
Abstract
Cytokine hemoadsorption might be beneficial in patients with sepsis. However, its effect on anti-infective agents' disposition remains largely unknown. We sought to determine the influence of hemoadsorption on the pharmacokinetics of common anti-infective agents. This is an interventional experimental study, conducted in 24 healthy pigs. Animals were randomly allocated to either hemoadsorption (cases) or sham extracorporeal circuit (controls) and to drug combinations (3 cases and 3 controls for each combination). Hemoadsorption was performed with CytoSorb (CytoSorbents Corporation, USA). We evaluated 17 drugs (clindamycin, fluconazole, linezolid, meropenem, piperacillin, anidulafungin, ganciclovir, clarithromycin, posaconazole, teicoplanin, tobramycin, ceftriaxone, ciprofloxacin, metronidazole, liposomal amphotericin B, flucloxacillin and cefepime). Repeated blood sampling from the extracorporeal circulation (adsorber inlet/outlet, sham circuit) was performed over six hours following administration. Total clearance and adsorber-specific clearance were computed. Hemoadsorption was associated with increased clearance of all study drugs, except ganciclovir. Its impact on total body clearance was considered as moderate for fluconazole (282%) and linezolid (115%), mild for liposomal amphotericin B (75%), posaconazole (32%) and teicoplanine (31%) and negligible for all other drugs. Hemoadsorber clearance declined over time, with even delayed desorption for beta-lactams. It was moderately correlated with drug's lipophilicity (p = 0.01; r2 = 0.43). Hemoadsorption with CytoSorb appears to increase to a clinically significant extent the clearance of five among 17 tested anti-infectives. Studies in human patients are required to confirm the need for dosage adjustment of these agents.
Similar content being viewed by others


Introduction
Sepsis is a major health issue worldwide, affecting close to 50 million individuals each year and leading to 11 million deaths1. Recognition of sepsis was identified as a global health priority by the World Health Organization in 2017. Sepsis results from a dysregulated host response to infection2. Activation of both complement and coagulation systems lead to the massive release of pro- and anti-inflammatory cytokines in the blood, a phenomenon sometimes referred to as "cytokine storm"3. This response through systemic hypotension, microcirculation alterations, endothelial lesions, as well as metabolism modulation, ultimately leads to cellular apoptosis, organ failure and death.
CytoSorb (CytoSorbents Corporation, NJ, USA), a hemoadsorption device, has been marketed and licensed for extracorporeal cytokine removal within the European Union since 20114. CytoSorb cartridges can easily be inserted in extra-corporeal circulation circuits. They contain biocompatible polystyrene divinylbenzene copolymer beads coated with polyvinylpyrrolidone capable of removing molecules of mid-molecular weight using a combination of hydrophobic or ionic interactions as well as hydrogen bonding5,6.
The level of evidence supporting the use of CytoSorb in septic shock remains low and largely observational7,8,9,10,11. In their latest statement, experts from the Surviving Sepsis Campaign urged for further research and did not recommend for or against blood purification techniques12. In this context, safety parameters are of particular importance for the decision to initiate such therapy in a patient. So far, post marketing surveillance and data from published literature has not suggested major adverse events apart from occasional thrombocytopenia. However, the potential removal of life-saving medications such as antibiotics in sepsis requires particular attention4. Indeed, little is known about the effect of CytoSorb on anti-infectives' pharmacokinetics. In vitro models have confirmed its ability to remove some anti-infectives from the blood13,14. However, these one-compartment models have numerous limitations, and their results might not be translatable to humans. Authors have reported cases suggesting a minor influence on serum blood levels15,16,17. However, these reports lack consistency and reproducibility.
Accordingly, we have designed an experimental animal study to determine the influence of hemoadsorption with CytoSorb on the pharmacokinetics of anti-infective agents commonly prescribed in sepsis.
Materials and methods
Ethics and legal aspects
The experimental study was conducted in a medical competence center located in Wendisch Rietz (Germany), in compliance with German law for animal protection.
