 10 mM PBS-N-Ethylamaleimide (NEM) before sacrificing. The heart tissue sections (5 µM) were incubated in ethanol containing 10 mM NEM for 30 min and washed three times with PBS prior to blocking. Slides were incubated at 4 °C overnight with the primary antibody mouse anti-Glutathione:N-Ethylamaleimide adduct (1:500 v/v; EMD Millipore Corp., Billerica, MA, US). Following the incubation with the secondary Ab conjugated to Alexa-Flour 488 and washing, imaging was performed using a Fluorescence microscope (Olympus BX43). The intensity of green fluorescence representing glutathione (GSH) was calculated using Image-J (NIH) in 6 images from 3 hearts per group.
Non-invasive cardiac imaging
Two-dimensional trans-thoracic echocardiography was performed in NTg and Tg mice at 6–7 months of age using the Vevo2100 system (Fujifilm Visual Sonics Inc., Ontario, Canada). In brief, animals were prepared for imaging as previously described25. Using a 38-MHz mechanical transducer, strain analysis in parasternal long-axis were acquired under isoflurane (1.5–2.0%) anesthesia. Structural and functional (systolic and diastolic) measurements were obtained using Vevo lab 3.1 software.
RNA and miRNA sequencing analysis
Details of the R coding scripts and other bioinformatics tools used in the current study are available as online Supplemental Methods and GitHub data repository: https://github.com/mepepin/ReductomiRs. High-throughput RNA-and miRNA-sequencing and analysis were performed at the University of Utah. Alignment of reads to the mm10 genome was accomplished using NovoAlign (Singapore, Malaysia), raw counts generated using Samtools26, and differential gene expression performed using DESeq227 (1.18.1) within the R (3.4.2) statistical computing environment as described previously28. Due to limited sample sizes (n = 3–4), dispersion estimates were first determined via maximum-likelihood, assuming that genes of similar average expression strength possess similar dispersion, as previously described27. Gene-wise dispersion estimates were then shrunken according to the empirical Bayes approach, providing normalized count data for genes proportional to both the dispersion and sample size. Differential expression was determined from normalized read counts via Log2 (fold-change) using the Wald test followed by Bonferroni-adjusted P-value (i.e. Q-value) for each aligned and annotated gene. Statistical significance was assessed with unpaired two-tailed Bonferroni-adjusted P-value (Q-value) < 0.05 used to identify differentially-expressed candidate genes, with a lower-stringency statistical threshold used for pathway enrichment (P < 0.01). Differentially expressed genes (DEGs) were generated with biological significance assumed when |Log2FoldChange|> 1 with normalized count sum > 1.
RNA isolation and real-time quantitative polymerase chain reaction
Total RNA was extracted from 10 to 25 mg of the heart’s apex using QIAzol lysis reagent (Qiagen, Cat. 79306) and the miRNeasy mini kit (Qiagen, Cat. 217004). After confirming sample purity through the NanoDrop One Spectrophotometer (ThermoFisher Scientific), the miScript II RT Kit including HiSpec Buffer (Qiagen, Cat. 218161) was used to reverse transcribe 1–2 μg RNA. A 10 μl reaction containing 20 ng cDNA template and miScript primer assays (Qiagen, Cat. miRs-361-3p:MS00025718, 491-5p:MS00002541, 34b-3p:MS00011900, 1983:MS00016884, 671-5p:MS00064385, 671-3p:MS00012467 5121:MS00043071, 215-5p:MS00001918, 200c-3p:MS00001827, 155-5p:MS00001701) and SYBR Green® (Qiagen Cat. 218075) was amplified according to the manufacturer’s instructions in a Roche LightCycler 480 (Roche Life Science). Sequences used to quantify relative transgene abundance were: forward primer (mCaNrf2-MHC-F), ACTTTACATGGAGTCCTGGTGGGA; reverse primer (mCaNrf2-MHC-R), AGGCATCTTGTTTGGGAATGTGGG. Endogenous Nfe2l2/Nrf2 transcript levels were measured using forward primer (mNrf2-F), CTGAACTCCTGGACGGGACTA; reverse primer (mNrf2-R), CGGTGGGTCTCCGTAAATGG. For each qPCR assay, equal amounts of cDNA were loaded for all control and experimental samples. Relative expression was determined according to the 2−ΔΔCt method using small nucleolar RNAs Snord42b or Snord72 as housekeeping genes.
