
Abstract
Ocean sediments are the largest sink for mercury (Hg) sequestration and hence an important part of the global Hg cycle1. Yet accepted global average Hg flux data for deep-ocean sediments (> 200 m depth) are not based on measurements on sediments but are inferred from sinking particulates2. Mercury fluxes have never been reported from the deepest zone, the hadal (> 6 km depth). Here we report the first measurements of Hg fluxes from two hadal trenches (Atacama and Kermadec) and adjacent abyssal areas (2–6 km). Mercury concentrations of up to 400 ng g−1 were the highest recorded in marine sediments remote from anthropogenic or hydrothermal sources. The two trench systems differed significantly in Hg concentrations and fluxes, but hadal and abyssal areas within each system did not. The relatively low recent mean flux at Kermadec was 6–15 times higher than the inferred deep-ocean average1,3, while the median flux across all cores was 22–56 times higher. Thus, some hadal and abyssal sediments are Hg accumulation hot-spots. The hadal zone comprises only ~ 1% of the deep-ocean area, yet a preliminary estimate based on sediment Hg and particulate organic carbon (POC) fluxes suggests total hadal Hg accumulation may be 12–30% of the estimate for the entire deep-ocean. The few abyssal data show equally high Hg fluxes near trench systems. These results highlight a need for further research into deep-ocean Hg fluxes to better constrain global Hg models.
Similar content being viewed by others

Introduction
The Minamata Convention on Mercury to reduce human emissions of this priority toxic metal was ratified and entered into force in 2017 (www.mercuryconvention.org)1 1,2,3. Attention is now turning to how rapidly Hg may decrease in the biosphere4,5, ultimately via storage in one of the planetary sinks—soils and ocean sediments6,7.
Oceans are crucial in understanding the global cycling and fate of anthropogenic Hg because they contain more Hg than the upper organic layers of soils and the atmosphere combined, and marine sediments are probably the largest long-term repository1,8. The rate of Hg accumulation on continental shelves (excluding estuaries) is constrained by numerous field studies and modeling3,9, and is estimated at 120–200 t Hg a−1 1,3. However, knowledge of Hg accumulation in deep-ocean sediments (91% of ocean area10) is limited. There are no measured Hg flux data from the deepest, hadal areas of the oceans. Consequently, global conceptual models and budgets used water column particulate Hg:POC data, scaled to global marine OC burial rates2, to infer deep-ocean Hg accumulation of 240–600 t a−1 1,3, equivalent to an average flux of 0.7–1.8 µg.m−2 a−1 over an area of 330 × 106 km2 10. Even measurements of Hg concentrations in deep-ocean sediments are sparse compared to coastal sediments (see Refs.11,12,13,14,15,16,17 for Hg concentration data on bathyal and abyssal sites; and Refs.16,18,19 for hadal sites).
The current calculations of deep-ocean Hg accumulation assumed a constant water column Hg:POC of 0.6 µg Hg g POC−1 2,20. However, measurements at many Pacific Ocean sites indicate that Hg:POC increases several-fold down to at least 500 m depth because sinking particles preferentially retain Hg relative to carbon during remineralization21. Also, measurements of the particulate:dissolved Hg partitioning co-efficient in the pelagic ocean show > 20 × higher values than models assume, possibly because Hg is “regeneratively scavenged” by decaying particulate organic matter22 (POM) similar to metals such as zinc23. Thus, Hg may penetrate deeper into the ocean at higher rates than models suggest. Given these discrepancies, and because deep-ocean sediments may be the largest oceanic Hg sink, there is a critical need to verify the inferred accumulation estimates with data based on sediment sampling.
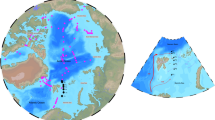
Here we report Hg concentration and flux data from 12 sediment cores from two hadal subduction trenches, and adjacent abyssal plains and slope, in the Pacific Ocean (Atacama Trench, N = 9 sites, off Chile; Kermadec Trench, N = 3, near New Zealand; Fig. 1 and Table S1). There are 47 subduction trenches, troughs and trench faults in the world’s hadal zone, concentrated mainly in the Pacific24. Together, the Atacama and Kermadec trench systems represent 2.2% of the total hadal area of about 3.44 × 106 km2 10,24. Because they are the deepest parts of the ocean basin, trenches are natural accumulators of the organic and inorganic matter that was deposited at the ocean’s surface or produced in surface waters25. Atacama underlies one of the most productive oceanic regions and has the highest estimated POC flux of any trench (3.2 ± 1.4 g OC m−2 a−1), whereas Kermadec has a moderate POC flux (1.6 ± 0.5 g OC m−2 a−1) which is similar to the mean of 1.3 ± 0.8 g OC m−2 a−1 across 21 major subduction trenches > 6.5 km deep26.

