
Abstract
Familial hypercholesterolaemia increases circulating LDL-C levels and leads to premature cardiovascular disease when undiagnosed or untreated. Current guidelines support genetic testing in patients complying with clinical diagnostic criteria and cascade screening of their family members. However, most of hyperlipidaemic subjects do not present pathogenic variants in the known disease genes, and most likely suffer from polygenic hypercholesterolaemia, which translates into a relatively low yield of genetic screening programs. This study aims to identify new biomarkers and develop new approaches to improve the identification of individuals carrying monogenic causative variants. Using a machine-learning approach in a paediatric dataset of individuals, tested for disease causative genes and with an extended lipid profile, we developed new models able to classify familial hypercholesterolaemia patients with a much higher specificity than currently used methods. The best performing models incorporated parameters absent from the most common FH clinical criteria, namely apoB/apoA-I, TG/apoB and LDL1. These parameters were found to contribute to an improved identification of monogenic individuals. Furthermore, models using only TC and LDL-C levels presented a higher specificity of classification when compared to simple cut-offs. Our results can be applied towards the improvement of the yield of genetic screening programs and corresponding costs.
Similar content being viewed by others


Introduction
Dyslipidaemia is one of the major cardiovascular risk factors and it is commonly associated with increased levels of serum low-density lipoprotein cholesterol (LDL-C) and/or reduced levels of high-density lipoprotein cholesterol (HDL-C), as well as high levels of triglycerides1,2. Once serum LDL particles exceed a threshold concentration, atherogenesis—an inflammatory process that precedes atherosclerosis—is stimulated, eventually leading to the development of fatty lesions (i.e. atheromatous plaques) on the lumen surface of large- and intermediate-sized arteries1,3. As a silent condition, dyslipidaemia usually produces no symptoms until the unexpected occurrence of an acute cardiovascular event1.
In addition to being secondary to other disorders or having nutritional causes, dyslipidaemia can occur as a consequence of specific genetic defects4. Familial hypercholesterolaemia (FH), an autosomal dominant disorder, is the most common monogenic dyslipidaemia, with an estimated heterozygous prevalence of 1/250 worldwide4,5. FH increases circulating LDL-C mainly by affecting LDL receptor function, with undiagnosed and untreated subjects being at extremely high risk of premature cardiovascular disease (CVD)3,6. These dyslipidaemic subjects present the most severe phenotype and prompt and accurate diagnosis is essential for CVD prevention, allowing earlier and/or more aggressive therapeutic measures, which have been shown to be effective at reducing cardiovascular morbidity and mortality in both adults and children6,7,8.
Given the silent nature and prevalence of FH, current guidelines support the testing of genes encoding the low-density lipoprotein receptor (LDLR), apolipoprotein B (APOB), and proprotein convertase subtilisin/kexin 9 (PCSK9) in patients that comply with clinical diagnostic criteria, and cascade screening of their family members9. However, most hyperlipidaemic subjects do not have a monogenic defect4,10. Rather, their disease is most likely established through a polygenic genetic background, with a variable environmental contribution modulating the phenotypic expression4,10. Although the lipid profile of polygenic subjects is usually less severe than that of FH subjects regarding total cholesterol (TC) and LDL-C levels, the differences are often subtle enough to prevent an accurate distinction between the two conditions3. As a consequence, the yield of FH genetic screening programs is relatively low, assuming significant costs for patients and/or national health systems.
The Portuguese FH study (PFHS) has been performing a systematic characterisation of FH cases in Portugal since 1999 and includes extended lipid profiles for a large number of index patients11. Previous work using data from this study revealed that the approximately 60% of children that complied with the Simon Broome (SB) clinical criteria for FH were negative for mutations in the hallmark genes, most likely corresponding to cases of polygenic hypercholesterolaemia12. FH-positive subjects (FH+, carrying a pathogenic/likely pathogenic variant) showed higher concentration of atherogenic (i.e. LDL-C) and lower concentration of anti-atherogenic particles (i.e. HDL-C)12. In contrast, most of FH-negative subjects (FH−, no causative variant found) presented higher levels of triglycerides (TG), apolipoprotein C-II (apoC-II), apolipoprotein C-III (apoC-III), apolipoprotein E (ApoE), as well as higher frequency of overweight/obesity12. This suggests that the integrated analysis of multiple biomarkers could be used to create a model that can effectively discriminate between these two populations, improving the selection of patients for genetic screening. Furthermore, a better understanding of the lipid profiles of FH+ and FH− patients may shed further light on the molecular and genetic basis of polygenic hypercholesterolaemia, eventually leading to the identification of novel biomarkers and/or therapeutic targets.