Data visualization
Functional and network gene set enrichment analysis (GSEA), along with curated literature-supported candidate upstream regulators, were performed using Enrichr, an interactive web-based tool for compiling multiple bioinformatics databases29. Within this software, Wikipathways30 and KEGG31 enrichment as well as ChIP Enrichment Analysis (ChEA) were done both on the individual datasets and as a combined comparative analyses to determine overlapping enriched pathways and associated transcriptional regulators, respectively. Heatmap generation and hierarchical clustering were performed using pheatmap package (1.0.8) within R, and VennPlex32 was used to create the Venn diagrams and determine overlapping gene lists.
Statistics
All data are represented as mean ± standard error of the mean (SEM) unless otherwise indicated. Statistical significance was determined using unpaired Student’s t-tests or, where appropriate, one-way (ANOVA) with Tukey post-test for multiple comparisons. Statistical analyses and data visualization were completed using GraphPad Prism (GraphPad Software, San Diego, CA) and R software, version 3.4.2 (R Foundation for Statistical Computing, Vienna, Austria). Statistical significance was assigned at the P < 0.05 level for two-way comparisons.
Consent for publication
All authors verified the content and approved the final version for submission and publication.
Results
Evidence of reductive stress and pathologic cardiac remodeling in CaNrf2-transgenic mice
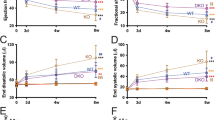
The concept myocardial reductive stress was first described in a mouse model of human cardiac disease8. However, the underlying molecular mechanisms remained unknown until the recent development of unique mouse models to study this phenomenon25,33. We recently described that constitutive activation of the transcription factor, Nrf2/NFE2L2, resulting in a dose-dependent increase of glutathione and other antioxidants, establishes a reductive and hyper-reductive redox condition in the heart33. Furthermore, a chronic hyper-reductive condition (i.e. “reductive stress”) was shown to cause pathological cardiac remodeling and diastolic dysfunction25. Here, we sought to identify a redox-responsive miRNA signature associated with reductive- and hyper-reductive-conditions in caNrf2 transgenic low (TgL) and high (TgL) hearts. Quantitative genotyping in TgL and TgH mice indicated dose-dependent transgene expression (respective Ct values) when compared to NTg, which did not amplify a PCR product for the primers that recognize only the truncated transgene (caNrf2) (Fig. 1A). Interestingly, qPCR using a primer that recognizes endogenous mouse Nrf2, but not caNrf2, also showed a dose-wise expression pattern consistent with reports of Nrf2 transcriptional auto-up-regulation (Fig. 1B)34. As Nrf2 is well-characterized to promote glutathione biosynthesis35, we performed immunofluorescent staining of glutathione-N-ethylmaleimide (GSH-NEM) adducts in TgL and TgH to confirm the biochemical effects of dose-dependent transgene expression (Fig. 1C). These results confirm our previous studies employing kinetic-based spectrophotometric measurements for glutathione in CaNrf2-Tg hearts33. To determine the consequences of Nrf2 expression on cardiac function, echocardiographic assessments (strain analysis) were performed (n = 10/group), which provided clear evidence of diastolic dysfunction in TgL and TgH relative to NTg mice (Fig. 1D). Taken together, these results suggest that Nrf2 expression leads to both reductive conditions and pathological cardiac remodeling.

Differential expression of transgene, myocardial glutathione, structural and functional remodeling in caNrf2 mice. Real-time qPCR-based quantitation of transgene (CaNrf2) expression in transgenic low (TgL) high (TgH) and non-transgenic (NTg) hearts using (A) genotyping primers (recognizes caNrf2) and (B) endogenous Nrf2 primers vs Gapdh (n = 4/group). (C) Representative immunofluorescent (IF) images and quantification of N-Ethylmaleimide-Glutathione adducts (green) and DAPI (blue) in heart sections of NTg, TgL and TgH mice (n = 3/group). (D) Two-dimensional trans-thoracic echocardiography (para-sternal long axis strain analysis) was used to estimate ejection fraction (EF%), mitral valve (early, E to late, A ratio) filling, end diastolic volume (EDV) and systolic volume (ESV) (n = 10/group).
Dose dependent effects of Nrf2 on cardiac miRNA expression
To determine whether transactivated Nrf2 expression is associated with differences in global cardiac microRNA expression, we used an unsupervised approach to cluster samples via principal components analysis (PCA) (Fig. 2A). Three distinct clusters emerged from the first 3 eigenvectors (71.7% of cumulative variance), which corresponded exactly to samples from Ntg, TgL, and TgH mice.

