
Abstract
Improvement of risk stratification through prognostic biomarkers may enhance the personalization of cancer patient monitoring and treatment. We used Ancer, an immunoinformatic CD8, CD4, and regulatory T cell neoepitope screening system, to perform an advanced neoantigen analysis of genomic data derived from the urothelial cancer cohort of The Cancer Genome Atlas. Ancer demonstrated improved prognostic stratification and five-year survival prediction compared to standard analyses using tumor mutational burden or neoepitope identification using NetMHCpan and NetMHCIIpan. The superiority of Ancer, shown in both univariate and multivariate survival analyses, is attributed to the removal of neoepitopes that do not contribute to tumor immunogenicity based on their homology with self-epitopes. This analysis suggests that the presence of a higher number of unique, non-self CD8- and CD4-neoepitopes contributes to cancer survival, and that prospectively defining these neoepitopes using Ancer is a novel prognostic or predictive biomarker.
Similar content being viewed by others


Introduction
Understanding mechanisms of cancer progression and identifying patients at high risk for recurrence are pivotal to the personalization of cancer care. Improvements in DNA sequencing techniques combined with cost reductions have enabled the routine mapping of the tumor genome and improved our mechanistic understanding of cancer progression and patients’ survival. Tumor mutational burden (TMB) has arisen as a potential cancer prognosis biomarker in numerous tumor types1,2. Higher TMB has been associated with improved survival, highlighting the link between immune recognition of tumor neoantigens and favorable clinical outcomes. In solid tumors, the generation of an adaptive anti-tumor immune response requires a complex coordination of events ultimately dependent on cross-presentation of tumor-associated or tumor-specific antigens and cytotoxic T lymphocyte recognition of antigenic peptides presented on class I major histocompatibility complex (MHC), or human leukocyte antigen (HLA), of tumor cells. Attention has shifted to neoantigens generated by somatic mutations, since their recognition by the immune system is less impacted by central tolerance mechanisms, and as they are the targets of effector CD8+ T cells post checkpoint blockade therapy3,4. An increase in the absolute quantity of tumor mutations and neoantigens has been associated with favorable response to checkpoint immunotherapy and has led to a tumor histology-agnostic regulatory drug approval2.
Importantly, the quality of each potential antigen may critically affect the likelihood of mounting an effector T cell anti-tumor immune response5,6,7. Thus, an individual patient’s prognosis is likely to be influenced not only by the quantity of neoantigens, but by the presence of neoantigens that are most likely to result in an effective anti-tumor immune response. Optimal neoantigens may be defined by several factors including the level of neoantigen gene expression, the processing of peptide fragments, the binding affinity of neoantigen peptide fragments to HLA, successful presentation on the surface of the cell, and the phenotype of the immune cell that responds to the neoantigen. The exclusion of potential regulatory T cell (Treg) epitopes in the tumor mutanome has been one focus of our cancer vaccine development program8. Other groups have also reported that tolerizing epitopes may arise during the mutational process, and that these suppressive epitopes have a deleterious effect on cancer vaccine efficacy9.
Because cancers arise from self, neoepitopes bound to HLA molecules must be sufficiently different from endogenous peptides in order to be recognized as non-self by the patient's existing T cell repertoire. A novel epitope where none previously existed is most readily identified as non-self, however, mutations that change the T cell receptor (TCR) facing portion of an existing epitope can also influence the immune response to an antigen. TCR-facing amino acids that contain sequences resembling the unaltered human genome have been shown to modulate immunity by activating a tolerizing response. This has been observed in the context of infectious disease antigens, where pathogen-derived epitopes presenting a TCR "face" homologous to self-derived epitopes elicited CD4+CD25+FoxP3+ (regulatory) T cell (Treg) responses10,11, which in turn led to the suppression of effector immune responses against co-administered epitopes11. Removal of self-like epitopes from vaccine formulations has shown to increase immunogenicity of H7N9 influenza and CT26 vaccines8,12 and protection against lethal H7N9 challenges13. Preliminary work from our group in oncology suggests these "self-like" inhibitory neoepitopes also exist in mutated antigens derived from the murine colon carcinoma CT26 cell line8. The presence of such Treg-inducing neoepitopes in tumors may camouflage cancerous cells from immune surveillance. Additionally, T cells recognizing neoepitopes with "self" TCR faces may have been rendered anergic during thymic selection or deleted before they can be released to the periphery. Therefore, self-like neoepitopes may reduce overall tumor immunogenic potential.
We hypothesized that the presence of a mutation alone is not sufficient to generate an immunogenic neoepitope, but that significant differences must exist at the HLA- and/or TCR-interfaces of the neoepitope as compared to (1) the non-mutated form of the neoepitope, and (2) to other self-epitopes, in order to be recognized as non-self by the immune system. Therefore, we hypothesized that individual patient outcomes may be determined by neoepitope analyses that integrates the consideration of self-epitopes into the analysis of tumor neoantigens. To test this assumption, we analyzed large scale bladder cancer genomic data using Ancer, an automated computational immunoinformatics pipeline that we developed for neoantigen screening and vaccine design. Ancer shares components with our commercial-grade screening platforms used routinely in immunogenicity assessments of infectious disease antigens14, such as the EpiMatrix algorithm for HLA class I and HLA class II neoepitope identification, and the JanusMatrix algorithm for tolerated, tolerogenic, and cross-reactive T cell epitope identification14,15. Animal proof-of-concept studies using RNA replicons revealed that neoantigen-based cancer vaccines designed with Ancer are immunogenic, induce multifunctional CD4+ and CD8+ T cell responses, and are effective in challenge experiments16. The prognostic value of Ancer is demonstrated here by our analysis of genomic and clinical data derived from bladder cancer patients. Our evaluation of patient survival with Ancer shows a marked improvement over other stratification approaches such as using TMB or a quantitative assessment of neoepitopes identified with NetMHCpan 4.0 and NetMHCIIpan 3.117,18, commonly used HLA class I and HLA class II T cell epitope prediction tools, respectively. Compared to existing tools, Ancer’s ability to assess self-like epitopes allows the identification of more immunologically relevant neoepitopes, which can also be employed to optimize personalized cancer vaccines.