In this work we used a machine learning approach to explore the paediatric subset of the PFHS 2018 dataset update (PFHS-ped) to develop novel models that can integrate data from multiple biomarkers and achieve a reliable discrimination between individuals. Our systematic exploration of available lipid parameters resulted in the development of several models that can robustly classify subjects into FH+ or FH− classes. Some of the models have parameters not routinely used in clinical practice but that are commercially available. Notwithstanding, models comprising only the standard lipid parameters used in the clinic also achieved a relatively good performance. Our results provide an approach for improving the yield of genetic screening programs while showing distinct biochemical backgrounds in monogenic and polygenic hypercholesterolaemia.
Subjects and methods
Patient selection, biochemical and clinical data
The work dataset—PFHS-ped—comprises a subset of 211 unrelated children (from 2 to 17 years old) from PFHS 11 that were not undergoing statin treatment at the time of referral and for which BMI and a basic set of lipid parameters were available (Supplementary Data S1). PFHS was approved by the National Institute of Health Ethic Committee and National Data Protection Commission. The study protocol conforms with the ethical guidelines of the 1964 Declaration of Helsinki and its later amendments. Written informed consent was obtained from parents or legal tutors. For this study, all data were fully anonymised before analysis.
The clinical criteria to be referred to the PFHS is the SB criteria. Between 2006 and 2011, patients with LDL-C or TC levels below the cut-offs established by SB criteria were admitted to the PFHS as long as TC was above the 95th percentile for age and sex of the Portuguese population and a family history of hypercholesterolaemia was present, aiming at a better definition of the clinical criteria for FH in Portugal11,13. For the purposes of this study, we decided to include these individuals in the PFHS-ped dataset to increase the number of available cases. Thus, 68% of the 211 individuals in PFHS-ped fulfil the SB clinical criteria for FH14, while the rest present TC above the 95th percentile for their age and sex and a family history of hypercholesterolaemia13. All the individuals were subjected to molecular study, resulting in the classification of 88 individuals as FH+ and 123 as FH−, defined respectively by presence or absence of known FH causal variants in LDLR, APOB or PCSK9 genes13.
Individuals presenting genetic variants of unknown significance according to the American College of Medical Genetics and Genomics guidelines15 were excluded from this study.
The PFHS-ped includes BMI, age and an extended characterization of lipid profiles, including quantification of small dense LDL (sdLDL), apolipoproteins (apo) A-I, A-II, B, C-II, C-III and E and a ‘Lipoprint’ profile measuring different subfractions of LDL-C (Table 1). The blood lipid profile was divided in three different levels: ‘Basic’, ‘Advanced’ and ‘Lipoprint’, for commonly determined, specialized and Lipoprint test lipid parameters, respectively (Table 1). Biochemical characterization of ‘Basic’ and ‘Advanced’ lipid profiles was performed as described before12. Briefly, fasting blood samples were collected from individuals and TC, direct LDL-C, HDL-C, TG, apoA-I, apoB, and lipoprotein (a) [Lp(a)] were determined for all individuals in a Cobas Integra 400 plus system (Roche) by enzymatic colorimetric and immunoturbidimetric methods. Serum levels of apoA-II, apoC-II, apoC-III, apoE, and sdLDL (sLDL-EX “SEIKEN” kit) were measured by direct quantification in an RX Daytona analyser (Randox Laboratories). The ‘Lipoprint’ profile was obtained using the ‘Lipoprint LDL subfractions test’ (Quantimetrix)16. This is a semiquantitative method that separates by polyacrylamide gel electrophoresis the different lipoprotein fractions as VLDL, IDL, LDL 1–7 subfractions (LDL subfractions 3–7 considered the sdLDL) and HDL16,17,18. For the purpose of this study, ratios that relate lipid parameters were calculated and included as additional variables to explore previous observations suggesting a differential contribution of TG and LDL metabolism and anti-atherogenic/pro-atherogenic factors to FH+ and FH− dyslipidaemic states (Table 1).