miRNA Sequencing Analysis of caNrf2 Transgene and Dose Effect. (A) Unsupervised three-dimensional principal components analysis of rlog-normalized counts from miRNA-sequencing. (B) Venn Diagram illustrating the overlap of differentially-expressed miRNAs (DEmiRs) following pooled transgene-and dose–effect analyses (P < 0.05) (n = 3/group). Hierarchial clustering of variance-stabilized counts according to regions illustrated by the venn diagram, with (C) 165 “Transgene-only” DEmiRs (Tables S3 and S4), (D) 22 “Transgene-Dose” overlapping DEmiRs (Tables S5 and S6), and (E) 40 “Dose-only” DEmiRs (Tables S7 and S8). Highlighted in bold are qPCR-validated DEmiRs. (F) Real-time quantitative PCR validation of up-regulated and (F) down-regulated DEmiRs from among “transgene-only” (↑miR-361-3p, ↑miR-491-5p, ↓miR-5121 and ↓miR-215-5p), “transgene-dose” (↑miR-34b-3p, ↓miR-200c-3p and ↓miR-155-5p) and “dose-only” (↑miR-1983) DEmiRs. Benjamini-hochberg (BH)-adjusted t-test was performed with ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05 (n = 6–8/group) reported as mean ± SEM.
To identify dose-responsive miRNA targets of caNrf2, we performed both a pooled analysis of caNrf2-Tg vs. NTg (Supplemental Table S1) and a dose-selective analysis of TgH vs. TgL differential miRNA expression (Supplemental Table S2). Comparing the pooled caNrf2-Tg vs. NTg with TgH vs. TgL analyses via Venn diagram (Fig. 2B, Supplemental Tables S3–S8) revealed three distinct differentially-expressed miRNA (DEmiR) subsets which were designated as follows: (1) “transgene-only” effect (P < 0.05 caNrf2-Tg vs. NTg; left), (2) “transgene-dose” effect (P < 0.05 caNrf2-Tg vs. NTg and TgL vs. TgL; middle), and (3) “dose-only” effect (P < 0.05 TgH vs. TgL; right). As anticipated, “transgene-only” DEmiRs displayed equivalent changes across TgL and TgH (Fig. 2C), “dose-only” DEmiRs displayed markedly distinct patterns of expression between TgH and TgL (Fig. 2D,E). The “transgene-dose” DEmiRs reported as significant in each independent analysis are expected to represent direct and dose-dependent targets of caNrf2, and thus putative effectors of myocardial reductive stress. Several miRNAs within each DEmiR subset were validated via qPCR, supporting their dysregulation in transgenic mouse hearts (Fig. 2F,G).
Nrf2-dependent cardiac mRNA expression
In a recent study, we analyzed mRNA-sequencing data from caNrf2 Tg mice to identify differences associated with the physiologic state of myocardial reductive stress36. However, this report lacked a comparative analysis of pooled vs. dose-responsive DEGs and the mRNA co-regulatory relationships with miRNAs. Thus, we analyzed differential expression for caNrf2-Tg vs. NTg (Supplemental Table S9) and TgL vs. TgH (Supplemental Table S10) to relate mRNA dysregulation with miRNA expression patterns. Principal Component Analysis (PCA) (Fig. 3A) and correlation heatmap (Fig. 3B) of DEGs revealed a striking pattern of mRNA expression of TgH compared to TgL mice. Consistent with our functional characterization (Fig. 1) and those of our earlier work37, GSEA of “dose-only” mRNA revealed pathways enriched for cardiac pathophysiology while “transgene-only” DEGs implicated noncanonical -possibly indirect- effects of transgenic Nrf2 expression including “longevity” and “FoxO signaling” (Fig. 3C). Notably, upregulated “transgene-dose” effect DEGs reflected quintessential functions of Nrf2 signaling such as “Glutathione” and “Xenobiotic Metabolism,” thereby suggesting that this subset of genes represent putative downstream targets of caNrf2 (Fig. 3C). Owing to its role as a positive transcriptional regulator, we were surprised to find that CaNrf2 decreased the expression of 206 “transgene-dose” DEGs (Fig. 3C). Because miRNAs silence gene expression at the post-transcriptional level, we hypothesized that this distinct DEG subset could represent miRNA targets induced downstream of Nrf2 transactivation. These DEGs thus became the focus of our study as putative targets of negative regulation via Nrf2-responsive miRNA, or cardiac “reductomiRs”.