Results
Neoepitope load is highly correlated with bladder cancer patient tumor mutational burden
Sequencing data from 412 chemotherapy-naïve bladder cancer (BLCA) tumors of the TCGA were downloaded and analyzed with Ancer for neoepitope identification and triaging. The TGCA's BLCA dataset was derived from a cohort of muscle-invasive bladder cancer patients who remained at large untreated prior to tumor collection19. HLA class I and class II types were first determined from the raw sequencing data using the xHLA, seq2HLA, and HLA-VBSeq in silico tools20,21,22. While predictions of HLA allele groups (i.e. two-digit HLA types) were largely consistent across the three HLA typing tools, some results varied when predicting specific HLA proteins (i.e. four-digit HLA types). Overall, 60% of HLA-A, 40% of HLA-B, and 67% of HLA-DRB1 protein predictions (four-digit HLA types) were concordant across the three HLA typing algorithms and a consensus approach was employed to resolve differing HLA mapping. Concordance rose to 85%, 81%, and 83% for HLA-A, HLA-B, and HLA-DRB1, respectively, when considering allele group (two-digit) results, highlighting a relatively high agreement between the HLA typing tools when predicting HLA families.
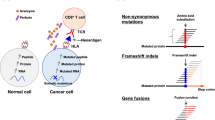
Cancer mutanomes were subsequently analyzed with the Ancer pipeline to evaluate HLA class I and HLA class II neoepitope burdens. Key steps in Ancer includes (1) identification of HLA class I and HLA class II mutation-bearing epitopes, or neoepitopes, with the EpiMatrix algorithm, (2) comparison of mutated and matched normal sequences for HLA/TCR-faces comparison to refine neoepitopes and discard ones where mutations do not significantly alter normal sequences, and (3) in-depth homology analysis of neoepitope TCR-faces against other self-antigens using the JanusMatrix algorithm to remove self-like cross-reactive, tolerated, or actively tolerogenic neoepitopes (Fig. 1). In this last step, each predicted HLA ligand is analyzed two ways: by evaluating its constitutive agretope (or HLA-facing interface) as well as its epitope (or TCR-facing interface). Ligands derived from the human proteome that have the identical TCR face and a similar-binding (but not necessarily sequence-identical) agretope are returned. We expect that T cells interacting with commonly observed TCR faces are deleted during thymic selection or are developed into cells that have a regulatory phenotype. Hence, epitopes presenting these commonly occurring TCR faces, or self-like epitopes, may be tolerated or actively tolerogenic.

BLCA mutanome analysis workflow. Mutations were retrieved for each patient sample and evaluated using three analysis workflows and then compared for overall survival and disease-free survival predictive accuracy. The three types of analyses were defined as follows: (A) “TMB analysis”: tumor mutational burden is evaluated from the count of mutations present in each tumor. (B) “NetMHCpan analysis”: mutation-bearing HLA class I and HLA class II ligands are identified with NetMHCpan 4.0 and NetMHCIIpan 3.1, respectively. This approach is similar to the one employed by the TCGA Research Network in their analysis of the BLCA cohort19. (C) “Ancer analysis”: mutation-bearing HLA class I and HLA class II ligands are identified with EpiMatrix, compared to matched normal sequences to identify Ancer-defined neoepitopes, and filtered with JanusMatrix to remove neoepitopes homologous to self.
We next aimed to identify the distribution of Ancer-derived neoepitopes across TCGA BLCA patients. HLA class I and class II Ancer neoepitopes were identified in all but one and three BLCA patients, respectively (Table 1, Supplementary Data 1). The median number of Ancer HLA class I neoepitopes was 400, and the median number of Ancer HLA class II neoepitopes was 54. As expected, patient total TMB was strongly correlated with the total counts of Ancer HLA class I (Pearson's r = 0.96, p < 0.0001) and class II neoepitopes (Pearson's r = 0.95, p < 0.0001) (Fig. 2a, b).

Association between mutational and neoepitope landscapes. BLCA mutanomes were analyzed with Ancer to determine counts of Ancer-defined HLA class I neoepitopes (a), Ancer-defined HLA class II neoepitopes (b), and Ancer-defined neoantigen candidates that could be used for precision immunotherapy purposes (c). Numbers of HLA class I neoepitopes, HLA class II neoepitopes, and neoantigen candidates observed in each patient is strongly correlated with observed tumor mutational burden (TMB). Each dot corresponds to one TGCA BLCA patient.
We then estimated the landscape of neoantigens that would be suitable for a hypothetical vaccine formulation using the Ancer tool. Candidate vaccine antigens were defined by Ancer based on a series of automated instructions that created optimal amino acid sequences, usually ranging between 15 and 25 amino acids, that contained overlapping HLA class I and class II neoepitopes of interest, while avoiding the inclusion of cross-conserved or otherwise detrimental epitopes, along with flanking residues. We again found that the number of Ancer-designed neoantigen candidates was strongly correlated with patient total TMB (Pearson's r = 0.99, p < 0.0001) (Fig. 2c). As most neoantigen-based vaccine trials employ up to 20 neoantigen candidates23,24, we determined that at least 20 optimal sequences could be generated for patients that have 1.46 mutations per megabase or more, corresponding to 95% of the BLCA cohort. Therefore, most bladder cancer patients present a sufficiently high number of mutations and would be eligible for standard neoantigen-based vaccinations designed by Ancer.