Modelling and data analysis
The full description of modelling and data analysis methods is available as supplementary methods. Briefly, the caret package for machine learning19 was used to train classification models based on logistic regression, and a resampling scheme of three times cross validation was applied to estimate model accuracy. Accordingly, data was randomly divided in two sets of 60% and 40% of the subjects defining the training and the testing sets, respectively. The training set was used for model generation and the testing set was used for posterior validation. Models were ranked according to a set of statistical criteria (see supplementary methods) and the top 10 models are discussed in more detail in the context of the biology of hypercholesterolaemia.
Results
Definition of PFHS-ped data subsets for exploratory modelling of extended lipid profiles
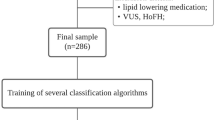
Given that the available information on lipid parameters varied between individuals and considering the three lipid profiles defined for this study—‘Basic’, ‘Advanced’, and ‘Lipoprint’, we began by establishing distinct data subsets regarding all the possible combinations of these profiles (Fig. 1). A detailed description of the seven data subsets is available as supplementary data (Supplementary Tables S1 and S2). As depicted in Fig. 1, the number of individuals across subsets varies between 78 and 211. Although relatively small, these numbers have been previously used in conjugation with machine learning approaches to derive valuable insights into complex biological problems20,21,22,23. We therefore set out to systematically search for the best model to discriminate between FH+ and FH− individuals using these different combinations of lipid parameters.

Data subsets used for model training. Figure shows how PFHS-ped was divided into smaller subsets, identified by a color-coded size (number of individuals) and name, according to the available biochemical parameters for each individual.
Systematic training of models to distinguish FH+ and FH− subjects using extended lipid profiles
We began by training models using all available parameters in each subset. These ‘pilot models’ provided a rough overview of the behaviour of the different parameters in our data subsets but presented a very low performance as assessed by their sensitivity and specificity values (Supplementary Data S2). This suggested an overfitting problem, which we attempted to correct through the use of three common methods to reduce the number of parameters considered for model training (see supplementary methods). This systematic approach resulted in a total of 35 models belonging to one of three categories: ‘cor models’, ‘Imp models’, and ‘RFE models’ (see Supplementary Fig. S1). Interestingly, a trend towards the selection of parameters from the ‘Advanced’ and ‘Lipoprint’ profiles as the most relevant for distinguishing FH+ from FH− subjects (Supplementary Data S2) was observed. Considering the relatively small size of the corresponding data subsets, we decided to investigate whether it could be influencing the perceived contribution of ‘Advanced’ and ‘Lipoprint’ parameters in our models.
For this purpose, we repeated our analysis (Supplementary Fig. S1) using the biochemical parameters available for each data subset restricting the number of individuals to 78. This number corresponds to the smaller sized subset used in this study (the ‘All’ subset), which comprises the subjects that present measures for all biochemical parameters. Two different approaches were followed: train all the models with the same 78 subjects from the ‘All’ subset; or use a random selection of 78 subjects. This analysis confirmed that parameters from the ‘Advanced’ and ‘Lipoprint’ profiles contribute to a better discrimination between FH+ and FH− status independently of the training set (Supplementary Data S2).