Animal preparation and monitoring
We included 24 healthy German landrace pigs (10 female, 14 male) with a body weight of 45–60 kg (mean 52.3 kg). The pigs were pre-medicated with an initial intramuscular injection of ketamine (15 mg/kg), midazolam (0.25 mg/kg) and azaperon (6 mg/kg). Sedation was maintained using a continuous intravenous infusion of ketamine (10 mg/kg/h) and midazolam (0.5 mg/kg/h). Pancuronium was administered for muscle relaxation as required. Anticoagulation with heparin was administered. Surgical tracheostomy was performed to allow conventional mechanical ventilation (Evita XL or Oxylog 3000; Draegerwerk AG & Co KG Luebeck, Germany). Surgical catheterization of the carotid artery and jugular vein was performed (respectively Avanti and Avanti plus sheath introducer; Cordis Miami, FL, USA) to allow for blood pressure monitoring and enable fluid and medication administration respectively. Vital signs were monitored throughout the experiment (Philips M3046A Philips IntelliVue MP5; Philips, Amsterdam, the Netherlands). Finally, a 12 Fr double lumen catheter (Dualyse expert; Vygon GmbH & Co KG, Aachen, Germany) was inserted into either the femoral or jugular vein to enable extracorporeal circulation. All animals were euthanized at the end of the experiment while fully sedated, by simultaneous intravenous potassium chloride and pancuronium administration.
Experimental design
Four experiments were conducted corresponding to four anti-infectives combinations. These combinations as well as drug dosing and injection patterns are presented in Table 1. We tested beta-lactams (meropenem, piperacillin, ceftriaxone, flucloxacillin and cefepime), antifungals (fluconazole, anidulafungin, posaconazole and liposomal amphotericin B), aminoglycosides (tobramycin) and other type of anti-infectives (linezolid, clindamycin, ganciclovir, clarithromycin, teicoplanin, ciprofloxacin and metronidazole).
During each experiment, six animals were prepared as described above. After the administration of the anti-infective combination, animals were randomly allocated to either hemoadsorption with CytoSorb (cases) or sham hemoperfusion (control group) on a one to one ratio.
Extracorporeal circulation
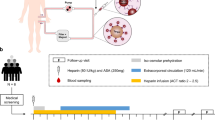
A schematic representation of the extracorporeal circulation (ECC) is shown in Fig. 1. It was established in all animals with a dedicated device [BM11a, Baxter, Deerfield, IL, USA) and corresponding circuit (BM11-Lines-BA-HP (tube system set BM11-hemoperfusion adult set; VE17; BLD-clamp/VE); Baxter, Deerfield, IL, USA]. For animals allocated to the intervention group (Table 1), a CytoSorb cartridge was inserted in the ECC. For those allocated to the control group no cartridge was integrated (sham hemoperfusion). ECC was started one hour after the start of study anti-infective administration. Blood flow rate was kept between 150 and 200 mL/min throughout the experiment.

Extra-corporeal circuit. According to study protocol, 1 h following anti-infective administration, study animals were randomly allocated to either CytoSorb hemoadsorption or sham procedure on a 1:1 basis. For animals allocated to the sham procedure, a similar circuit without hemoadsorber was used.
Blood samples and laboratory analysis
Blood samples were collected before the initiation of the anti-infective infusion, then respectively 5, 30, 90, 250 and 330 minutes after adsorber/sham procedure initiation. For animals allocated to the intervention group, we collected two samples per time point: before (inlet) and after (outlet) the adsorber. For those allocated to the control group, we collected a single systemic sample (Fig. 1). All samples (approx. 9 mL) were drawn into EDTA (ethylenediaminetetraacetic acid) monovettes (Sarstedt AG & Co KG, Nuembrecht, Germany). They were then centrifuged at 30,000 rpm for 10 min, split into two 2 mL Eppendorf tubes (Eppendorf AG, Hamburg, Germany) and frozen at -20°C. Drugs plasma levels were determined quantitatively by liquid chromatography tandem mass spectrometry (LC-MS/MS) (MS/MS: API 2000 or API 4000; Sciex, Nieuwerkerk aan den Ijssel, The Netherlands; HPLC pump: Agilent Technologies, Santa Clara, CA, USA; CTC autosampler: Sciex) in a human medical laboratory (Bioscientia GmbH, Ingelheim, Germany).
Pharmacokinetic parameters calculations
Pharmacokinetics parameters were computed individually for each study drug and each animal using standard non-compartmental calculations and considering first order elimination kinetics. All calculations were performed using Microsoft Excel 2016 (Microsoft Corp., Redmond, WA, USA).