Number of Ancer-derived neoepitopes is a prognostic biomarker for bladder cancer
We next evaluated whether neoepitope count was a prognostic biomarker in bladder cancer and compared the performance of the Ancer pipeline analysis with TMB or neoepitope counts determined with NetMHCpan 4.0 and NetMHCIIpan 3.1. As no standardized NetMHCpan-based neoantigen computational pipeline exists, we employed an approach that was similar to the one employed by the TCGA Research Network in their analysis of the BLCA cohort19, where neoepitopes were defined as mutated HLA ligands identified with default NetMHC- pan cutoff values. Our latter analysis, employing NetMHCpan 4.0 and NetMHCIIpan 3.1, is referred to as the "NetMHCpan" analysis in this manuscript (Fig. 1).
The cohort's median TMB was employed to identify BLCA patients with high (TMBhi) or low TMB (TMBlo). Similarly, patients with high and low neoepitope burdens were defined using the median class I or class II neoepitope counts, based on the Ancer analysis or the NetMHCpan analysis (Fig. 1). Patients with overall high neoepitope burdens were defined as having both a higher than median class I neoepitope burden and a higher than median class II neoepitope burden (CD8hiCD4hi patients). These patients' survival was compared to the remainder of the cohort, which includes (1) patients with high class I neoepitope burden but lower than median class II neoepitope burden (CD8hiCD4lo patients), and (2) patients with lower than median class I neoepitope burden, regardless of their class II neoepitope burden (CD8lo patients, i.e. CD8loCD4lo and CD8loCD4hi patients). The use of categorical variables was motivated by the desire to generate distinct patient subgroups combining CD8 and CD4 neoepitope information without losing information about the source of the neoepitopes (class I vs class II). Adding counts of CD8 and CD4 neoepitopes to generate an "overall" neoepitope burden would obscure information that we believe is important to consider and would collapse epitopes that are associated with different immunological functions, such as promoting either cytotoxic (class I) or helper (class II) T cell responses.
While the difference in DFS between TMBlo and TMBhi patients was not significant (Fig. 3a), CD8hiCD4hi patients, defined by Ancer or NetMHCpan, had a significantly prolonged DFS (Fig. 3c, e). The maximum difference in median DFS was achieved by defining neoepitopes and removing tolerated or tolerizing neoepitopes with the Ancer pipeline. Ancer-derived neoepitope quantification resulted in a DFS difference of 32 months (log-rank p = 0.0028), compared with 27 months when using NetMHCpan-derived neoepitopes (log-rank p = 0.0157). Improved patient stratification with Ancer was also confirmed using Cox proportional-hazards models when considering the CD8hiCD4hi, CD8hiCD4lo, CD8loCD4hi, CD8loCD4lo patient subgroups (Fig. 4a).

Stratification of cancer patients according to TMB analysis, NetMHCpan analysis, and the Ancer pipeline. TCGA bladder cancer patients were separated based on their median TMB (a,b), NetMHCpan neoepitope burden (c,d), or Ancer pipeline-defined neoepitope burden (e,f). Median disease-free survival (DFS) (a,c,e) and median overall survival (OS) (b,d,f) were evaluated with the Kaplan–Meier estimator. Double-ended arrows define the differences in median survival times between the groups for each of the analyses. Statistical significance was evaluated using the log-rank test. The largest differential in median overall survival, 70 months, was obtained with the Ancer pipeline and was more than double the difference in median overall survival observed when stratifying patients using NetMHCpan (34 months).

Univariate survival analysis forest plots. Univariate survival analyses were conducted separately for the TMB, NetMHCpan, and Ancer analyses while considering either disease free survival (DFS) (a) or overall survival (OS) (b). BLCA patients were separated based on their median TMB or neoepitope burdens. Association with DFS and OS is improved with Ancer compared to the other analysis. Hazard ratios (HR), confidence intervals (CI) and p-values (p) were calculated using univariate Cox proportional-hazard models. Overall log-rank p-value is provided for each model on the left.
Univariate analyses focusing on overall survival showed that TMBhi patients or CD8hiCD4hi patients, based on the NetMHCpan or the Ancer analyses, had statistically prolonged survival compared to the remainder of their respective cohorts (Fig. 3b, d, f). However, improved patient cohort differentiation was again achieved using the Ancer pipeline (log-rank p < 0.0001), when compared to stratifications performed using median TMB (log-rank p = 0.0003) or median NetMHCpan neoepitope burden (log-rank p = 0.0024). The largest differential in median overall survival was obtained with the Ancer pipeline and was more than double the difference in median overall survival observed when stratifying patients using NetMHCpan (70 versus 34 months). Cox proportional-hazards models for OS confirmed improved hazard ratios for the CD8hiCD4hi patients with the stratification performed with Ancer compared to the other analyses (Fig. 4b).
HLA- and TCR-face assessments using the Ancer pipeline improves neoepitope quality
The unique method of neoepitope characterization is one of the key differentiating features of the mutanome analysis that is performed using the Ancer pipeline. First, for each predicted neoepitope, Ancer performs a comparison of its HLA- and TCR-facing portions against its respective normal sequence to evaluate the impact of the underlying mutation on either faces (Ancer pipeline step 2, Fig. 1). Once the unique neoepitope is confirmed to be truly "neo" (i.e. not matching to the normal sequence), the JanusMatrix algorithm filters out any neoepitope that shares a high degree of homology, at the TCR interface, with other self-epitopes (Ancer pipeline step 3, Fig. 1). These two filters have the effect of removing from consideration neoepitopes that may not contribute to productive anti-tumor immune responses.