Study areas in the Kermadec Trench and the Atacama Trench regions in the Pacific Ocean (a). Detailed bathymetry data with specific sampling sites in the Kermadec Trench (b) and Atacama Trench (c). The sites were visited during two cruises with RV Tangaroa (TAN1711, 2017) and RV Sonne (SO261, 2018), respectively. Black line in (b) and (c) indicates the 6000 m depth contour. Black line in b and c indicates the 6000 m depth contour. Map adapted from Ref.46. Figure Copyright British Geological Survey UKRI 2021. Bathymetry data were obtained from the Global Multi-Resolution Topography Synthesis47.
Results and discussion
Sediment dynamics in hadal trenches are governed by two processes: (1) continuous pelagic deposition and (2) infrequent mass wasting/deposition events that translocate large quantities of sediment from the trench slope to the trench interior (typically driven by seismic activity27,28). The profiles of sediment core excess 210Pb (210Pbex: defined as the 210Pb fraction in secular disequilibrium from 226Ra daughters derived from the atmosphere and the water column) have clear indications of both processes (Fig. S1). Periods of monotonic exponential decreases of 210Pbex shown in most core sections reflect relatively constant sediment deposition. Sections of some Atacama cores (At 2, 3, 5, 7, 9 and 10) exhibit short-term 210Pbex trend reversals and plateaus that reflect dilution of 210Pbex by episodic mass deposition of low 210Pbex sediment. We calculated constant linear sedimentation rates (i.e., the sedimentation rates were unvarying down-core) from the periods of continuous deposition, excluding mass deposition events, and thereby provide a conservative estimate of overall sediment deposition rates at the trench axis. The Atacama cores exhibiting mass deposition events have similar calculated sedimentation rates to the other Atacama sites (Table S1).
For our purposes, we grouped Hg concentration data from the top 5–6 cm (4–5 slices; Fig. S2) of each core, where 210Pbex was present, as “recent” sediments which, based on the vertical accretion rates, represent material accumulated over about the last 60–190 years (median 120 years). The bottom five slices of each core, including sediments from 10 to 40 cm depth where 210Pbex was absent (see Fig. S1), were grouped as “historical” sediments. Although the historical samples can not be accurately dated, they occur at core depths well below the point of 210Pbex extinction, which would typically place their deposition prior to about the middle of the nineteenth century given 210Pb’s half-life of 22.3 years.
Atacama sediments overall contained significantly higher Hg concentrations (P < 0.001) than Kermadec (Fig. 2). Sites in the trenches at both locations contained similar recent Hg concentrations to sites on the adjacent abyssal plains and slope (i.e., At1, At7, At9, and Kc7; the difference between trench and abyssal sites is non-significant at P > 0.05). Recent Atacama sediments on average had significantly higher Hg concentrations (188 ± 56 ng g−1; P < 0.001) than historical sediments (137 ± 51 ng g−1). The exception to this pattern was At2, in which recent sediments had lower Hg concentrations (P < 0.01) than historical. Recent Kermadec sediment Hg concentrations averaged across all sites (44 ± 26 ng g−1) were not significantly different from historical sediments (41 ± 16 ng g−1; P > 0.05), although they were higher at Kc7 (P < 0.001).

Plots of average (A) Hg concentrations and (B) Hg fluxes at the deep-ocean study sites. Left hand panels show concentration and fluxes for recent and historical sediments at individual sites. Right hand panels show concentrations and fluxes at all At and all Kc sites combined, and the concentrations and fluxes in recent and historical sediments of all sites. For individual sites, in (A) bars represent mean ± standard deviation (SD) Hg concentrations in the upper 5 cm (“recent”, N = 5) and bottom 5 slices (“historical”, N = 5) of sediment cores. In (B), bars represent mean ± SD Hg fluxes calculated from the mean and SD concentrations and sediment accumulation rates. Significant differences between recent and historical Hg concentrations and fluxes indicated by: *P < 0.05, **P < 0.01, ***P < 0.001. The Deep-Ocean Average line represents the inferred average flux over the entire deep-ocean based on Outridge et al.1 and Zhang et al.3.