The areas under the plasma drug concentration curves (AUC) were calculated using the log-trapezoidal rule based on the last measurement (AUCo-last) and with extrapolation to infinity assuming a constant elimination rate (AUCo-inf). Total clearance (CLtot) could be deduced from the dose (D):
Total amount eliminated during the study period (DTE) was calculated using the following formula:
In addition, for animals allocated to the adsorber arm, adsorber's instantaneous removal amount (DIR) were evaluated based on the following formula:
where Qp is the effective plasmatic flow (hematocrit considered to be 40%), [inlet] the drug concentration in the pre-adsorber sample and [outlet] the drug concentration in the post adsorber sample. Cumulative removal attributable to the adsorber during study period (DCC) was calculated using the log trapezoidal rule on instantaneous removal rates. Finally, overall adsorber clearance during study period (CLc) could be estimated:
For each drug, we computed the mean clearances with standard deviation by study group. Finally, we calculated the relative increase in overall animal total clearance brought by the hemoadsorber clearance (percent):
Clearance interpretation
We assessed the impact of the adsorber on drug clearance based on the classification proposed by the WHO Committee for Proprietary Medicinal Products (CPMP) for drug inhibitors and inducers according to potency19. Weak inducers increase clearance by >25%, moderate inducers by >100% and strong inducers by >400%. A mild effect was thus retained when the adsorber increased the baseline drug clearance by >25%, a moderate effect by >100% and a strong effect by >400%.
Determinants of hemoadsorber clearance
To evaluate the impact of drugs’ physico-chemical properties on adsorber removal, we assessed their correlation with calculated adsorber clearance. For each study drug, we tested octanol–water partition coefficient (logP), octanol–water distribution coefficient (logD), acid dissociation constant at logarithmic scale (pKas), molecular electric charge (positive, negative, neutral or zwitterion), protein binding or molecular weight. These values were obtained using the Chemaxon online molecular library20.
Statistical analyses
Between-groups comparisons were conducted using bilateral Student’s t-test. Correlation analyses were performed using the Pearson’s test. The strength of associations was assessed according to the correlation coefficient (ρ), considered to be strong (ρ > 0.8), fair (0.6 < ρ ≤ 0.8), moderate (0.4 < ρ ≤ 0.6) or weak (ρ ≤ 0.4). For all analyses, a p value < 0.05 was considered to be statistically significant. All analyses were performed using Graphpad Prism 8.3 (Graphpad Software Inc.).
Ethics approval and consent to participate
The study was approved by the State Office for Occupational Safety, Consumer Protection and Health—Department of Consumer Protection (Brandenburg, Potsdam, Germany), approval number 2347-3-2018. According to the German animal protection law, no additional approval by an Ethics Committee was necessary. The present report was prepared following the ARRIVE guidelines for animal research18.
Results
Measured plasma concentrations and calculated clearances for all study drugs are reported in the supplemental material (Figs. S1–S4).
Overall hemoadsorber clearance
Fig. 2 depicts mean overall hemoadsorber clearance measured during the study period for each study drug under our experimental conditions. Mean clearance of almost all study drugs (except for ganciclovir) was positive indicating removal of the drug by the procedure. Mean clearance was > 3L/h for linezolid (4.6 L/h, SD 0.4), posaconazole (4.2 L/h SD 0.7), fluconazole (4.0 L/h SD 0.4), clindamycin (3.9 L/h SD 0.2) and clarithromycin (3.3 L/h SD 0.8). It was less than 2 L/h for all other molecules. A negative clearance (-0.3L/h) was observed for ganciclovir.

Total Adsorber Clearance. Clearance attributable to CytoSorb (mean and SD). Values were calculated based on pre and post adsorber measurements at different timepoints. AmphoB lipo: liposomal amphotericin B.
Relative part of hemoadsorber over total body clearance
Figure 3 presents for each study drug the impact of the hemoadsorber's associated clearance on total clearance. The hemoadsorber was associated with an increase in total clearance for all tested drugs (range 1 to 282%) except for ganciclovir (-3%). This increase was considered as moderate for fluconazole (282%) and linezolid (115%), mild for liposomal amphotericin B (75%), posaconazole (32%) and teicoplanin (31%) and negligible for all other drugs.

Additional clearance provided by adsorber. The white areas of the bars represent endogenous drug clearance (without adsorber) while the grey areas represent the additional clearance provided by the adsorber under the experimental conditions. Percentage refer to the relative increase in clearance associated with adsorber insertion. AmphoB lipo: liposomal amphotericin B.