Identification of Direct and Indirect mRNA Targets. (A) Unsupervised two-dimensional principal component analysis in R (3.4.2) using normalized counts from non-transgenic (NTg, n = 4), caNrf2 transgenic low (TgL, n = 3) and high (TgH, n = 3) mice. (B) Unsupervised correlation heatmap according to pearson’s ρ. (C) Venn Diagram illustrating the overlap of differentially-expressed genes (DEGs) following pooled “transgene-only”, “transgene-dose” “dose-only” mRNAs (P < 0.05). The underlying bar graph displays FDR-corrected p-values (left y-axis) and gene enrichment ratio (right y-axis) of the top 3 most-enriched KEGG pathways (ranked by p-value) across the venn diagram regions. Enrichment ratio reflects the percentage of a pathway’s genes significantly altered in the current dataset.
In-silico antioxidant response element (ARE)-based microRNA target prediction
As a transcriptional regulator, Nrf2 promotes the expression of genes bearing proximal AREs18. While GSEA analysis of transgene-dose mRNAs implicated upregulated DEGs as direct targets of Nrf2, we hypothesized that the suppression of 206 DEGs in this subset correlates with the relative induction of intermediary miRNAs, as illustrated in Fig. 4A. To identify DEmiRs containing upstream AREs (“ReductomiRs”), we used the position-weighted matrix (PWM) developed by Malhotra et al.38 within PWMscan39 to identify genome-wide (Mus musculus, mm10) Nrf2 consensus elements. The resulting genomic coordinates were then used to locate a total of 142 AREs within the promoter most-proximal to the coding sequence (+ 5 kB) of 40 upregulated Nrf2-dependent DEmiRs (Fig. 4B). From this, approximately half (22/40) of the DEmiRs were found to exhibit a transgene dose-responsive pattern of expression in caNrf2 TgL and TgH hearts. To determine whether ARE bearing dose-responsive miRNAs accounted for downregulated mRNAs within the transgene-dose mRNA subset, we used multiMiR40 as a computational tool to systematically compile predicted mRNA targets from the following 8 algorithms: DIANA-microT, ElMMo, MicroCosm, miRanda, miRDB, PicTar, PITA, and TargetScan. Targets were then compiled using cross-algorithm validation to predict miRNA–mRNA interactions41. From this analysis, 19 miRNAs exhibited sequence complementarity to seed sequences within 61 down-regulated DEGs. Gene-set enrichment of these DEGs using KEGG pathways disproportionately represented “hypoxia-inducible factor-1α signaling” and “cardiac muscle contraction” (Fig. 4C). Enrichment analysis using the ARCHS442 gene expression atlas revealed a cardiac-specific signature among these DEGs (Fig. 4D). Taken together, these preliminary findings implicate “reductomiRs” as possible Nrf2-responsive mediators of myocardial reductive stress.

Elucidation of Antioxidant Response Element (ARE)-containing DEmiRs and their Putative mRNA Targets. (A) Graphical model representing Nrf2-dependent regulation of miRNA and downstream mRNA. (B) The 40 induced DEmiRs* (Tg Vs. NTg) containing at least one proximal (+ 5kB) upstream ARE. (C) Gene set enrichment (GSEA) of KEGG pathways and (D) ARCHS4 gene atlas-based enrichment using DEGs identified as putative targets of ARE-containing DEmiRs. *P < 0.05.
Integrative Nrf2-responsive miRNA–mRNA functional network
It became apparent from our in silico analysis that multiple miRNAs shared one or more putative downstream mRNA targets. To understand these interactions further, we generated a bipartite network of the 19 “dose-responsive” reductomiRs (P < 0.01) found to possess a downstream seed sequence among the suppressed mRNAs (Fig. 5A). The resulting network of interactions suggested that Nrf2-mediated miRNAs display a varied capacity to influence mRNA transcript abundance in caNrf2-Tg hearts. Whereas miR-301a-5p and miR-31-3p were predicted to target relatively few mRNAs, the seed sequences of miR-449a-5p, miR-96-5p, miR-298-5p, miR-491-5p, and miR-671-5p matched a larger number of cardiac-specific mRNAs within this network.

Cardiac miRNA:mRNA Reductive Stress Regulatory Network. (A) Bipartite network generated in R (3.4.2) illustrating the complex predicted interaction between ARE-containing induced reductomiRs and suppressed “transgene-dose” effect DEGs, with box-width defined by the relative number of miRNA-mRNA interactions. Line thickness indicates frequency of detection across miRNA target prediction algorithms. (B) Ingenuity Pathway Analysis (IPA)-derived signaling network diagram of repressed “transgene-dose” effect DEGs identified as putative downstream targets of ARE-containing DEmiRs. Cross-validated predictions (red dotted lines) using multiMiR (1.12.0) represent miRNA-mRNA interactions predicted by at least 3 independent algorithms.