The recent and historical Hg concentrations at Atacama were markedly higher than those in surface sediments in other oceanic regions remote from anthropogenic or hydrothermal sources, where Hg concentrations were usually < 80 ng g−1 9,15. They were also higher than in other hadal and abyssal sites where mean concentrations ranged from 12 ± 6 to 132 ± 20 ng g−1 (Fig. 3A)16,18,19. The mean Atacama values were even higher than in some regions affected by anthropogenic activities. For example, the most contaminated deep-ocean site in the Mediterranean Basin exhibited a mean Hg concentration of 77 ng g−1 13. Maximum concentrations in recent Atacama sediments, which ranged up to 401 ng g−1 (see Fig. S2) are the highest reported from remote locations anywhere including in the Kamchatka Trench and abyssal plains (158 ng g−1 16) and bathyal areas of the Southern Ocean (87 ng g−1 15). They approach the maxima reported from some of the most contaminated shelf sites world-wide including the Bohai Sea, China (574 ng g−129) and the Laurentian Trough, Canada (520 ng g−1 11).

Plots of sediment Hg concentrations (A) and fluxes (B), and particulate organic carbon (POC) flux in hadal trench and abyssal plain sites world-wide. (Plots show mean ± SD values, for the top few cm of sediment cores or surface sediment grab samples. Study locations and Hg data sources in panel (A): 1—Marianna/Yap Trenches18, 2—Marianna Trench19, 3—Kermadec Trench and abyssal plain (this study), 4—New Britain Trench19, 5—Kuril-Kamchatka Trench and abyssal plain16, 6—Atacama Trench and abyssal plain and slope (this study). In panel (B), the locations and sources for data points 3 and 6 are the same as in panel A, but for point 5 the flux is from one core on the Kuril Basin abyssal plain17. Linear regression equations are shown as dashed lines, and in (B) the regression was forced through the origin. POC flux data are from Longhurst26 as tabulated by Stewart and Jamieson24 and Jamieson35.
POM scavenging and sedimentation of Hg from the euphotic zone, which is ultimately sourced from atmospheric deposition, is likely the main source of Hg in deep-ocean sediments. This is evidenced by Hg isotope analyses of hadal sediments18, ocean Hg-primary productivity modeling3, and geochemical associations between Hg and POM especially diatoms in particle traps and sediments15,17,22. An analysis of the few Hg concentration and flux data available for hadal and abyssal sediments showed positive though non-significant correlations between estimated POC flux and sediment Hg concentrations and fluxes (Fig. 3) which are also suggestive of an upper-ocean source of POM-associated Hg.
There are two other possible sources or processes that can contribute to high Hg concentrations in deep-ocean sediments. First, post-depositional redistribution of Hg could occur following the decomposition of Hg-containing POM, releasing dissolved Hg to porewater. Its subsequent diffusion and adsorption onto authigenic Fe and Mn hydroxides in oxic surface sediments can result in Hg enrichment9,12. Diagenetic redistribution of Hg is typically negligible in lake and coastal sediments11,30, but is less studied and could be important in ocean basins due to different sedimentation and mixing regimes12,13. As porewater Hg concentrations were not measured in this study, we cannot directly estimate the magnitude of redistribution. Significant correlations between concentrations of solid phase Hg and reactive Fe (see Fig. S3), an association suggestive of Hg redistribution12, were observed in four of nine Atacama cores (At1, At5, At7, At9; Table S2). To test the effect of possible redistribution, we compared mean recent Hg concentrations with those in the whole cores. Whole core data represent the integrated averages of increased surface sediment concentrations and decreased lower core concentrations due to possible redistribution. Recent sediment Hg concentrations were only 16.7% higher on average than whole core concentrations at Atacama (188 vs 162 ng g−1), and 1.9% higher at Kermadec (44.3 vs 43.5 ng g−1). Higher Hg values in recent Atacama sediments but not in Kermadec could also be the result of elevated deposition of anthropogenic atmospheric Hg into the eastern Pacific Ocean, compared to lower levels of anthropogenic Hg in ocean waters near Kermadec3. While we cannot resolve the two alternate explanations, the absence of significant Hg-reactive Fe correlations at the majority of Atacama sites, and the small increase in recent Hg concentrations relative to whole cores at both trenches, suggests that any effect of redistribution on recent sediment Hg concentrations and fluxes was small.