To test the effect of these filters, we first determined the number of "raw" Ancer class I and class II neoepitopes contained within mutated sequences of bladder cancer patients, i.e. without comparing predicted neoepitopes to their matched normal sequence or other self-antigens (i.e. step 1 only from the Ancer pipeline outlined in Fig. 1 and skipping steps 2 and 3). Then, we determined the number of "non-matching" Ancer class I and class II neoepitopes that significantly differed from their matched normal sequences, but without filtering them using the JanusMatrix algorithm (i.e. steps 1 and 2 from the Ancer pipeline detailed in Fig. 1 and skipping step 3). Finally, stratification of bladder cancer patients was performed based on (1) median "raw" Ancer neoepitope burdens (step 1 of the Ancer pipeline), (2) median "non-matching" Ancer neoepitope burdens (steps 1 and 2 of the Ancer pipeline), and (3) median Ancer neoepitope burdens (all steps of the Ancer pipeline).
Significant associations with DFS (Fig. 5a) and OS (Fig. 5b) were observed when stratifying bladder cancers based on their raw Ancer class I and class II neoepitope burdens (Ancer step 1 only; DFS HR = 0.69, p = 0.033 OS HR = 0.59, p < 0.001). Gradual improvements were obtained in subsequent steps of the analysis pathway, which considered comparisons with matched normal sequences (Ancer steps 1–2; DFS HR = 0.64, p = 0.007; OS HR = 0.56, p < 0.001), and other self-antigens (Ancer steps 1–3; DFS HR = 0.61, p = 0.003; OS HR = 0.52, p < 0.001). The incremental enhancement in hazard ratios suggests that the quality of neoepitopes retained after each filtering step is improved by removing sequences that do not contribute to the tumor's immunogenicity. Consequently, using all steps of the pipeline best predicted patients’ survival.

Effect of Ancer's neoepitope and homology filters on survival analyses. Ancer neoepitope and homology filters improve association with disease free survival (DFS) (a) and overall survival (OS) (b). Hazard ratios (HR), confidence intervals (CI) and p-values (p) were calculated using univariate Cox proportional-hazard models. BCLA patients were separated based on their Ancer neoepitope burden (Ancer, all steps), similarly to Fig. 3, "non-matching" Ancer neoepitope burden without considering the JanusMatrix filter (Ancer, steps 1–2), or based on their "raw" Ancer neoepitope burden (Ancer, step 1 only).
Improved neoepitope quality is associated with enhanced five-year survival prediction of bladder cancer patients
To evaluate Ancer's ability to identify long-term survivors based on their genomic data, we hypothesized that bladder cancer patients with high Ancer class I and class II neoepitope burdens (Ancer CD8hiCD4hi patients) would be more likely to survive more than five years while other patients would survive less than five years. Predicted survival status was compared to observed overall survival for 220 BLCA patients, after removing 192 individuals lost to follow-up before the five-year mark and for which survival status could not be precisely assessed.
For the cohort of 220 bladder cancer patients with known five-year OS outcomes, Ancer neoepitope burden, as determined by the full Ancer pipeline, was a more accurate predictor of five-year survival (Fig. 6a, 65% accuracy) than TMB (59% accuracy) or NetMHCpan neoepitope burden (61% accuracy). The Ancer analysis also achieved higher Positive Predictive Value (PPV) and Negative Predictive Value (NPV) statistics (PPV = 34%, NPV = 88%) as compared to TMB- (PPV = 29%, NPV = 86%) or NetMHCpan-based predictors (PPV = 29%, NPV = 85%) (Fig. 6b). The elevated NPV obtained with Ancer suggests that our analysis may be better suited to identify patients at a greater risk of earlier mortality (~ 9 out of 10 correct predictions).

Prediction of bladder cancer patient five-year survival rate. Ancer pipeline analysis improves prediction of five-year survival compared to TMB- or NetMHCpan-based predictors. BLCA patients were predicted to survive more or less than five years based on their mutational or neoepitope burdens. Predicted survival status was compared to observed overall survival. (a) Accuracy of the five-year survival predictions. *p-value < 0.05, McNemar's test. (b) Positive (PPV) and negative predictive values (NPV) obtained for the TMB, NetMHCpan, and Ancer predictors. (c) PPVs and NPVs obtained using truncated and full versions of the Ancer pipeline. The increased NPV as compared to NetMHCpan and TMB suggests that an analysis that identifies tolerated or tolerogenic epitopes (Ancer) may be better suited to identify patients at a greater risk of earlier mortality (~ 9 out of 10 correct predictions) than the other types of analyses.
By isolating each of the Ancer pipeline steps in this five-year survival analysis, we confirmed the additive importance of Ancer's unique homology filters (steps 2 and 3 from Fig. 1) which demonstrated gradual improvements in PPV and NPV upon their integration into the five-year survival predictor (Fig. 6c). This result further showcases the improvement in predictive capacity that results from refinement of neoepitopes by using JanusMatrix to eliminate putatively tolerated or tolerogenic neoepitopes that may not contribute to tumor immunogenicity. Since Ancer employs more than one variable (i.e. both HLA class I and class II neoepitope burden), generating Receiver Operating Characteristic (ROC) curves was not possible. Instead we replicated our five-year survival analysis for various survival intervals, every 3 months between t = 0 and t = 14 years, and calculated PPVs and NPVs for each analysis and time interval. The resulting values were plotted to calculate AUCs for each analysis (Supplementary Fig. 1). The AUC obtained for Ancer (AUC = 0.6506) was greater than those obtained for the TMB (AUC = 0.6270) and NetMHCpan (AUC = 0.5991) analyses, again demonstrating superior classification using Ancer across a range of survival periods.