Once putative miRNA–mRNA interactions were developed, the curated database from Ingenuity Pathway Analysis (IPA) was used to incorporate known miRNA–mRNA interactions into a caNrf2-dependent reductomiR–mRNA network, which we then compiled to identify nodal DEGs (Fig. 5B). This approach identified several key mRNAs, which together comprise established mediators of pathological cardiac remodeling: sarcomeric genes myomesin (Myom2), myosin light chain kinase 4 (Mylk4), voltage gated calcium and chloride channels subunits (Cacnad2d2, Clcn1), the striated muscle isoform subunit VIa of cytochrome C oxidase (Cox6a2), and adrenergic alpha1a receptor (Adra1a). Furthermore, reductomiR targets of caNrf2 were predicted to mediate the negative regulation of SET and MYND domain containing 1 (Smyd1) and myocyte enhancer factor 2A (Mef2a), two transcriptional regulators essential for cardiac development (Fig. 5B)43,44,45. Although future work should verify direct post-transcriptional regulatory interaction and resultant changes in proteins encoded by these mRNAs, these data implicate miRNAs as putative mediators of Nrf2 signaling and, ultimately, myocardial reductive stress effectors.
Bidirectional association of cardiac Nrf2 and miR-671
Our data suggest that constitutive activation of cardiac Nrf2 dose-dependently promotes expression of several ARE-bearing DEmiRs capable of silencing cardiac-specific transcripts. While this result highlights the sufficiency of Nrf2 for cardiac miRNA induction, it does not indicate which loci require Nrf2-mediated transactivation. Therefore, we analyzed the expression of the 40 reductomiRs in hearts from age-matched Nrf2−/− mice (Table S11) (Fig. 6A)22. Although 17 miRNAs were inversely trending, only three were inversely co-regulated: miR-378a-3p, miR-455-5p, and miR-671-3p (Fig. 6B). Of these, miR-671-3p was the most robustly altered in both animal models. While only miR-671-3p was reported as significant in Nrf2−/− miRNA sequencing22, both the 5′ (miR-671-5p) and 3′ (miR-671-3p) mature miRNA products of the precursor miR-671 hairpin (pre-miR-671) were significantly increased in caNrf2-Tg analysis (Fig. 6C). Taken together, these results support that Nrf2 expression is both necessary and sufficient for the cardiac expression of miR-671.

Nrf2 is both necessary and sufficient to induce reductomiR expression. (A) Quantitative realtime PCR (qPCR)-based candidate validation of 3 miRNAs found to reach significance (n = 3, P < 0.05) with inverse expression between Nrf2 knockout (Nrf2−/−, Table S11) and CaNrf2-Tg (Table S1) mouse models: miR-378a-3p, miR-455-5p, and miR-671-3p. (B) qPCR validation of caNrf2-Tg miRNA sequencing analysis. Benjamini-hochberg (BH)-adjusted t-test was performed with ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05 reported as mean ± SEM.
Discussion
Since their initial discovery in 199346, two and a half decades of research have shown miRNAs to be central determinants of human biology and disease. As such, miRNA therapies are continuously being patented, and a few have entered clinical trials47. The heart’s susceptibility to miRNA dysregulation is well-known, with aberrant miRNA induction found to directly cause pathological cardiac remodeling48,49. Nevertheless, endogenous miRNA activity is essential for cardiac morphogenesis12,13 and postnatal functions50,51. While efforts to identify candidate miRNAs in human patients14,15,16 have led to the identification of key miRNAs governing calcium handling, extracellular matrix remodeling, and vascularization52, their role in the regulation of myocardial reductive potential has remained elusive. In this context, our recent discovery of suppressed miRNAs in Nrf2 knockout (Nrf2−/−) hearts supports the notion that Nrf2-dependent miRNA expression offers a mechanistic link between redox disequilibrium and cardiac pathophysiology22.
In the absence of oxidative conditions, Nrf2 activation is generally inhibited through its interaction with the cytosolic repressor protein Keap153,54,55. Thus, despite its cardioprotective roles in response to pathological oxidative stress stimuli, such as pressure overload56 and ischemia–reperfusion injury19, the adaptive phase of Nrf2 signaling is generally transient in an unstressed adult myocardium. Our laboratory has shown that this basal restriction of Nrf2 is required, since its sustained activation leads to myocardial reductive stress characterized by accumulation of reducing equivalents and attenuation of physiological oxidant signaling8,20,21,37. In addition to our observations in mouse models, others have reported that human germline gain-of-function mutations in Nrf2 produce early cardiac pathologies57. Sariam and colleagues have reported that a subset of human heart failure patients exhibit a hyper-reductive redox state9. This suggests that divergent patient populations may contribute to the insufficiency and/or adverse effects of exogenous antioxidant supplementation in cardiac patients4,5.
