
Abstract
Retinitis pigmentosa (RP) is a heterogenous hereditary disorder leading to blindness. Despite using next-generation sequencing technologies, causal variants in about 60% of RP cases remain unknown. The heterogeneous genetic inheritance pattern makes it difficult to pinpoint causal variants. Besides, rare penetrating variants are hardly observed in general case–control studies. Thus, a family-based analysis, specifically in a consanguineous family, is a clinically and genetically valuable approach for RP. We analyzed a Japanese consanguineous family with a member suffering from RP with a typical autosomal recessive pattern. We sequenced five direct descendants and spouse using Whole-exome sequencing (WES) and Whole-genome sequencing (WGS). We identified a homozygous pathogenic missense variant in CNGA1 (NM_000087.3, c.839G > A, p.Arg280His) in the proband, while we found no homozygous genotypes in the other family members. CNGA1 was previously reported to be associated with RP. We confirmed the genotypes by the Sanger sequencing. Additionally, we assessed the homozygous genotype in the proband for the possibility of a founder mutation using homozygosity analysis. Our results suggested the two copies of the variant derived from a founder mutation. In conclusion, we found the homozygotes for c.839G > A in CNGA1 as causal for RP.
Similar content being viewed by others
Introduction
Retinitis pigmentosa (RP) is a genetic disorder causing blindness due to the degeneration of photoreceptor cells. RP has various inheritance patterns, of which 15–25%, 5–10%, and 5–15% are autosomal dominant, autosomal recessive (AR), and X-linked, respectively1. There are more than 2 million patients with RP worldwide2, and RP is the second cause of visual impairment in Japan3. Various treatment approaches have been developed or proposed, including drug treatment such as 9-cis-retinyl acetate4, cell transplantation5 (stem or retinal cells), and gene therapy5 (e.g., targeting to RPE65 mutations6). As the latest treatments can be applied only in confirmed diagnosis cases with specific genetic backgrounds, genetic analysis has become clinically meaningful for RP patients.
Since Next-generation sequencing (NGS) became a standard tool for genetic research of RP7,8, more than 70 RP-related genes have been reported to date (Retnet: http://www.sph.uth.tmc.edu/retnet/ June 20, 2020). However, despite the increase of extensive large cohort studies using the NGS, approximately 60% of RP cases' genetic etiology has remained unknown9. In addition to a cohort study, a family-based analysis has also been an informative and meaningful approach to exploring genetic etiology. Specifically, the analysis with consanguineous families provides more rigorous assessments of heritability and pathogenicity.
Here, we describe a Japanese consanguineous family showing a typical autosomal recessive pattern of RP (arRP) caused by a homozygous rare CNGA1 variant (NM_000087.3, c.839G > A, p.Arg280His). Besides, we revealed the variant was in the homozygous identical-by-descent segments, suggesting a founder mutation effect.
Results
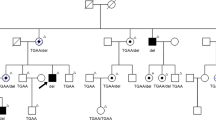
The family history showed a typical AR inheritance pattern (Fig. 1). We performed the whole genome sequence (WGS) for the proband (II-1) and the whole exome sequence (WES) for five unaffected members (II-2, III-1, III-2, III-3, III-4). The coverage of the targeted exon site was × 38 and × 106 ± 2.3(mean ± SD) for WGS and WES, respectively.

Genetic analysis of the consanguineous family with retinitis pigmentosa. (A) Family pedigree with variant in CNGA1. Circles represent females and squares represent males. Black filled squares represent patients with RP. Proband is indicated by black arrow. (B) Electropherograms around codon 280 in the CNGA1 gene in the peripheral blood DNA. The result of the proband showed c.839G > A in homozygosity (II-1). The result of his partner showed wild (II-2). The result of the other unaffected members showed c.839G > A in heterozygosity.