Kinetics of adsorber associated clearance
As depicted in Figs. 4 and 5, anti-infective clearance by the adsorber was not constant throughout the study period. For beta-lactams (piperacillin, flucloxacillin, ceftriaxone, cefepime and meropenem), it decreased progressively, even reaching negative values at the end of the experiment. For most other drugs (fluconazole, posaconazole, liposomal amphotericine B, linezolid, clarithromycin and teicoplanin), adsorber clearance decreased slowly but remained positive throughout the experiment. The clearance of some molecules became almost null respectively after one (anidulafungin, tobramycin, ganciclovir), two (metronidazole) or six (ciprofloxacin) hours of therapy and remained so for the rest of the experiment.

Kinetics of adsorber clearance of beta-lactams (a) and antifungals (b). Bars represent instantaneous clearance at the different study time points: 30 min (0.5 h), 1, 2 and 3 h after therapy initiation as well as the last measure obtained (6 h). Dotted lines represent total clearance (also represented in Fig. 2). Reported values are mean and standard deviation. AmphoB lipo: liposomal amphotericin B.

Kinetics of adsorber clearance of other anti-infective agents. Bars represent instantaneous clearance at the different study time points: 30 min (0.5 h), 1, 2 and 3 h after therapy initiation as well as the last measure obtained (6 h). Dotted lines represent total clearance (also represented in Fig. 2). Reported values are mean and standard deviation.
Factors associated with overall hemoadsorber clearance
Overall hemoadsorber clearance appeared to increase proportionally with the octanol–water partition coefficient of drugs (logP) i.e. their lipophilicity (p= 0.01; r2=0.43), and to a lesser degree with the logD value (p=0.01; r2=0.36) (Fig. S5 of the supplementary material). There was no significant association with pKa, molecular electric charge, protein binding or molecular weight (all r2<0.1).
Discussion
Key findings
We have conducted an experimental study on 24 pigs to assess the impact of hemoadsorption with CytoSorb on the pharmacokinetics of a panel of anti-infective drugs commonly utilized in the management of patients with sepsis. We have found that for most drugs the procedure was associated with relatively high clearance. However, when endogenous clearance was considered, we found that the additional clearance provided was pharmacologically significant for only a few drugs (fluconazole, linezolid, liposomal amphotericin, posaconazole and teicoplanin) but negligible for other tested drugs. Importantly, we observed that the kinetics of clearance were not stable over time, with the highest clearance observed during the first hours of therapy, often followed by a rapid decline and even desorption for some drugs (in particular beta-lactams). Finally, we found that lipophilicity was the only pharmacokinetic factor moderately associated with overall hemoadsorber clearance.
Comparison with previous studies
These first systematic data from in vivo experiments have to be compared to in vitro studies. Koenig et al. observed strong adsorption of anti-infectives to the CytoSorb adsorber with normal saline or human albumin as the perfusion fluid. Adsorption was markedly reduced for two anti-infectives, namely meropenem and ciprofloxacin, if reconstituted blood was used for perfusion. However, Koenig et al. integrated the adsorber into continuous veno-venous hemodialysis, so the results do not resemble pure hemoadsorption alone14. In addition, such data does not consider endogenous clearance and therefore the real impact of CytoSorb on in vivo pharmacokinetics is difficult to infer.
Other available data consist of case reports. The low clindamycin clearance observed in a young patient with refractory septic shock caused by Panton Valentin leucocidin producing methicillin resistant Staphylococcus aureus is consistent with the minimal effect of CytoSorb on clindamycin’s overall clearance (+4.7%) observed in our study16. Similarly, immediate removal of teicoplanin with a saturable process was described by Dimski et al17. This is in full agreement with our data, which shows high initial removal followed by progressive decline. Other observations have reported substantially lower linezolid peak levels during CytoSorb adsorption15,21. This is consistent with our finding of doubled clearance of this molecule during therapy.
Our finding of a paradoxical reduction of the systemic clearance of ganciclovir by the adsorber, corresponding to an apparent “creation” of drug in the cartridge, is in line with a release of drug from red blood cells, already observed during hemodiafiltration22.
A high affinity of CytoSorb for lipophilic molecules particularly in the range up to approximately 55 kDa has also been suspected14,23. We found a weak but significant correlation between the logP and hemoadsorber associated clearance. However, this property alone cannot be used to predict the impact of the hemoadsorber on drugs' pharmacokinetics. Indeed, when total clearance is considered, CytoSorb' s effect on total clearance was limited for some drugs with high logP values (i.e. posaconazole, clarithromycin, flucloxacillin and clindamycin) while it was relatively important for some drugs with low logP values (fluconazole, linezolid or liposomal amphotericine B). This further highlights the need to consider hemoadsorber clearance relative to overall clearance.