Multivariate analysis indicates Ancer neoepitope burden is independently predictive of DFS and OS
The robustness of Ancer as a prognostic biomarker was evaluated in multivariate analyses to test if the association between Ancer CD8 and CD4 neoepitope burdens and patient outcome remained significant after adjusting for common cofactors, such as TMB, age, sex, PD-L1 expression, smoking status, and disease stage. A comparative analysis was performed with NetMHCpan CD8 and CD4 neoepitope burdens.
We first evaluated each clinical cofactor in separate univariate analyses to identify which of them were significantly associated with patient survival. Cofactors significantly associated with survival in univariate analyses were subsequently included in multivariate analyses. Of those, only disease stage was significantly associated with DFS (Table 2, p < 0.001). With respect to OS, age (p < 0.001) and disease stage (p < 0.001) were significant cofactors. The significance of the TMB, NetMHCpan, and Ancer factors in these univariate analyses were maintained whether considered as categorical or continuous variables. Other factors taken into consideration (sex, PD-L1 expression, smoking status) did not reach statistical significance in any of the univariate analyses.
Multivariate survival models were subsequently generated to investigate whether Ancer or NetMHCpan neoepitope burdens remained associated with DFS when adjusting for TMB and disease stage, or associated with OS when adjusting for TMB, age, and disease stage. We expected to lose association with survival as we had previously observed strong correlations between TMB and counts of HLA class I and class II neoepitopes (Fig. 2). Ancer and NetMHCpan neoepitope burdens' association with DFS was lost when adjusting for TMB and disease stage (data not shown). Nonetheless, Ancer neoepitope burden remained a significant cofactor associated with OS once adjusted for TMB, age, and disease stage (Fig. 7a). Independence was maintained in analyses where Ancer neoepitope burden was considered as a continuous variable. TMB was no longer significantly associated with OS in this model. Multivariate survival analyses including NetMHCpan neoepitope burden no longer showed significant association with OS after a similar adjustment (Fig. 7b).

Multivariate survival analysis forest plots. Ancer neoepitope burden remains a significant co-factor associated with overall survival when adjusting for TMB, age, and disease stage (a). NetMHCpan neoepitope burden's association with overall survival is lost when adjusting for TMB, age, and disease stage (b). Hazard ratios (HR), confidence intervals (CI) and p-values (p) were calculated using multivariate Cox proportional-hazard models. Ancer neoepitope burden remained a significant cofactor associated with overall survival once adjusted for TMB, age, and disease stage.
Discussion
In this study, we report the application of Ancer, a novel multistep immunoinformatic pipeline for the identification and refinement of neoepitopes most likely to generate an effector T cell immune response. We demonstrate that Ancer improved prediction of clinical outcomes in bladder cancer compared with existing neoepitope identification tools. While these findings have broader relevance, we focused our analyses in this study on bladder cancer given that this disease has long been known to elicit an endogenous anti-tumor response with known individual tumor variability at the patient level. Furthermore, this is a cancer of growing worldwide importance causing serious morbidity and mortality worldwide.
In the landmark study of the TCGA bladder cancer (BLCA) cohort, Robertson et al. showed that TMB and HLA I neoepitopes burden, identified with NetMHCpan 3.0, were associated with BLCA patient survival19, even though these patients did not undergo checkpoint inhibition immunotherapy as this form of therapy was not approved in bladder cancer at the time the samples were collected. In addition to quantity, other groups have focused on the quality of predicted neoepitopes, highlighting a link between patient outcome and the presence of high quality neoepitopes, ones homologous to other known immunogenic epitopes derived from infectious agents5,6. While many T cell epitope induces immunogenic effector T cell responses, some may instead engage regulatory T cells (Tregs) leading to tolerance and immunosuppression25,26,27. Treg epitopes, or Tregitopes, have been documented in both biologics and pathogens10,11,28,29. Mutations generating Treg neoepitopes, or neo-Tregitopes, may camouflage tumors from the immune system. Hence, they should be filtered or removed when optimally evaluating tumor T cell neoepitope burdens or when designing novel neoantigen-based precision immunotherapies.
Ancer integrates EpiMatrix, an extensively validated HLA class I and HLA class II T cell epitope prediction algorithm, in addition to JanusMatrix, a specialized homology tool to identify putative tolerated, cross-reactive, or tolerogenic (i.e. Treg) epitopes (Fig. 1). These tools have been well validated in the biologics and infectious disease fields10,11,12,14,29,30,31, and have been employed in tumor associated antigen and neoantigen-based vaccine studies16,32. Analysis of genomic data from the TCGA BLCA cohort with Ancer found that both Ancer HLA class I and HLA class II neoepitope loads were strongly correlated with patient TMB (Fig. 2a, b), similarly to other reports using alternative T cell epitope prediction tools19. Our analysis also suggests that Ancer can be used as a feasible adjunct for developing personalized vaccines for bladder cancer patients (Fig. 2c), despite the relatively lower number of mutations compared to tumors traditionally investigated for neoantigen-based therapy, such as melanoma23,33.
When we stratified BLCA patients based on their HLA I and HLA II neoepitope burden, we observed significantly prolonged disease free and overall survival in patients whose tumor contains both high numbers of HLA I and HLA II neoepitopes (CD8hiCD4hi patients), compared to other individuals (Fig. 3e, f). Stratifications performed with Ancer were superior to comparative analyses performed with TMB or with neoepitopes counts determined by commonly used T cell epitope prediction tools (Figs. 3, 4). In addition, we showed that Ancer's precise epitope filtering and characterization steps contributed to this increased association with survival, by removing from consideration neoepitopes that should not support T cell-based recognition of the tumor based on homology with matched normal and other self-sequences (Fig. 5). Refining tumor neoepitope burdens by discarding putative non-immunogenic or putative inhibitory T cell neoepitopes provided a clear advantage at improving our understanding of patient outcomes. Follow-up analyses investigating how the balance between putative effector and regulatory neoepitopes affected survival did not yield conclusive results (data not shown).