In the present investigation, we observed that constitutive Nrf2 activation led to profound and dose-responsive miRNA changes in the mouse heart. We specifically found multiple patterns of Nrf2-dependent miRNA expression, with 165 “transgene-only” miRNAs, 22 “transgene-dose” miRNAs, and 40 “dose-only” miRNAs were differentially expressed in caNrf2-Tg mice. Our in silico search for ARE-containing caNrf2-dependent miRNAs, or “reductomiRs”, identified 40 candidate miRNAs likely to be trans-activated by cardiac Nrf2, 22 of which also exhibited dose-responsive induction in TgH and TgL. Lastly, our mRNA target prediction identified 61 putative downstream transcriptional targets for 19 (86%) reductomiRs. While a causal role of these Nrf2-mediated reductomiRs remains untested, several miRNAs have already been implicated in the failing human myocardium (e.g. miR-326-3p and miR-214-5p) and plasma (e.g. miR-671-5p)58,59. Therefore, this “reductomiR” signature may provide important future insights into redox disequilibrium during cardiac pathophysiology.
Owing to the bioinformatic tools used in this study, mechanistic studies are still needed to confirm our analyses. Since RNA was isolated from heart homogenates, we cannot exclude the possibility that miRNA could originate from non-myocyte cells. Furthermore, future work should differentiate between indirect and effector reductomiRs in myocardial reductive stress as the current investigation does not provide causal evidence that ARE-harboring miRNAs contribute to cardiac pathology or altered redox state.
Nevertheless, our data strongly implicate Nrf2 as regulator of the myocardial transcriptome, from which direct and indirect regulation of numerous mRNAs and miRNAs are likely to occur. While Nrf2-independent changes in the caNrf2-Tg model remain unexplained, we demonstrate a strong link between Nrf2-mediated miRNA activity and post-transcriptional regulation of genes involved in pathological cardiac remodeling. The current study therefore identifies a novel network of Nrf2-responsive miRNAs that amy represent mediators of myocardial reductive stress.
Data availability
All data generated for this study are provided as open-access on NCBI Gene Expression Omnibus (GEO) (miRNA: GSE120087; mRNA: GSE120088), with all coding scripts and quality control available from the following GitHub repository: https://github.com/mepepin/ReductomiRs.
Abbreviations
- Nrf2 :
-
Nuclear factor, erythroid 2 like 2
- CaNrf2-Tg:
-
Constitutively active Nrf2 transgenic
- TgL:
-
Low-expressing caNrf2 mouse line
- TgH:
-
High-expressing caNrf2 mouse line
- ARE:
-
Antioxidant response element
- DEG:
-
Differentially-expressed genes
- DEmiR:
-
Differentially-expressed microRNA
- GSEA:
-
Gene set enrichment analysis
References
Tsutsui, H., Kinugawa, S. & Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol.-Heart Circ. Physiol. 301, H2181–H2190 (2011).
van der Pol, A., van Gilst, W. H., Voors, A. A. & van der Meer, P. Treating oxidative stress in heart failure: past, present and future. Eur. J. Heart Fail. 21, 425–435 (2019).
Seddon, M., Looi, Y. H. & Shah, A. M. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 93, 903–907 (2007).
Jenkins, D. J. et al. Supplemental vitamins and minerals for CVD prevention and treatment. J. Am. Coll. Cardiol. 71, 2570–2584 (2018).
Steinhubl, S. R. Why have antioxidants failed in clinical trials?. Am. J. Cardiol. 101, S14–S19 (2008).
Brewer, A. C., Mustafi, S. B., Murray, T. V., Rajasekaran, N. S. & Benjamin, I. J. Reductive stress linked to small HSPs, G6PD, and Nrf2 pathways in heart disease. Antioxid. Redox Signal. 18, 1114–1127 (2013).
Narasimhan, M. & Rajasekaran, N. S. Reductive potential—A savior turns stressor in protein aggregation cardiomyopathy. Biochimica et Biophysica Acta (BBA)-Mol. Basis Disease 1852, 53–60 (2015).
Rajasekaran, N. S. et al. Human αB-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 130, 427–439. https://doi.org/10.1016/j.cell.2007.06.044 (2007).
Sairam, T. et al. Evidence for hyper-reductive redox in a subset of heart failure patients. J. Transl. Med. 16, 130. https://doi.org/10.1186/s12967-018-1503-x (2018).