After joint calling, we identified a total of 98,841 variants in at least one of the six subjects. Next, we filtered out variants with either DP < 20 or GQ < 20. As a result, 61,634 variants remained including 57,430 Single-Nucleotide Polymorphisms (SNPs), 1906 insertions, and 2301 deletions. Functional annotations revealed 13,331 (21.6%) missense, and 156 (0.25%) nonsense variants. Transition-transversion ratio (Ts/Tv) was 2.6. The Ts/Tv has been used as a QC metric in WES and is expected to above 2.0 within coding regions10,11. Supposing a rare pathogenic causal variant in this family, we further filtered variants with a maximum minor allele frequency < 0.01 in 1000 Genomes project12, NHLBI Exome Variant Server (https://evs.gs.washington.edu/EVS/), and gnomAD13 (Fig. 2). We then excluded variants for which proband’s genotypes contained alternative alleles (i.e., not homozygous for a reference allele, 0/0). After the filtering, the 2725 variants remained. We further restricted candidates of a causal variant in 790 genes related to retinal diseases in the RetNet database (https://sph.uth.edu/retnet/, updated on June 20, 2020), resulting in 43 variants. Since there was no loss of function variants, we focused on the 10 missense variants with the functional impact of “High” or “Moderate”. We could not identify the genotype of the I-1, as we could not obtain the sample. However, we speculate I-1 may have this variant as a homozygous manner due to possible consanguineous marriage of his parents, as the regional characteristics of their living area. Thus, with the consideration of the AR inheritance pattern, we identified homozygous suspected-pathogenic variants in the two genes, namely, the CNGA1 (NM_000087.3, c.839G > A, p.Arg280His) and KCNV2 (NM_133497.4, c.1063T > C, p.Phe355Leu). CNGA1 is a well-known gene associated with RP. CNGA1 is a well-known gene associated with RP, and according to HDMG (http://www.hgmd.org), the variant was “disease causing” to RP (reported to be disease-causing in the corresponding literature)9. The variant in the KCNV2 was found in the Clinvar database with conflicting interpretations of pathogenicity for non-RP disorders (likely benign for dystrophy with supernormal rod response and uncertain significance for Retinal dystrophy). According to ACMG/AMP guidelines14, the variant in the CNGA1 was classified as pathogenic (Supplemental Table S1), and the variant in the KCNV2 was classified as Uncertain Significant (Supplemental Table S1). From these annotation analyses, we considered the variant in the CNGA1 as a causal variant for arRP in this family.

Variant filtering schemes. This figure shows the overview of variant filtering steps. The number shows the sum of variants detected in the jointed data of six family members. DP depth, GQ genotype quality, Lof loss of function, MAF minor allele frequency, VEP Ensembl Variant Effect Predictor.
Then, to analyze whether the homozygote variants derived from a common ancestor, we performed ROH analysis. As a result, the homozygous regions in the affected proband (II-1) had a total of 31 (129 Mb) homozygous regions that contained the homozygous variants in the CNGA1. The unaffected four descendants had no homozygous regions, and the mother had 1 homozygous part on chromosome 3 (3.4 M). These findings confirmed strong consanguinity in the proband and indicated that the homozygous variants in the CNGA1 derived from a founder mutation.
We performed direct sequencing for the variant in the CNGA1 in all subjects using the Sanger Sequence. We confirmed that the affected proband was the only one who carried two copies of the variant (Fig. 1).
Discussion
In the present study, we identified a homozygous disease-causing variant c.839G > A in the CNGA1 in a consanguineous Japanese family with arRP using NGS sequencing and Sanger sequencing.
Our study showed two important novel points. First, this is the first time to classify the variant in the CNGA1 (NM_000087.3, c.839G > A, p.Arg280His) as pathogenic according to the ACMG/AMP guideline14 (Supplementary Table S1). Although the variant has already been found to be associated with RP in a large sequencing study in a Japanese population9, to predict the pathogenicity, the previous study did not follow the ACMG/AMP guideline14 but depended on in-silico prediction algorithms and conservation scores. The evaluation of pathogenicity based on the ACMG/AMP guideline is clinically important since clinicians see their patients and diagnose genetic diseases in the clinical settings based on evaluations of variants by the ACMG/AMP guideline. Furthermore, it is essential to validate a variant's pathogenicity in independent samples. Otherwise, a single case with the variant is used to define its pathogenicity, and the definition will affect many potential patients. Second, in our study, we found that the variant in the family of the current analysis derived from a founder mutation.