These observations led us to test the assumption that long-term bladder cancer survivors could be identified by evaluating their tumor for immunogenic neoepitope content. Five-year survival classification of BLCA patients based on Ancer HLA I and HLA II neoepitope contents appeared again to be superior compared to classifications based on NetMHCpan neoepitope content or based on TMB (Fig. 6a, b, Supplementary Fig. 1). Furthermore, we showed that each filtering steps embedded in Ancer incrementally refined neoepitopes quality which subsequently improved five-year survival assessments (Fig. 6c). Lastly, our analysis suggests Ancer neoepitope content remained a significant factor in patient overall survival even when adjusted for TMB, and other clinical covariates such as age at diagnosis and disease stage (Fig. 7). It was initially unexpected that Ancer remained significant when adjusting for TMB, given the high correlation observed between TMB and counts of class I neoepitopes (Fig. 2a) and class II neoepitopes (Fig. 2b). However, Ancer employs a combination of both counts of class I and class II neoepitopes, which increases the precision of the classifier over TMB. Upon close inspection of the results, 54 BLCA TCGA patients, or 13% of the cohort, are classified differently by the TMB and Ancer analyses, with 14 TMBlo patients classified as CD8hiCD4hi patients by Ancer, and 40 TMBhi patients classified as CD8hiCD4lo/CD8lo patients by Ancer. These observations further support the concept of evaluating both class I and class II neoepitope content in prognostic analyses.
There are some limitations to our data. While bladder cancer patients with "high" and "low" neoepitope burdens were identified according to median number of neoepitopes identified in the TCGA BLCA cohort, alternative cutoffs may be more appropriate to further identify specific patients that are at an even higher risk of disease recurrence or death based on their mutanome, and for whom more aggressive treatment options may be considered. Nonetheless, a similar improvement with Ancer over traditional methods was observed when using continuous variables. Furthermore, our current analysis focuses on patients who did not undergo checkpoint inhibitor (CPI) therapy and follow-up analyses are ongoing to determine whether the multi-step filtering process used in the Ancer pipeline will also predict for CPI-treated patients. We hypothesize that filtering for ‘true’ neoepitopes and removing tolerated neoepitopes may also be critical for understanding response to checkpoint therapy and for determining predicted outcomes of patients treated with a CPI agent.
In summary, our report suggests that optimal host-immune recognition of CD8, CD4, and Treg neoepitopes plays a key role in endogenous cancer control and duration of survival. These results suggest that defining the number of true neoepitopes using Ancer may represent a novel prognostic or predictive biomarker. In addition to biomarker identification, using Ancer when designing novel precision immunotherapies, such as neoantigen-based vaccines or TCR-based therapies, offers the advantage of prioritizing immunogenic CD8 and CD4 neoepitopes, while discarding self-like or inhibitory neoepitopes. Therapies that include these design considerations should promote an optimal immune response in cancer patients, leading to improved clinical outcomes when combined with checkpoint inhibitors. The advantage of Ancer-designed precision immunotherapies will be determined in forthcoming clinical trials.
Methods
HLA typing for TCGA samples
In order to perform HLA typing, the full set of normal sequencing data was obtained from The Cancer Genome Atlas (TCGA) for its bladder cancer (BLCA) cohort including blood-derived normal samples and solid tissue normal samples, for each patient. HLA genes all occur in a continuous, approximately 5 megabase region of chromosome 6, and for efficiency only this segment of the aligned read files was retrieved from the genomic data source. Performance using entire BAM file versus the segment was validated and found to be similar. Biobambam2 (version 2.0.89) was used to convert aligned reads to paired read FASTQ files. Bwa (version 0.7.17) was used to align reads to HLA allele references as input for HLA-VBSeq. We first used xHLA and seq2HLA. These two tools represented alternative methodologies for calling HLA types. xHLA called the three class I types (HLA-A, HLA-B, and HLA-C) and 3 of the class II genes (HLA-DPB1, HLA-DQB1, HLA-DRB1), while seq2HLA called three class I genes, 6 class II genes, and 9 non-classical class II genes. The majority of calls were in agreement for class I and class II, except for HLA-DPB1. We then used HLA-VBSeq to form a consensus classification of HLA class I and II types.
Tumor mutational burden (TMB) analysis
Counts of silent and non-silent mutations per megabase for the TCGA bladder cancer cohort were retrieved from NCI's Genomic Data Commons34. Patients whose combined silent and non-silent TMB fell above or below the cohort median were defined as having high (TMBhi) or low (TMBlo) TMB, respectively.
Neoepitope analysis
Somatic mutations were retrieved from the TCGA for the bladder cancer cohort. Mutations identified through all available variant callers (Muse, Mutect, SomaticSniper, and VarScan) were first merged for each patient. Mutations were subsequently analyzed with two independent analyses: (1) with an internally designed neoantigen pipeline, Ancer, that uses proprietary T cell epitope prediction tools and (2) with publicly available T cell epitope prediction tools, NetMHCpan 4.0 and NetMHCIIpan 3.117,18.
Ancer, an end-to-end computational platform that analyzes mutanome data, identifies patient-specific T cell neoepitopes, and subsequently rank them for immunotherapy design. Ancer neoantigen analyses can be performed through collaborations with EpiVax Therapeutics, Inc. Readers are encouraged to contact the authors if they wish to use Ancer in their research.