Touyz, R. M., Anagnostopoulou, A., Camargo, L. D. L. & Montezano, A. C. Novel biosensors reveal a shift in the redox paradigm from oxidative to reductive stress in heart disease. Circ. Res. 119, 969–971 (2016).
Bartel, D. P. Metazoan microRNAs. Cell 173, 20–51 (2018).
Chen, J.-F. et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. 105, 2111–2116 (2008).
Zhao, Y. et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 129, 303–317 (2007).
Thum, T. et al. MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation 116, 258–267 (2007).
Sucharov, C., Bristow, M. R. & Port, J. D. miRNA expression in the failing human heart: functional correlates. J. Mol. Cell. Cardiol. 45, 185–192 (2008).
Yang, K.-C. et al. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNA in failing human heart and remodeling with mechanical circulatory support. Circulation 129(9), 1009–1021 (2014).
Kumarswamy, R. & Thum, T. Non-coding RNAs in cardiac remodeling and heart failure. Circ. Res. 113, 676–689 (2013).
Kensler, T. W., Wakabayashi, N. & Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47, 89–116. https://doi.org/10.1146/annurev.pharmtox.46.120604.141046 (2007).
Calvert, J. W. et al. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 105, 365–374 (2009).
Kannan, S. et al. Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy. Cardiovasc. Res. https://doi.org/10.1093/cvr/cvt150 (2013).
Rajasekaran, N. S. et al. Sustained activation of nuclear erythroid 2-related factor 2/antioxidant response element signaling promotes reductive stress in the human mutant protein aggregation cardiomyopathy in mice. Antioxid. Redox Signal. 14, 957–971. https://doi.org/10.1089/ars.2010.3587 (2011).
Quiles, J. M. et al. Differential regulation of miRNA and mRNA expression in the myocardium of Nrf2 knockout mice. BMC Genom. 18, 509. https://doi.org/10.1186/s12864-017-3875-3 (2017).
Shanmugam, G., Narasimhan, M., Tamowski, S., Darley-Usmar, V. & Rajasekaran, N. S. Constitutive activation of Nrf2 induces a stable reductive state in the mouse myocardium. Redox Biol. 12, 937–945. https://doi.org/10.1016/j.redox.2017.04.038 (2017).
Miller, V., Lawrence, D., Mondal, T. & Seegal, R. Reduced glutathione is highly expressed in white matter and neurons in the unperturbed mouse brain—Implications for oxidative stress associated with neurodegeneration. Brain Res. 1276, 22–30 (2009).
Shanmugam, G. et al. Reductive stress causes pathological cardiac remodeling and diastolic dysfunction. Antioxid. Redox Signal. 32(18), 1293–1312 (2020).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Pepin, M. E. et al. Antiretroviral therapy potentiates high-fat diet induced obesity and glucose intolerance. Mol. Metab. 12, 48–61 (2018).
Chen, E. Y. et al. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 14, 128 (2013).
Kelder, T. et al. WikiPathways: Building research communities on biological pathways. Nucleic Acids Res. 40, D1301-1307. https://doi.org/10.1093/nar/gkr1074 (2012).
Kanehisa, M. The KEGG database. Novartis Found Symp. 247, 91–101 (2002) (discussion 101–103, 119–128, 244–152).
Cai, H. et al. VennPlex—A novel Venn diagram program for comparing and visualizing datasets with differentially regulated datapoints. PloS One 8(1), e53388 (2013).
Shanmugam, G., Narasimhan, M., Tamowski, S., Darley-Usmar, V. & Rajasekaran, N. S. Constitutive activation of Nrf2 induces a stable reductive state in the mouse myocardium. Redox Biol. 12, 937–945 (2017).
Kwak, M.-K., Itoh, K., Yamamoto, M. & Kensler, T. W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 22, 2883–2892 (2002).
Shih, A. Y. et al. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 23, 3394–3406 (2003).
Quiles, J. M. et al. Identification of transcriptome signature for myocardial reductive stress. Redox Biol. 13, 568–580. https://doi.org/10.1016/j.redox.2017.07.013 (2017).
Shanmugam, G. et al. Reductive stress causes pathological cardiac remodeling and diastolic dysfunction. Antioxid. Redox Signal 32, 1293–1312. https://doi.org/10.1089/ars.2019.7808 (2020).
Malhotra, D. et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718–5734 (2010).
Ambrosini, G., Groux, R. & Bucher, P. PWMScan: A fast tool for scanning entire genomes with a position-specific weight matrix. Bioinformatics 1, 2 (2018).
Ru, Y. et al. The multiMiR R package and database: Integration of microRNA–target interactions along with their disease and drug associations. Nucleic Acids Res. 42, e133–e133 (2014).