A second possible Hg source in deep-ocean sediments is seafloor hydrothermal vents associated with mid-ocean ridges. Globally, vents are thought to contribute minor amounts of Hg to oceans, ≤ 100 t a−1 1,22, but could be locally important. The Atacama study area is several thousand km east of the nearest known hydrothermal field31 indicating that, in this area with the highest Hg concentrations, hydrothermal influences are negligible. The Kermadec trench is located relatively close to a hydrothermal field31, but no studies have been conducted on vent releases or seawater Hg concentrations in this region. However, there is little evidence of significant hydrothermal impacts on Hg concentrations in seawater32,33,34 or biota18 in the vicinity of vent plumes elsewhere, suggesting that the possibility is unlikely in this study. Overall, therefore, the Hg concentrations in these trench sediments are likely to predominantly reflect POM-associated transport of Hg from the euphotic zone.
Mean total Hg fluxes in recent sediments at Atacama (63.6 ± 30.8 µg m−2 a−1) were significantly higher (P < 0.001) than in historical sediments (44.4 ± 19.5 µg m−2 a−1), and also significantly higher (P < 0.001) than recent Kermadec sediments (10.4 ± 7.5 µg m−2 a−1; see Fig. 2B). There was no significant difference between average fluxes measured in trenches and in abyssal plain and slope sites at either location (P > 0.10). Combined mean recent and historical Hg fluxes were 50.6 ± 36.8 (median 39.1) µg m−2 a−1 and 35.9 ± 23.8 (median 31.5) µg m−2 a−1, respectively. A similar range of fluxes (30–44 µg m−2 a−1) was reported in post-1900 sediments in a dated core from the abyssal Kuril Basin17, supporting these findings.
To place these results in context, the relatively low recent Hg flux measured at Kermadec (10.4 µg m−2 a−1) is 6–15 times higher than the currently estimated deep-ocean average of 0.7–1.8 µg m−2 a−1 1,3. Because the POC flux at Kermadec is similar to the average of all of the world’s trenches > 6.5 km depth26, Kermadec may approximate the global average Hg flux in the hadal. The median recent flux across our study sites (39.1 µg m−2 a−1) was 22–56 times larger than the inferred deep-ocean average Hg flux, but the median includes Atacama with the highest POC flux in the world, and which therefore may have a substantially higher Hg flux than most other trenches. Overall, however, these results indicate that some hadal trenches and adjacent abyssal areas are intense focal points of Hg accumulation. The question arises as to what impact these high hadal fluxes might have on global deep-ocean Hg accumulation rates.
To tentatively calculate the total hadal zone Hg flux, the few available data from hadal and abyssal sites (the present study, and Aksentov and Sattarova17) were used to approximate Hg fluxes for each trench system using the much larger global trench POC database. Individual trench estimates were summed to arrive at a total hadal Hg accumulation rate. POC flux data are available for 18 of the deepest trenches > 6.5 km depth, including Atacama and Kermadec24,35, representing 92% of the global hadal area below 6.5 km. Applying a Hg:POC regression slope (± standard error, SE) of 16.1 ± 2.9 μg Hg g POC−1 (see Fig. 3B), we arrive at a preliminary estimate of total Hg burial of 16.9 ± 1.0 t a–1 in this zone (see Table S3). Scaling up to the much larger hadal area bounded by the 6 km isobath10, the total Hg flux is estimated to be 73 ± 4.4 t a–1. The error terms for the 18 trenches and for the total hadal zone were propagated from the SE of the Hg:POC regression slope applied to each trench’s POC data. We emphasize the uncertainty associated with the 73 t a−1 value because of the few hadal or abyssal sites in total that have been sampled for Hg fluxes (N = 3), the large variation in fluxes between cores within the study trenches, and the relatively large confidence intervals associated with the Hg:POC regression (see Fig. 3B).