CNGA1 is known to be a susceptibility gene to RP, especially arRP. The estimated prevalence of RP with variants in the CNGA1 is approximately 2–5% with a slight deviation to the Asian population15,16,17. Among the causative gene with arRP, the CNGA1 was estimated to account for a similar proportion17 to the EYS gene (5–16%)18,19 The first report of patients with arRP with variants in the CNGA1 was in 199520. To date, 28 missense/nonsense, 10 small deletion mutations, and 1 splicing substitutions in the CNGA1 are found in the HDMG database (http://www.hgmd.org). The variant c.839G > A in the CNGA1 is too rare (allele frequency: 0.0032% from gnomAD) to be found in the general population. Interestingly, this variant has also been identified homozygous in a Japanese RP patient9. However, there are no details about clinical and family information, and no functional analysis was conducted other than in silico analysis9.
Despite the genetic evidence that the homozygous genotype of this variant leads to RP, its detailed mechanism is still unknown. The CNGA1 encodes the α subunit of cyclic nucleotide-gated (CNG) channels, one of the cGMP-binding transmembrane channels of cone cells20. CNG channel is essential for maintaining the structure and function of photoreceptor cells21,22,23. In the UniProtKB (P29973), there are four known functional domains CNGA1 protein as follows: P-helix (residues 350–360), Selectivity filter (residues 361–369), C-linker (residues 402–484), Cyclic nucleotide-binding domain (residues 485–612) and C-terminal coiled-coil domain (residues 623–666). However, the region containing c.839G > A (p.Arg280His) does not overlap with the functional domains. Therefore, further molecular experiments are necessary to understand the molecular mechanism of this variant.
We found another rare homozygous candidate variant c.1063T > C in the KCNV2. The KCNV2 is known as causing Cone dystrophy retinal 3B (RCD3B)24. RCD3B is a rare disease with supernormal rod responses, which is distinct from RP with peripheral retina atrophy25. We found that KCNV2 was also in the homozygous region, which implicated this variant also derived from the same ancestor in spite of unknown functional significance.
In conclusion, we identified a homozygous rare pathogenic variant c.839G > A in CNGA1 in a consanguineous Japanese family with arRP using NGS sequencing and the Sanger Sequence. Additionally, this is the first study to classify the variant's pathogenicity according to the ACMG/AMP guideline. Our results also suggested the variant c.839G > A derived from a founder mutation. Furthermore, future functional studies are necessary to conclude the effects of the variant in this study as well as the other known pathogenic variants.
Methods
Ethics
This study complied with the standards of the Declaration of Helsinki and the ethics committee approved this study of Shizuoka General Hospital, and we obtained written informed consent from all the participants.
Subject
A total of six members participated in this study (Fig. 1). An ophthalmologist diagnosed the proband (II-1) with RP based on comprehensive ophthalmologic examinations, slit-lamp biomicroscopy, color fundus photography, fundus autofluorescence, and ISCEV Standard electroretinogram (The imaging findings of the proband was shown in Fig. 3). All the six participants have performed a fundus examination and electroretinogram. The interviews with the proband determined the affection of the other family members, including their information on daily visual matters. The proband (II-1) is 67 years old. He was diagnosed with RP at the age of 56 and has suffered from visual impairment since then.

Retinitis pigmentosa phenotype of the proband at 68 years old. (a) Color images; (b) red free images; (c) autofluorescence images; (d) macula section with Optical coherence tomography; (e) Electroretinography test.
DNA sequence using the Next-generation sequencer
According to the manufacturer's instruction, the genomic DNA of each member was extracted from peripheral blood using PAXgene Blood DNA Kit (Qiagen). The samples were sent to Macrogen Japan Corp. at the University of Kyoto to perform NGS using Novaseq 6000 (Illumina, San Diego, CA, United States) on a 150-bp paired-end read protocol. We conducted the whole-exome sequence (WES), using the SureSelect Human All Exon Kit v6 (Agilent, Santa Clara, CA, United States). We conducted the whole-genome sequence (WGS), using the TruSeq DNA PCR-Free Library Preparation Kit (Illumina, San Diego, CA, United States).