Peterson, S. M. et al. Common features of microRNA target prediction tools. Front. Genet. 5, 23 (2014).
Lachmann, A. et al. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun. 9, 1366 (2018).
Naya, F. J. et al. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat. Med. 8, 1303 (2002).
Gottlieb, P. D. et al. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat. Genet. 31, 25 (2002).
Rasmussen, T. L. et al. Smyd1 facilitates heart development by antagonizing oxidative and ER stress responses. PLoS ONE 10, e0121765 (2015).
Lee, R. C., Feinbaum, R. L. & Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854 (1993).
Chakraborty, C., Sharma, A. R., Sharma, G., Doss, C. G. P. & Lee, S.-S. Therapeutic miRNA and siRNA: Moving from bench to clinic as next generation medicine. Mol. Therapy-Nucleic Acids 8, 132–143 (2017).
Cheng, Y. et al. MicroRNAs are aberrantly expressed in hypertrophic heart: Do they play a role in cardiac hypertrophy?. Am. J. Pathol. 170, 1831–1840 (2007).
Van Rooij, E. et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. 103, 18255–18260 (2006).
Rao, P. K. et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ. Res. 105, 585–594 (2009).
da Costa Martins, P. A. et al. Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation 118, 1567–1576 (2008).
Thum, T. & Condorelli, G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ. Res. 116, 751–762 (2015).
Itoh, K. et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86. https://doi.org/10.1101/gad.13.1.76 (1999).
McMahon, M., Itoh, K., Yamamoto, M. & Hayes, J. D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 278, 21592–21600 (2003).
Zhang, D. D. & Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 23, 8137–8151. https://doi.org/10.1128/MCB.23.22.8137-8151.2003 (2003).
Li, J. et al. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arterioscler. Thromb. Vasc. Biol. 29, 1843–1850 (2009).
Huppke, P. et al. Activating de novo mutations in NFE2L2 encoding NRF2 cause a multisystem disorder. Nat. Commun. 8, 818. https://doi.org/10.1038/s41467-017-00932-7 (2017).
Akat, K. M. et al. Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc. Natl. Acad. Sci. 111, 11151–11156 (2014).
Wong, L. L. et al. Circulating microRNAs in heart failure with reduced and preserved left ventricular ejection fraction. Eur. J. Heart Fail. 17, 393–404 (2015).
Acknowledgements
Authors thank Jennifer Hong, Nancy Atieno and Dr. Sellamuthu S. Gounder for their assistance with animal breeding and maintenance.
Funding
This study was supported by funding from NHLBI (2HL118067 and HL118067), NIA (AG042860), the AHA (BGIA 0865015F), the University of Utah Center for Aging Pilot Grant (2009), the Division of Cardiovascular Medicine/Department of Medicine, University of Utah, and the start-up funds (for N.S.R.) by the Department of Pathology and School of Medicine, the University of Alabama at Birmingham, AL, and UABAMC21 Grant by the University of Alabama at Birmingham, AL. Funding support for bioinformatics software and analysis was provided by A.R.W. using NIH R01 HL133011. Training support was provided to J.M.Q. by an NIH T32 (T32HL007918), and to M.E.P. by an NIH F30 (HL137240) and Postdoctoral fellowship through the Alexander von Humboldt Foundation.
Author information
Authors and Affiliations
Contributions
N.S.R. and J.M.Q. conceived the idea and designed the experiments. J.M.Q., S.S., S.S.B., A.K.C., and N.S.R. conducted the experiments. J.M.Q., M.E.P., S.S., and N.S.R. interpreted the data, created figures, and drafted the manuscript. B.D. has performed the NGS runs and prepared the raw data sets. M.E.P. developed the bioinformatics analysis. S.M.P. interpreted the echocardiography results. A.R.W. and N.S.R. oversaw the computational design and methods, interpretation, and manuscript preparation. A.K.C. and J.R.H. were involved in interpreting the data and critical discussions. All authors read and approved the final version of this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Quiles, J.M., Pepin, M.E., Sunny, S. et al. Identification of Nrf2-responsive microRNA networks as putative mediators of myocardial reductive stress. Sci Rep 11, 11977 (2021). https://doi.org/10.1038/s41598-021-90583-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90583-y
This article is cited by
-
The interplay between microRNAs and Nrf2 signaling in human cancers
Cancer Cell International (2024)
-
Pulmonary artery embolism: comprehensive transcriptomic analysis in understanding the pathogenic mechanisms of the disease
BMC Genomics (2023)
-
Systematic characterization of seed overlap microRNA cotargeting associated with lupus pathogenesis
BMC Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