Conclusions
This study has substantially expanded the knowledge base concerning Hg flux in the deep-ocean. These results increase by six-fold the number of dated cores analyzed from this zone and provide the first estimates of hadal Hg flux, which is shown to be high at both study sites relative to the currently accepted deep-ocean average flux. Although still limited in number and geographical scope, the results can be placed in the context of the current estimate of deep-ocean Hg accumulation. Our first order estimate of 73 t a−1 for hadal Hg burial represents 12–30% of the putative deep-ocean accumulation of 240–600 t a−1 1,3, occurring within an area equal to 1.1% of the total deep-ocean. This estimate omits Hg flux in the much larger abyssal zone which is 85% of total ocean area10. Given the evidence of similarly high Hg fluxes in some abyssal sediments (this study and Ref.17), and even higher fluxes in high productivity bathyal areas of the Great Southern Ocean15, it is possible that the total deep-ocean Hg accumulation is higher than is presently believed. Our results thus call for extensive additional sampling of the deep-ocean to better constrain these preliminary estimates and to improve global Hg modeling.
Methods
Sampling
Sediment was for most part recovered by a multi-corer36, but in some cases we were successful using box coring (site Kc7) or an autonomous lander system for core recovery (Kc4). Recovered sediment cores were transferred to an onboard laboratory that maintained in situ bottom water temperature (2–4 °C). For the Kermadec Trench system, sediment cores were sectioned in 1 cm slices down to 2 cm depth, 2 cm slices to 10 cm and subsequently in 5 cm slices to the core bottom. For the Atacama Trench system, the cores were sectioned in 1 cm slices down to 10 cm, 2.5 cm slices to 20 cm, followed by 5 cm slices until the bottom of the core. Samples were homogenized in plastic bags and stored at -20 °C until laboratory analysis onshore.
Excess 210Pb (210Pbex) analysis and sedimentation rate calculation
The procedures for 210Pbex analysis and sediment accumulation rate calculation are well established, and here followed previous studies on hadal sediments37,38,39. Sediment was dried in an oven at 80 °C for 48 h, then milled with a mortar. 2.0 g of sediment powder were transferred in a plastic tube and hermetically sealed. The tubes were left for more than 2 months to establish secular equilibrium between 226Ra and 222Rn. To obtain 210Pbex activity, counting peaks of the specific gamma rays from 210Pb (46.5 keV) and 214Pb (351.9 keV) were measured with well-type HPGe detectors (GWL120230 and GWL120-15, ORTEC) connected to a multichannel spectrum analyzer (APV8002, TechnoAP or MCA-7a, SEIKO EG&G). Counting peak areas of the target nuclides were calculated with Gaussian curve fitting with KaleidaGraph 4.5 (Synergy Software). To quantify these activities, Reference Uranium–Thorium ore DL-1a (CANMET) was used as the standard material. 210Pbex activity was calculated by subtracting 214Pb from the 210Pb based on an assumption of secular equilibrium between 226Ra and the daughter nuclei in the sediment. Sediment accumulation rates were calculated based on the Constant Initial Concentration model40,41 from the 210Pbex activity (Bq g−1) in the core sections displaying exponential decreases in the plots. Instead of the core depth (cm), cumulative mass depth (g cm−2) obtained from the core depth and the dry bulk density (g cm−3) was used to express the sediment accumulation rate (initially as g cm−2 a−1).
Hg analysis
Mercury in the sediment samples was determined as total Hg on a Hydra IIC direct mercury analyzer (Teledyne Leeman Labs, Hudson, NH, USA) using thermal decomposition, amalgamation and atomic absorption spectrophotometry following EPA Method 747342. 10–50 mg of individual freeze-dried sediment samples were weighed into quartz boats. Mercury concentrations were quantified by the external standard method with calibration curves ranging from 1 to 1500 μg g−1. Within every batch of 10 samples, 3 blanks, 1–2 pairs of duplicates and 2–3 certified reference materials (CRMs: MESS-3, DOLT-5; and NIST 2709a and PACS-3, obtained from the National Research Council of Canada and NIST, respectively) were analyzed. In order to be deemed valid, the CRMs must fall within the range of the certified mean ± standard deviation. If CRM concentrations were outside the acceptable range, 2 more CRMs were analyzed. If the following CRMs were still outside the acceptable range, the instrument was re-calibrated. The mean CRM recoveries were 101.9 ± 9.0% and were within the expected ranges.