Finally, in vitro studies have described CytoSorb 's adsorptive capacities as saturable17 and even subject to desorption (i.e. release of an adsorbed drug)24. Our study confirms these findings for the first time in vivo. The ability of polyvinylpyrrolidone, the substance covering CytoSorb's beads, to adsorb and desorb molecules has in principle been described in technical settings other than hemoadsorption, and has even been proposed for use as a component of pharmaceutical drug delivery systems25.
Strengths and limitations
To the best of our knowledge, this is the first in vivo experimental study conducted to evaluate the impact of CytoSorb on the pharmacokinetics of anti-infective drugs. We tested a wide panel of medications commonly used in clinical practice. We have conducted sound analyses with thorough pharmacokinetics models. However, it has several limitations worth discussing.
First, our experimental study was conducted in healthy animals and our results might not directly be translatable to humans with septic shock. Indeed, drugs pharmacokinetics might vary from species to species. For instance, the clearances of ceftriaxone and teicoplanin measured in our model were higher than values previously reported in humans (respectively 8.4 vs 1.0 L/h and 5.4 vs 0.7-1.0 L/h) while that of linezolid was lower (8.8 vs 3.7 L/h)26,27. In addition, and more importantly, pharmacokinetic parameters are known to be massively altered in sepsis (increased volume of distribution, decreased protein binding etc.) particularly in cases of associated acute kidney injury or liver failure. In addition, in patients with sepsis, CytoSorb’s adsorptive capacities might be modified, typically by competitive adsorption of other molecules such as pro-inflammatory mediators. The net effect of such competition is unknown and might lead to desorption or decreased drug adsorption. Our model was designed to evaluate the isolated impact of hemoadsorption and minimize such confounders as much as possible. Further studies in humans with sepsis are therefore required to confirm or refute our findings.
Second, our protocol, enabled us to compute clearances obtained during the first 6 hours of CytoSorb adsorption, a duration lower than the manufacturer's recommended therapy (24 h). Hence, a delayed effect of CytoSorb therapy for instance total desorption (i.e. beta-lactams) or more significant binding (i.e. posaconazole) cannot be ruled out. However, our kinetics analyses strongly suggest that the majority of the adsorption process takes place in the first hours of therapy.
Third, medications were not administered separately but as a group, and their pharmacokinetics might have influenced each other through binding competition. However, our findings for drugs classes (beta-lactams and azoles) were robust and consistent, even if these medications were administered in different group combinations.
Last, administered doses of anti-infectives were higher than those typically recommended in clinical practice. We aimed to maximize serum concentration, based on the assumption that removal by the adsorber might be concentration dependent. This choice is likely to have biased our results toward an increased clearance attributable to the adsorber.
Implications for clinicians and policy makers
Based on our data, some tentative recommendations can be formulated. Obviously, human studies should be performed before these recommendations are implemented in clinical practice.
The net influence of hemoadsorption with CytoSorb on beta-lactams' pharmacokinetics appears to be minimal. In addition, the initial adsorption followed by desorption might even be beneficial in terms of pharmacodynamics. Indeed, the intervention might optimize antimicrobial exposure and the activity of such time dependents agents. Hence, no dose adaptation would theoretically be required for this class of medications.
On the other hand, the observed initial removal of tobramycin, a concentration dependent drug, might be associated with a decrease in its antibacterial clinical efficacy. Here, a dose increase, accounting for the additional clearance, would be recommended. Temporary interruption of the adsorption therapy prior to drug administration could represent an alternative solution. The latter would have the theoretical advantage of enabling a high peak level (efficacy) and rapid removal (decreased toxicity).
For other drugs included in this study, biologic activity is related to the area under curve (AUC) divided by the minimum inhibitory concentration. This parameter was significantly decreased by CytoSorb hemoadsorption for three study drugs: fluconazole, linezolid and liposomal amphotericin B. Hence, decreased anti-infective activity is likely and drug dosage adaptation appears advisable. However, any required dose modification is likely to be minor. For all other drugs, the impact of the therapy on their total clearance was small and no major impact on pharmacodynamics would be expected and no dose adaptation would be required at least in patients with normal renal and liver function.