Ancer makes use of the EpiMatrix and JanusMatrix algorithms for T cell epitope mapping and removal of putative inhibitory or cross-reactive epitopes, respectively. Both tools have been previously described for the immunogenicity analysis of biologics (ISPRI) and other non-mutated vaccine antigens (iVAX)14. Briefly, Ancer parses mutated and matched normal amino acid sequences into overlapping 9- and 10-mer frames. Each frame is then assessed with EpiMatrix to determine its likelihood of binding to one of a patient HLA class I (HLA-A, HLA-B) or class II (HLA-DRB1) alleles. Mutated and normal matched sequences are then compared to identify tumor-specific neoepitopes that significantly differ from their normal matched counterparts at the HLA- and/or TCR-interfaces. Neoepitopes are then screened with JanusMatrix to remove sequences cross-conserved at the TCR interface with epitopes present in self, non-mutated, proteins which may be recognized by natural regulatory T cells (Tregs) or otherwise tolerated due to negative selection of lymphocytes recognizing self-antigens. JanusMatrix has previously been employed to identify Treg epitopes in HCV and H7N9 influenza10,11,12 among other targets. For immunotherapy design, EpiMatrix and JanusMatrix results are compiled for each patient and then reviewed by Ancer to computationally design neoantigen sequences that only contain neoepitopes with limited potential to cross-react with self-epitope sequences.
HLA Class I and Class II neoepitope counts were calculated for each patient. For the Ancer analysis, counts were obtained before and after filtering neoepitopes with the JanusMatrix algorithm, which removes putative tolerated, tolerogenic, or cross-conserved epitopes.
For the NetMHCpan analysis, neoepitopes were defined as mutated epitopes predicted to bind to patients' HLA according to recommended thresholds (Class I/NetMHCpan 4.0: below a percentile rank of 2; Class II/NetMHCIIpan 3.1: below a predicted binding affinity of 500 nM), similarly to the methodology employed by the TCGA Research Network in their analysis of the same cohort of patients19. As neoepitopes analyses are restricted to the HLA-A, HLA-B, and HLA-DBR1 genes within the Ancer pipeline, we applied the same restrictions when analyzing mutations with NetMHCpan and NetMHCIIpan for comparative purposes.
Median neoepitope counts were employed to define patients with high and low neoepitope burdens, similarly to the TMB analysis. Patients whose count of Class I neoepitopes fell (1) at or above or (2) below the cohort median were defined as having high (CD8hi) or low (CD8lo) Class I burden, respectively. Patients whose count of Class II neoepitopes fell above or below the cohort median were defined as having high (CD4hi) or low (CD4lo) Class II burden, respectively.
PD-L1 expression
RNA sequencing data for the TCGA bladder cancer cohort was downloaded from the TCGA. PD-L1 expression was obtained by retrieving FPKM (Fragments Per Kilobase Million) values for the ENSG00000120217.12 Ensembl Gene ID.
Survival analysis
Clinical data, including disease free survival (DFS) and overall survival (OS), for the TCGA bladder cancer cohort was retrieved from the TCGA. Survival curves were plotted using the Kaplan–Meier estimator for the TMB (TMBhi vs TMBlo patients), NetMHCpan (CD8hiCD4hi vs CD8hiCD4lo/CD8lo patients), and Ancer (CD8hiCD4hi vs CD8hiCD4lo/CD8lo patients) analyses. Differences in median overall survival were evaluated with the Log-rank test. Cox proportional hazards models were employed to obtain hazard ratios for each subgroup. Clinical covariates (age, sex, PD-L1 expression, smoking status, and disease stage) were individually evaluated with Cox proportional hazards models to identify variables significantly associated with either DFS or OS. Significant cofactors were included in multivariate survival analyses that considered TMB and Ancer neoepitope groupings, or TMB and NetMHCpan neoepitope groupings. All statistical analyses were performed with GraphPad Prism and R.
Data availability
Data used in this study, including clinical outcomes, tumor mutational burdens, and neoepitope counts, are provided in the Supplementary Source Data.
References
Cristescu, R. et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 362, eaar3593 (2018).
Samstein, R. M. et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 51, 202–206 (2019).
Gubin, M. M. et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014).
Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in Melanoma. N. Engl. J. Med. 23, 2189–2199. https://doi.org/10.1056/NEJMoa1406498 (2014).
Łuksza, M. et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 551, 517–520 (2017).
Balachandran, V. P. et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, S12–S16 (2017).
McGranahan, N. & Swanton, C. Neoantigen quality, not quantity. Sci. Transl. Med. 11, 11–13 (2019).
Richard, G. et al. Abstract 943: Filtering out self-like neoantigens improves immune response to cancer vaccines. Cancer Res. 79, 943–943 (2019).
Lam, H. et al. An empirical antigen selection method identifies neoantigens that either elicit broad anti-tumor T cell responses or drive tumor growth. Cancer Discov. https://doi.org/10.1158/2159-8290.cd-20-0377 (2021).
Losikoff, P. T. et al. HCV epitope, homologous to multiple human protein sequences, induces a regulatory T cell response in infected patients. J. Hepatol. 62, 48–55 (2015).
Liu, R. et al. H7N9 T-cell Epitopes that mimic human sequences are less immunogenic and may induce Treg-mediated tolerance. Hum. Vaccines Immunother. 11, 2241–2252 (2015).
Wada, Y. et al. A humanized mouse model identifies key amino acids for low immunogenicity of H7N9 vaccines. Sci. Rep. 7, 1283 (2017).
Jang, H. et al. Immune-engineered H7N9 influenza hemagglutinin improves protection against viral influenza virus challenge. Hum. Vaccines Immunother. 16, 2042–2050 (2020).
De Groot, A. S. et al. Better epitope discovery, precision immune engineering, and accelerated vaccine design using Immunoinformatics tools. Front. Immunol. 11, 1–13 (2020).
Moise, L. et al. The two-faced T cell epitope: examining the host-microbe interface with JanusMatrix. Hum. Vaccin. Immunother. 9, 1577–1586 (2013).