Reactive Fe analysis
Two reactive Fe extraction procedures were employed on frozen sediments—Fe-DCA (Fe species solubilized in a dithionite-citrate-acetic acid solution (50 mg mL−1 sodium dithionite in 0.35 M sodium citrate, 0.2 M acetic acid, pH 4.8 for 1 h)), and Fe-HCl (Fe species solubilized by a mild HCl extraction (0.5 M HCl for 1 h)). Fe-DCA extracts the non-silicate-bound Fe fraction (oxides and authigenic Fe(II) phases excluding pyrite), whereas Fe-HCl releases poorly crystalline oxides and authigenic Fe(II) but also leaches some Fe from silicates43,44. Fe in the extract solutions was determined as total Fe using Ferrozine45. Concentrations were calculated on a dry weight basis using the water content.
Statistics
Concentration and flux data in the text are represented as mean ± standard deviation (SD) values. Mercury concentration and flux data were not normally distributed in some cores, and differences between recent and historical sediments in these cases were tested with a Mann–Whitney Rank sum test. Those with data that were normally distributed and which satisfied the assumption of equal variance were analysed with t-tests. Linear regressions analysed relationships between mean Hg concentrations and fluxes and POC fluxes because the combined data were normally distributed. We used SigmaPlot 14.0 Software (Systat Software Inc.). Error propagation was calculated with the online calculator at www.julianibus.de.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Outridge, P. M. et al. Updated global and oceanic mercury budgets for the United Nations Global Mercury Assessment 2018. Environ. Sci. Technol. 52, 11466–11477 (2018).
Sunderland, E.M., Mason, R.P. Human impacts on open ocean mercury concentrations. Global Biogeochem. Cycles, 21, GB4022. https://doi.org/10.1029/2006GB002876 (2007).
Zhang, Y. et al. Six centuries of changing oceanic mercury. Global Biogeochem. Cycles 28, 1251–1261 (2014).
Evers, D. C. et al. Evaluating the effectiveness of the Minamata Convention on Mercury: Principles and recommendations for next steps. Sci. Total Environ. 569–570, 888–903 (2016).
Wang, F. et al. How closely do mercury trends in fish and other aquatic wildlife track those in the atmosphere? Implications for evaluating the effectiveness of the Minamata Convention. Sci. Total Environ. 674, 58–70 (2019).
Mason, R. P. et al. Mercury biogeochemical cycling in the ocean and policy implications. Environ. Res. 119, 101–117 (2012).
Obrist, D. et al. A review of global environmental mercury processes in response to human and natural perturbations: Changes of emissions, climate, and land use. Ambio 47, 116–140 (2018).
Amos, H. M. et al. Legacy impacts of all-time anthropogenic emissions on the global mercury cycle. Global Biogeochem. Cycles 27, 410–421 (2013).
Fitzgerald, W. F. et al. Marine biogeochemical cycling of mercury. Chem. Rev. 107, 641–662 (2007).
Harris, P. T. et al. Geomorphology of the oceans. Mar. Geol. 352, 4–24. https://doi.org/10.1016/j.margeo.2014.01.011 (2014).
Gobeil, C. & Cossa, D. Mercury in sediments and sediment porewater in the Laurentian Trough. Can. J. Fish. Aquat. Sci. 50, 1794–1800 (1993).
Gobeil, C. et al. Mercury profiles in sediments of the Arctic ocean basins. Environ. Sci. Technol. 33, 4194–4198 (1999).
Ogrinc, N. et al. Distribution of mercury and methylmercury in deep-sea surficial sediments of the Mediterranean Sea. Mar. Chem. 107, 31–48 (2007).
Astakhov, A. S. et al. Distribution and accumulation rate of Hg in the upper quaternary sediments of the Deryugin Basin Sea of Okhotsk. Geochem. Int. 45, 47–61 (2007).
Zaferani, S. et al. Diatom ooze - A large marine mercury sink. Science 361, 797–800 (2018).
Sattarova, V. V. & Aksentov, K. I. Geochemistry of mercury in surface sediments of the Kuril Basin of the Sea of Okhotsk, Kuril-Kamchatka Trench and adjacent abyssal plain and northwest part of the Bering Sea. Deep-Sea Res. Part I(154), 24–31 (2018).
Aksentov, K. I. & Sattarova, V. V. Mercury geochemistry of deep-sea sediment cores from the Kuril area, northwest Pacific. Prog. Oceanogr. 180, 102235. https://doi.org/10.1016/j.pocean.2019.102235 (2020).