Data analysis
We performed quality control of fastq files and adapter trimming of the reads, using fastp v0.20.126. In the process of filtering by the fastp, each read was filtered out with > = 40% of unqualified bases (phred quality < = Q15) and low complexity (< 30%) (the complexity was the proportion of bases which were different from next ones (base[i] ! = base[i + 1])). In the process of adapter trimming, we trimmed polyG tails (a minimum length was 10). The sequence reads were then aligned to the human GRCh38 reference genome using BWA-MEM (BWA 0.7.17-r1188 package)27,28. Duplicates of the reads were marked and removed from the mapped reads using Picard (http://broadinstitute.github.io/picard). We conducted recalibration of base quality scores using GATK 4.129. We performed SNV and indel variant calling separately using the GATK HaplotypeCaller for each sample. We created an interval bed file using the protein-coding region information (Consensus Coding Sequence: CCDS) in the UCSC genome browser (https://genome.ucsc.edu/).
GVCF files were combined and processed with joint-call using GATK (CombineGVCFs and GenotypeGVCFs) based on the family pedigree information. After the joint calling, we performed variant filtering using GATK (VariantFiltration and SelectVariant) with depth > 20 and GQ > 20, followed by Genotype refinement process (CalculateGenotypePosteriors). We annotated variants using SnpEff 4.3t30. Information on allele frequency was also added with VEP (Ensembl Variant Effect Predictor)31 based on 1000 Genomes project12, NHLBI GO Exome Sequencing Project (https://evs.gs.washington.edu/EVS/), and Genome Aggregation Database (gnomAD)13. The candidate genes were searched from the RetNet database (https://sph.uth.edu/retnet/). In silico analysis was conducted using SIFT (Dec 6, 2019; http://sift.jcvi.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), MutationTaster232, and CADD33. The ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) dbSNP database (https://www.ncbi.nlm.nih.gov/snp/), and Human Gene Mutation Database (HDMG; http://www.hgmd.org) were also referred to obtain functional annotation of the variants.
The runs of homozygosity analysis were performed using PLINK v1.90 (option: homozyg)34.
PCR
To validate genotypes of a pathogenic variant in CNGA1, we conducted direct sequencing. A part of exon 11 of CNGA1 was amplified by PCR using primers of 3′ GGACTGCTGGTAAAGGAAGAACTTAAACTC 5′ for sense primer and 3′ CACCAATGGTAGTCAAAGTCAGTGTAGACC 5′ for antisense primer. The sequencing was carried out by using a sense primer. with a 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Data availability
The list of variants analyzed in this study is available upon reasonable request to the corresponding author. Data not available due to ethical restrictions.
References
Dias, M. F. et al. Molecular genetics and emerging therapies for retinitis pigmentosa: basic research and clinical perspectives. Prog. Retin. Eye Res. 63, 107–131 (2018).
Chizzolini, M. et al. Good epidemiologic practice in retinitis pigmentosa: from phenotyping to biobanking. Curr. Genomics 12, 260–266 (2011).
Morizane, Y. et al. Incidence and causes of visual impairment in Japan: the first nation-wide complete enumeration survey of newly certified visually impaired individuals. Jpn. J. Ophthalmol. 63, 26–33. https://doi.org/10.1007/s10384-018-0623-4 (2019).
Koenekoop, R. K. et al. Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: an open-label phase 1b trial. The Lancet 384, 1513–1520. https://doi.org/10.1016/S0140-6736(14)60153-7 (2014).
Lipinski, D. M., Thake, M. & MacLaren, R. E. Clinical applications of retinal gene therapy. Prog. Retin. Eye Res. 32, 22–47. https://doi.org/10.1016/j.preteyeres.2012.09.001 (2013).
Cideciyan, A. V. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog. Retin. Eye Res. 29, 398–427 (2010).
Wang, F. et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum. Genet. 133, 331–345 (2014).
Dan, H., Huang, X., Xing, Y. & Shen, Y. Application of targeted panel sequencing and whole exome sequencing for 76 Chinese families with retinitis pigmentosa. Mol. Genet. Genomic Med. 8, e1131. https://doi.org/10.1002/mgg3.1131 (2020).