Maine, C. J. et al. Self-replicating RNAs drive protective anti-tumor T cell responses to neoantigen vaccine targets in a combinatorial approach. Mol. Ther. 29, 1186–1198 (2021).
Jurtz, V. et al. NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 199, 3360–3368 (2017).
Andreatta, M. et al. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 67, 641–650 (2015).
Robertson, A. G. et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 171, 540-556.e25 (2017).
Xie, C. et al. Fast and accurate HLA typing from short-read next-generation sequence data with xHLA. Proc. Natl. Acad. Sci. USA. 114, 8059–8064 (2017).
Boegel, S. et al. HLA typing from RNA-Seq sequence reads. Genome Med. 4, 102 (2012).
Nariai, N. et al. HLA-VBSeq: accurate HLA typing at full resolution from whole-genome sequencing data. BMC Genom. 16, 1–6 (2015).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
Keskin, D. B. et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 (2019).
Shevach, E. M. Mechanisms of Foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645 (2009).
Cousens, L. P. et al. Tregitope update: mechanism of action parallels IVIg. Autoimmun. Rev. 12, 436–443 (2013).
Cousens, L., Najafian, N., Martin, W. D. & De Groot, A. S. Tregitope: immunomodulation powerhouse. Hum. Immunol. 75, 1139–1146 (2014).
De Groot, A. S. et al. Activation of natural regulatory T cells by IgG Fc – derived peptide “ Tregitopes ” T Reg depletion. Blood 112, 3303–3311 (2008).
Jawa, V. et al. T-cell dependent immunogenicity of protein therapeutics pre-clinical assessment and mitigation–updated consensus and review 2020. Front. Immunol. 11, 1301 (2020).
Scholzen, A. et al. Promiscuous Coxiella burnetii CD4 epitope clusters associated with human recall responses are candidates for a novel T-cell targeted multi-epitope Q fever vaccine. Front. Immunol. 10, 1–22 (2019).
Scholzen, A. et al. Coxiella burnetii epitope-specific T-cell responses in patients with chronic Q fever. Infect. Immun. 87, 1–12 (2019).
Hoffmann, P. R. et al. Multi-antigen vaccination with simultaneous engagement of the OX40 receptor delays malignant mesothelioma growth and increases survival in animal models. Front. Oncol. 9, 1–11 (2019).
Sahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017).
Hoadley, K. A. et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291-304.e6 (2018).
Acknowledgements
The results published here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. The authors wish to thank Leonard Moise and Dominique Bridon for fruitful discussions of the results, and Heather Stuart for her careful review of the manuscript.
Author information
Authors and Affiliations
Contributions
Designed the study: G.R., A.D.G., G.D.S., W.D.M., G.B., M.F.P., A.V.B., R.F.S. Data acquisition: G.R., R.F.S. Data analysis: G.R., T.I.G., A.K., M.A. Data interpretation: G.R., A.D.G., G.D.S., W.D.M., G.B., M.F.P., A.V.B., R.F.S. Created new software: M.A., W.D.M. Drafted the manuscript: G.R., A.D.G., R.F.S. Made substantial review of the manuscript: G.R., A.D.G., G.D.S., R.F.S.
Corresponding authors
Ethics declarations
Competing interests
ADG and WDM are senior officers and majority shareholders, and MA is an employee of EpiVax, Inc, a privately owned immunoinformatics and vaccine design company. All three of these authors are also involved in developing the Ancer pipeline. These authors acknowledge that there is a potential conflict of interest related to their relationship with EpiVax and attest that the work contained in this research report is free of any bias that might be associated with the commercial goals of the company. GB was previously a senior officer of EpiVax Therapeutics, Inc., MFP is a senior officer and GR is currently an employee of EpiVax Therapeutics, Inc., a precision immunotherapy company and subsidiary of EpiVax, Inc. MFP and GR have equity in EpiVax Therapeutics. These authors acknowledge that there is a potential conflict of interest related to their relationship with EpiVax Therapeutics and attest that the work contained in this research report is free of any bias that might be associated with the commercial goals of the company. EpiVax, Inc. and EpiVax Therapeutics, Inc. own patents to technologies utilized by associated authors in the research reported here. RFS reports honoraria from Aduro, AstraZeneca, BMS, Exelixis, Eisai, Janssen, Mirati, Pfizer, and Puma. GDS is a member of Clinical Trial Protocol Committees for the following companies: Merck, BMS, Janssen, Cold Genesys, Pfizer, PhotoCure, Fidia, is or has been a scientific advisor/consultant within the past 5 years for the following companies: Heat Biologics, Cold Genesys, PhotoCure, Merck, Roche/Genentech, Ciclomed, Taris Biomedical, MDxHealth, Fidia Farmaceuticals, Urogen, Ferring, Aduro, Boston Scientific, Bristol Myers Squibb, Astra Zeneca, Pfizer, Janssen, EpiVax Therapeutics, Natera, FKD, Ferring, EnGene Bio, SesenBio, BioCanCell, Nucleix, Ipsen, Combat Medical, Astellas, Fergene, Dendreon, Abbvie, Seattle Genetics, and has equity stock/options in EpiVax Therapeutics and Urogen. AVB reports equity stock/options in EpiVax Therapeutics. TIG and AK declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Richard, G., De Groot, A.S., Steinberg, G.D. et al. Multi-step screening of neoantigens’ HLA- and TCR-interfaces improves prediction of survival. Sci Rep 11, 9983 (2021). https://doi.org/10.1038/s41598-021-89016-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-89016-7
This article is cited by
-
A Novel Multiepitope Vaccine Against Bladder Cancer Based on CTL and HTL Epitopes for Induction of Strong Immune Using Immunoinformatics Approaches
International Journal of Peptide Research and Therapeutics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