Sun, R. et al. Methylmercury produced in upper oceans accumulates in deep Mariana Trench fauna. Nat. Commun. 11, 3389. https://doi.org/10.1038/s41467-020-17045-3 (2020).
Liu, M. et al. Methylmercury bioaccumulation in deepest ocean fauna: implications for ocean mercury biotransport through food webs. Environ. Sci. Technol. Lett. 7, 469–476 (2020).
Mason, R. P. et al. The biogeochemical cycling of elemental mercury: Anthropogenic influences. Geochim. Cosmochim. Acta 58, 3191–3198 (1994).
Munson, K. M. et al. Mercury species concentrations and fluxes in the central tropical Pacific Ocean. Global Biogeochem. Cycles 29, 656–676. https://doi.org/10.1002/2015GB005120 (2015).
Lamborg, C. H. et al. An examination of the role of particles in oceanic mercury cycling. Phil. Trans. R. Soc. A 374, 20150297 (2016).
John, S. G. & Conway, T. M. A role for scavenging in the marine biogeochemical cycling of zinc and zinc isotopes. Earth Planet. Sci. Lett. 394, 159–167. https://doi.org/10.1016/j.epsl.2014.02.053 (2014).
Stewart, H. A. & Jamieson, A. J. Habitat heterogeneity of hadal trenches: Considerations and implications for future studies. Prog. Oceanogr. 161, 47–65 (2018).
Danovaro, R. et al. A depocenter of organic matter at 7800 m depth in the SE Pacific Ocean. Deep-Sea Res. Part I(50), 1411–1420 (2003).
Longhurst, A. Ecological Geography of the Sea 2nd edn. (Academic Press, 2007).
Bao, R. et al. Tectonically-triggered sediment and carbon export to the Hadal zone. Nat. Commun. 9, 121. https://doi.org/10.1038/s41467-017-02504-1 (2017).
Oguri, K. et al. Hadal disturbance in the Japan Trench induced by the 2011 Tohoku-Oki earthquake. Sci. Rep. 3, 1915 (2013).
Xiao, C. et al. Toxic metal pollution in the Yellow Sea and Bohai Sea, China: Distribution, controlling factors and potential risk. Mar. Pollut. Bull. 119, 381–389 (2017).
Feyte, S. et al. Mercury dynamics in lake sediments. Geochim. Cosmochim. Acta 82, 92–112 (2012).
German, C. R. et al. Hydrothermal impacts on trace element and isotope ocean biogeochemistry. Phil. Trans. R. Soc. A 374, 20160035 (2016).
Lamborg, C. H. et al. Mercury and monomethylmercury in fluids from Sea Cliff submarine hydrothermal field Gorda Ridge. Geophys. Res. Lett. 33, L17606. https://doi.org/10.1029/2006GL026321 (2006).
Bowman, K.L., et al. Mercury in the North Atlantic Ocean: The U.S. GEOTRACES zonal and meridional sections. Deep Sea Res. Part II, 116, 251−261 (2015).
Bowman, K.L., et al. Distribution of mercury species across a zonal section of the eastern tropical South Pacific Ocean (U.S. GEOTRACES GP16). Marine Chemistry, 186, 156−166 (2016).
Jamieson, A. J. The Hadal Zone: Life in the deepest oceans. Cambridge University Press https://doi.org/10.1017/CBO9781139061384 (2015).
Barnett, P. R. O. et al. Multiple corer for talking virtually undisturbed samples from shelf, bathyal and abyssal sediments. Oceanol. Acta 7, 399–408 (1984).
Glud, R. N. et al. High rates of microbial carbon turnover in sediments in the deepest oceanic trench on Earth. Nat. Geosci. 6, 284–288 (2013).
Turnewitsch, R. et al. Recent sediment dynamics in hadal trenches: Evidence for the influence of higher-frequency (tidal, near-inertial) fluid dynamics. Deep-Sea Res. Part I(90), 125–138 (2014).
Wenzhöfer, F. et al. Benthic carbon mineralization in hadal trenches: Assessment by in situ O2 microprofile measurements. Deep-Sea Res. Part I(116), 276–286 (2016).