In acute kidney injury, or liver failure, the impact of hemoadsorption on total clearance is likely to increase making drug adaptation necessary. In these situations, therapeutic drug monitoring is strongly advised.
Conclusions
Hemoadsorption with CytoSorb appears to have limited effect on the pharmacokinetics of the majority of drugs tested. However, the clearance of fluconazole, linezolid and liposomal amphotericin B appears to be increased by the procedure. These observations need to be confirmed during clinical trials conducted in humans with septic shock, to devise appropriate recommendations for dosage adjustment. In the meantime, therapeutic drug monitoring remains advisable.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- HA:
-
Hemoadsorption
- ECC:
-
Extracorporeal circulation
- logP:
-
Octanol–water partition coefficient
- LogD:
-
Octanol–water distribution coefficient
References
Rudd, K. E. et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 395, 200–211. https://doi.org/10.1016/S0140-6736(19)32989-7 (2020).
Singer, M. et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315, 801–810. https://doi.org/10.1001/jama.2016.0287 (2016).
Tisoncik, J. R. et al. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 76, 16–32. https://doi.org/10.1128/MMBR.05015-11 (2012).
Poli, E. C., Rimmele, T. & Schneider, A. G. Hemoadsorption with CytoSorb((R)). Intensive Care Med. https://doi.org/10.1007/s00134-018-5464-6 (2018).
Rimmelé, T. & Kellum, J. A. Clinical review: Blood purification for sepsis. Crit. Care 15, 205–205. https://doi.org/10.1186/cc9411 (2011).
Malard, B., Lambert, C. & Kellum, J. A. In vitro comparison of the adsorption of inflammatory mediators by blood purification devices. Intensive Care Med. Exp. 6, 12. https://doi.org/10.1186/s40635-018-0177-2 (2018).
Friesecke, S. et al. International registry on the use of the CytoSorb® adsorber in ICU patients. Med. Klin. https://doi.org/10.1007/s00063-017-0342-5 (2017).
Friesecke, S., Stecher, S.-S., Gross, S., Felix, S. B. & Nierhaus, A. Extracorporeal cytokine elimination as rescue therapy in refractory septic shock: A prospective single-center study. J. Artif. Organs 20, 252–259. https://doi.org/10.1007/s10047-017-0967-4 (2017).
Kogelmann, K., Jarczak, D., Scheller, M. & Drüner, M. Hemoadsorption by CytoSorb in septic patients: A case series. Crit. Care 21, 74. https://doi.org/10.1186/s13054-017-1662-9 (2017).
Brouwer, W. P., Duran, S., Kuijper, M. & Ince, C. Hemoadsorption with CytoSorb shows a decreased observed versus expected 28-day all-cause mortality in ICU patients with septic shock: A propensity-score-weighted retrospective study. Crit. Care 23, 317. https://doi.org/10.1186/s13054-019-2588-1 (2019).
Schadler, D. et al. The effect of a novel extracorporeal cytokine hemoadsorption device on IL-6 elimination in septic patients: A randomized controlled trial. PLoS ONE 12, e0187015. https://doi.org/10.1371/journal.pone.0187015 (2017).
Rhodes, A. et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock: 2016. Crit. Care Med. 45, 486–552. https://doi.org/10.1097/CCM.0000000000002255 (2017).
Reiter, K. et al. In vitro removal of therapeutic drugs with a novel adsorbent system. Blood Purif. 20, 380–388. https://doi.org/10.1159/000063108 (2002).
Konig, C. et al. In vitro removal of anti-infective agents by a novel cytokine adsorbent system. Int. J. Artif. Organs 42, 57–64. https://doi.org/10.1177/0391398818812601 (2019).
Zoller, M. et al. Can cytokine adsorber treatment affect antibiotic concentrations? A case report. J. Antimicrob. Chemother. 70, 2169–2171. https://doi.org/10.1093/jac/dkv068 (2015).
Poli, E. C. et al. Clindamycin clearance during Cytosorb((R)) hemoadsorption: A case report and pharmacokinetic study. Int. J. Artif. Organs 42, 258–262. https://doi.org/10.1177/0391398819831303 (2019).
Dimski, T., Brandenburger, T., MacKenzie, C. & Kindgen-Milles, D. Elimination of glycopeptide antibiotics by cytokine hemoadsorption in patients with septic shock: A study of three cases. Int. J. Artif. Organs 43, 753–757. https://doi.org/10.1177/0391398820917151 (2020).