Oishi, M. et al. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Invest. Ophthalmol. Vis. Sci. 55, 7369–7375. https://doi.org/10.1167/iovs.14-15458 (2014).
Wang, G. T., Peng, B. & Leal, S. M. Variant association tools for quality control and analysis of large-scale sequence and genotyping array data. Am. J. Hum. Genet. 94, 770–783. https://doi.org/10.1016/j.ajhg.2014.04.004 (2014).
Abecasis, G. R. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073. https://doi.org/10.1038/nature09534 (2010).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74. https://doi.org/10.1038/nature15393 (2015).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. https://doi.org/10.1038/s41586-020-2308-7 (2020).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423. https://doi.org/10.1038/gim.2015.30 (2015).
Paloma, E. et al. Novel homozygous mutation in the alpha subunit of the rod cGMP gated channel (CNGA1) in two Spanish sibs affected with autosomal recessive retinitis pigmentosa. J. Med. Genet. 39, e66–e66 (2002).
Dryja, T. P. et al. Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. 92, 10177–10181 (1995).
Katagiri, S. et al. Whole exome analysis identifies frequent CNGA1 mutations in Japanese population with autosomal recessive retinitis pigmentosa. PLoS ONE 9, e108721. https://doi.org/10.1371/journal.pone.0108721 (2014).
Littink, K. W. et al. Mutations in the EYS gene account for approximately 5% of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology 117, 2026–2033 (2010).
Hosono, K. et al. Two novel mutations in the EYS gene are possible major causes of autosomal recessive retinitis pigmentosa in the Japanese population. PLoS ONE 7, e31036. https://doi.org/10.1371/journal.pone.0031036 (2012).
Dryja, T. P. et al. Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. U. S. A. 92, 10177–10181. https://doi.org/10.1073/pnas.92.22.10177 (1995).
Trudeau, M. C. & Zagotta, W. N. Dynamics of Ca2+-calmodulin–dependent inhibition of rod cyclic nucleotide-gated channels measured by patch-clamp fluorometry. J Gen. Physiol. 124, 211–223 (2004).
Pagès, F., Ildefonse, M., Ragno, M., Crouzy, S. & Bennett, N. Coexpression of α and β subunits of the rod cyclic GMP-gated channel restores native sensitivity to cyclic AMP: role of D604/N1201. Biophys. J. 78, 1227–1239 (2000).
Bradley, J., Frings, S., Yau, K.-W. & Reed, R. Nomenclature for ion channel subunits. Science (New York, NY) 294, 2095 (2001).
Wissinger, B. et al. Cone dystrophy with supernormal rod response is strictly associated with mutations in KCNV2. Invest. Ophthalmol. Vis. Sci. 49, 751–757. https://doi.org/10.1167/iovs.07-0471 (2008).
Michaelides, M. et al. A detailed phenotypic study of “cone dystrophy with supernormal rod ERG”. Br. J. Ophthalmol. 89, 332–339. https://doi.org/10.1136/bjo.2004.050567 (2005).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. https://doi.org/10.1093/bioinformatics/btp698 (2010).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. https://doi.org/10.1101/gr.107524.110 (2010).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. https://doi.org/10.4161/fly.19695 (2012).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362. https://doi.org/10.1038/nmeth.2890 (2014).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. https://doi.org/10.1038/ng.2892 (2014).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Acknowledgements
We sincerely thank the patient and his family members for participating in this study.
Funding
This study is supported by Medical Research Support Project of Shizuoka Prefectural Hospital Organization.
Author information
Authors and Affiliations
Contributions
K.S. and C.T.; Designed and performed data analysis, and drafted the manuscript. I.K. and T.U.; designed the sequencing experiment. N.G.; evaluated the patients and acquired sample data and obtained funding. T.U., T.S., N.G., C.T.; conceived the study design. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saito, K., Gotoh, N., Kang, I. et al. A case of retinitis pigmentosa homozygous for a rare CNGA1 causal variant. Sci Rep 11, 4681 (2021). https://doi.org/10.1038/s41598-021-84098-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-84098-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