Krishnaswami, L. D. et al. Geochronology of lake sediments. Earth Planet. Sci. Lett. 11, 207–414 (1971).
Robbins, J. A. & Edgington, D. N. Determination of recent sedimentation rates in Lake Michigan using Pb-210 and Cs-137. Geochim. Cosmochim. Acta 39, 285–307 (1975).
U.S. EPA. Method 7473 (SW-846): Mercury in solids and solutions by thermal decomposition, amalgamation, and atomic absorption spectrophotometry, Revision 0. Washington, DC. www.epa.gov/sites/production/files/2015-07/documents/epa-7473.pdf (2007).
Canfield, D. E. Reactive iron in marine sediments. Geochim. Cosmochim. Acta 53, 619–632 (1989).
Kostka, J. E. & Luther, G. W. I. I. I. Partitioning and speciation of solid phase iron in saltmarsh sediments. Geochim. Cosmochim. Acta 58, 1701–1710 (1994).
Thamdrup, B. et al. Manganese, iron, and sulfur cycling in a coastal marine sediment, Aarhus Bay Denmark. Geochim. Cosmochim. Acta 58, 5115–5129 (1994).
Glud, R. N. et al. Hadal trenches are dynamic hotspots for early diagenesis in the deep-sea. Commun. Earth Environ. 2, 21. https://doi.org/10.1038/s43247-020-00087-2 (2021).
Ryan, W. B. F. et al. Global multi-resolution topography synthesis. Geochem. Geophys. Geosyst. 10, Q03014. https://doi.org/10.1029/2008GC002332 (2009).
Acknowledgements
We would like to thank the captains, crews, Ashley Rowden and other scientific personnel of the RV Tangaroa (TAN1711; 24.11.2017–14.12.2017) and RV Sonne (SO261; 02.03.2018–02.04.2018). Ship time for RV Tangaroa was funded by the HADES-ERC Advanced grant “Benthic diagenesis and microbiology of hadal trenches” #669947 and the Coasts & Oceans Centre of New Zealand’s National Institute of Water & Atmospheric Research (NIWA, New Zealand), while ship time for RV Sonne was provided by BMBF (Germany). The study was financially supported by the HADES-ERC Advanced grant “Benthic diagenesis and microbiology of hadal trenches” #669947, and by the Max Planck Society. The Danish National Research Foundation grant DNRF145 established the Danish Center for Hadal Research. Furthermore, we would like to thank Johannes Lemburg, Per Martensen, Ashley Rowden, Diana Macpherson, Matthias Zabel, Anni Glud, Axel Nordhausen and Volker Asendorf for technical assistance, and Alan J. Jamieson for discussions about POC fluxes. Heather A. Stewart is thanked for refining Figure 1 from the original references. Michael Parsons reviewed this manuscript for the Geological Survey of Canada.
Author information
Authors and Affiliations
Contributions
All authors contributed directly and substantially in analysis, interpretation, and writing of this manuscript. H.S. and R.N.G. initiated this project. F.We., and R.N.G. led the seagoing expeditions and co-organized the sediment sampling, H.S. and P.M.O. contributed equally to the bulk of the data interpretation and writing. G.S. conducted Hg analysis, K.O. analysed the 210Pb and interpreted the sediment accumulation rates, B.T. conducted and interpreted the Fe analysis, F.Wa. contributed to discussion of Hg redistribution and estimate of Hg fluxes, R.N.G. led the project and contributed to interpretation of hadal systems. All authors discussed the results, their implications and commented on the manuscript as it progressed.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sanei, H., Outridge, P.M., Oguri, K. et al. High mercury accumulation in deep-ocean hadal sediments. Sci Rep 11, 10970 (2021). https://doi.org/10.1038/s41598-021-90459-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90459-1
This article is cited by
-
Globally distributed marine Gemmatimonadota have unique genomic potentials
Microbiome (2024)
-
Deep sea cold seeps are a sink for mercury and source for methylmercury
Communications Earth & Environment (2024)
-
Organic matter degradation causes enrichment of organic pollutants in hadal sediments
Nature Communications (2023)
-
A new chloro-azaphilone derivative with pro-angiogenesis activity from the hadal trench-derived fungus Chaetomium globosum YP-106
Journal of Oceanology and Limnology (2023)
-
The hadal zone is an important and heterogeneous sink of black carbon in the ocean
Communications Earth & Environment (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