Kilkenny, C. et al. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. J. Cereb. Blood Flow Metab. 31, 991–993. https://doi.org/10.1038/jcbfm.2010.220 (2011).
European_Medicine_Agency. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf. (2012).
ChemAxon. (https://chemaxon.com/products/chemicalize)
Morris, C., Gray, L. & Giovannelli, M. Early report: The use of Cytosorb haemabsorption column as an adjunct in managing severe sepsis: Initial experiences, review and recommendations. J. Intensive Care Soc. 16, 257–264. https://doi.org/10.1177/1751143715574855 (2015).
Perrottet, N. et al. Disposition of valganciclovir during continuous renal replacement therapy in two lung transplant recipients. J. Antimicrob. Chemother. 61, 1332–1335. https://doi.org/10.1093/jac/dkn102 (2008).
De Schryver, N., Hantson, P., Haufroid, V. & Dechamps, M. Cardiogenic Shock In A Hemodialyzed Patient On Flecainide: Treatment with intravenous fat emulsion, extracorporeal cardiac life support, and CytoSorb(R) hemoadsorption. Case Rep. Cardiol. 2019, 1905871. https://doi.org/10.1155/2019/1905871 (2019).
Lang, C. N. et al. Use of the CytoSorb adsorption device in MDMA intoxication: A first-in-man application and in vitro study. Intensive Care Med. Exp. 8, 21. https://doi.org/10.1186/s40635-020-00313-3 (2020).
Franco, P. & De Marco, I. The use of poly(N-vinyl pyrrolidone) in the delivery of drugs: A review. Polymers (Basel) 12, 1114. https://doi.org/10.3390/polym12051114 (2020).
Goodman, L., Brunton, L., Chabner, B. & Knollmann, C. Goodman & Gilman’s Pharmacological Basis of Therapeutics (McGraw-Hill, New York, 2011).
Targocid_information. https://www.swissmedicinfo.ch/Accept.aspx?Lang=EN. Swissmedicinfo (2017).
Acknowledgements
The authors want to thank Dr Michael Zoller (Munich, Germany) for advising on the interpretation of the results, Dr Sven Giersdorf from CytoSorbents Europe for his support and, finally, Harriet Adamson for language editing.
Funding
The study was funded by CytoSorbents Europe, Berlin, Germany. The company was involved with the study design. However, data interpretation was conducted independently by AS, PA, TB and DKM. The company was allowed to read the draft of the manuscript before submission but had no influence on its content or decision for submission.
Author information
Authors and Affiliations
Contributions
A.G.S.: Participated in study design, data interpretation and analyses and drafted the manuscript. P.A.: Performed pharmacokinetic analyses, participated in data interpretation and critically reviewed the manuscript. J.S.: Participated in study design, drew Fig. 1 and critically reviewed the manuscript. M.S.: Performed the animal experiment and critically reviewed the manuscript. H.Z.: Planned and monitored the animal experiment and critically reviewed the manuscript. T.B.: Participated in pharmacokinetic analyses and critically reviewed the manuscript. D.K.M.: Participated in study design, data interpretation and critically reviewed the manuscript. All authors read and approved the final manuscript and agree to be personally accountable for their contribution
Corresponding author
Ethics declarations
Competing interests
JS is Senior Director Medical & Clinical of CytoSorbents Europe. AS and DKM have received speaking honorarium from CytoSorbents Europe. All other authors stated that they have no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schneider, A.G., André, P., Scheier, J. et al. Pharmacokinetics of anti-infective agents during CytoSorb hemoadsorption. Sci Rep 11, 10493 (2021). https://doi.org/10.1038/s41598-021-89965-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-89965-z
This article is cited by
-
Ineffectiveness of hemoadsorption in large animals with abdominal sepsis: a randomized controlled porcine study
Intensive Care Medicine Experimental (2024)
-
Extracorporeal Elimination of Pro- and Anti-inflammatory Modulators by the Cytokine Adsorber CytoSorb® in Patients with Hyperinflammation: A Prospective Study
Infectious Diseases and Therapy (2024)
-
The phenomenon of desorption: What are the best adsorber exchange intervals?
Critical Care (2024)
-
CytoSorb hemoperfusion markedly attenuates circulating cytokine concentrations during systemic inflammation in humans in vivo
Critical Care (2023)
-
Cytokine hemoadsorption with CytoSorb® in post-cardiac arrest syndrome, a pilot randomized controlled trial
Critical Care (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