Through careful inspection of all models regarding variable importance and correlation, we noticed that a group of four parameters (LDL1, apoC-III, TC/HDL-C and sdLDL.Day) consistently appeared as highly relevant for the discrimination between FH+ and FH− individuals. However, none of the trained models used this small group of parameters as the only predictors. Such models could be relevant for clinical purposes given their comparative simplicity. Therefore, we decided to train two additional models including only these selected parameters (Sel1 and Sel2, Supplementary Table S3). Given that BMI and age are likely to influence the lipid profile of subjects12,24, we further conjugated these parameters with them (models Sel3 and Sel4, Supplementary Table S3). Given the fact that these ‘selected models’ comprise parameters from different lipid profiles, they were trained on the ‘All’ subset.
Altogether, a total of 67 models were generated during this analysis (Supplementary Data S2). Given that the presence of models with highly correlated parameters does not contribute substantially to new insights into the biological background of dyslipidaemia, we identified all models containing any pair of parameters whose correlation was equal to or higher than |0.6|. For this purpose, we generated a correlation plot for all parameters used during modelling analysis (Fig. 2). A total of 14 pairs of highly correlated parameters were identified, 12 of which belong to the ‘Basic’ profile. These pairs were found in 32 out of 67 trained models and were thus discarded from further analysis.

Correlation plot for the dataset parameters. Negative and positive correlations are presented in red and blue, with darker colours corresponding to higher absolute values, according to the scale.
Extended lipid profiles contribute to an improved distinction between FH+ and FH− subjects
Following model training, testing datasets were used to assess model performance and corresponding descriptive statistics were determined. We established a set of ranking criteria to apply to the 35 final models, with cut-off values defined considering the properties and observed range for each statistic (see supplementary methods). We used this approach to retain only the top 10 models (Table 2).
The two best ranked models were the Imp_B and RFEct_BL models, trained with the ‘Basic’ and the ‘Basic & Lipoprint’ subsets, respectively. Among the top 10, these models presented the highest AUC values combined with the best k metrics (Table 2), revealing a substantial agreement between observed and predicted classification of subjects25. These models further displayed the best association between sensitivity and specificity, with Imp_B performing better for sensitivity and RFEct_BL for specificity. Of note, eight of the top 10 models were trained using at least one parameter of the ‘Advanced’ and/or ‘Lipoprint’ profiles. The Lipoprint measurement for LDL1 is present in six of these models. The other models (RFE78t_Ad and RFE78ct_Ad) include sdLDL.Day, ApoA-II, ApoC-II and ApoC-III values from the ‘Advanced’ profile. The models that were trained using only parameters from the ‘Basic’ profile include the ApoB/ApoA-I ratio in addition to LDL-C (Imp_B and RFE78t_B). The Imp_B model further includes the TG/ApoB ratio.
In summary, the comparative analysis of model performance revealed that the integration of lipid parameters from different profiles through machine learning can support a robust discrimination between FH+ and FH− subjects (Table 2). Moreover, our results suggest that biochemical parameters not commonly used in clinical practice, but available commercially, may provide important information towards this distinction, namely contributing to a higher specificity.
Modelling of TC and LDL-C levels improves identification of FH+ individuals in comparison to clinical cut-offs
The biochemical parameters and cut-offs of the SB criteria—defined as blood TC values ≥ 260 mg/ml or LDL-C values ≥ 155 mg/ml for children—are widely used to identify candidate FH individuals and refer them for therapy and genetic testing12. Of note, only ~ 60% of the PFHS-ped individuals that fulfilled these criteria were actually FH+, whereas 3 FH+ individuals were found among the 67 that had TC or LDL-C values below these cut-offs.
Given that the SB criteria are based on two simple biochemical parameters, we decided to train two models exclusively using TC and LDL-C and assess their ability to correctly distinguish between FH+ and FH− individuals (‘SB models’, Table 3). These models were trained using all the PFHS-ped subjects or just the ‘Basic & Lipoprint’ subset (Table 2). The resulting models had a weaker performance when compared to top 10 models trained on the same subsets (cf Tables 2 and 3). To explore the differences between SB models and the two best ranked models, we used them to classify 50 individuals randomly selected from the ‘Basic & Lipoprint’ subset (Supplementary Table S4). Specificity, sensitivity and the positive and negative predictive values (PPV and NPV, respectively) were calculated for the predictions made by these models, as well as for the FH+/FH− classification according to SB criteria cut-offs (Supplementary Table S4). As expected, SB criteria have a very high sensitivity and NPV. However, they are extremely unspecific, with a high likelihood of selection of FH− patients for genetic testing. SB models can considerably improve on this, although they present a lower sensitivity in comparison to SB cut-offs. However, in contrast with SB cut-offs, these models present a very good balance between sensitivity and specificity (Supplementary Table S4). The two top-ranked models trained with the extended lipid profile can achieve very good PPVs while keeping acceptable values for sensitivity and NPV.
These results emphasize how modelling approaches can improve patient classification compared to the use of strict cut-off values. The reduced performance of SB models in comparison to top 10 models supports our suggestion that extended lipid parameters contain relevant biological information for an improved classification of FH+ and FH− individuals.
Implementing the best-ranking models in a clinical setting
Our top 10 models can be easily used in clinical practice to prioritize patients for genetic testing. Clinicians can access the different models and select the one that better suits their practice, in the following link: https://github.com/GamaPintoLab/FH-Models-.git. Models can be grouped into three different categories, depending on the availability of parameters required to run them. A first set of models, including the best ranked model, require biochemical parameters that can be provided by most clinical laboratories. Other models include additional values for ApoA-II, ApoC-II, ApoC-III, sdLDL.Day, which are only available in more specialized clinical laboratories, while the final set of models relies on ‘Lipoprint’ parameters LDL1, MIDC or MIDB, a method that is currently for research use only. We provide an Excel file for simple implementation of the two best ranked models (Table 2) and the SB_B model, which classifies patients as FH+ or FH− upon introduction of the required parameter values. In addition, all top 10 models can be downloaded and applied to a new dataset using R software.
Discussion
Given the high risk for severe CVD at an early age and the benefits of early therapeutic intervention, the identification of children carrying monogenic FH mutations is of extreme importance. Biochemical identification of dyslipidaemic subjects in clinical practice usually relies on the analysis of serum levels for total cholesterol, HDL-C, TG, LDL-C and eventually apoA-I and apoB11,26. Although these biochemical markers allow for a relatively sensitive screening of individuals at risk for CVD, including FH candidates, their specificity in distinguishing monogenic individuals is very low27. In addition, several studies show that many children do not comply with multiple parameters of clinical diagnostic criteria, including the presence of family history of hypercholesterolaemia/CVD or LDL-C levels above the defined cut-offs9,11. Screening for genetic mutations was therefore recommended as standard of care for patients with definite or probable FH by an international Expert Consensus Panel9. However, the diagnostic yield of these screening programs is low28, ranging between 20 and 80%29, as a high number of suspected patients suffer from polygenic conditions9. Thus, the development of robust approaches that can contribute to increase this yield is critical to support a widespread use of FH genetic testing, with a considerable reduction of the resulting burden on health systems.
In this study, we have applied machine learning-based methods to perform a thorough analysis of the extended lipid profiles of the PFHS-ped dataset. We hypothesized that using an extended lipid profile would confer an additional layer of information, supporting a more accurate identification of FH+ subjects, leading to the identification of novel clinically relevant biomarkers. Multiple ‘training’ sets comprising different combinations of biochemical parameters were used to train classification models to distinguish FH+ and FH− individuals, followed by an assessment of performance on independent ‘testing’ sets. For comparison purposes, similar models using only TC and LDL-C were trained. Predictions of FH+ and FH− status for the same group of patients were performed using the two best models, SB models and standard SB criteria cut-offs (Supplementary Table S4). Results show that modelling can considerably improve the specific identification of FH+ individuals and the PPV, with a limited impact on the high sensitivity afforded by SB cut-off criteria. Furthermore, the inclusion of extended lipid parameters contributes to an improved patient identification.
The best ranking model Imp_B uses ApoB/ApoA-I and TG/ApoB ratios, in addition to LDL-C levels, to generate predictions with the highest sensitivity values. Of note, LDL-C levels used in this study were directly determined and thus their accuracy is not affected by TG levels. The current guidelines for dyslipidaemia already recommend the determination of LDL-C, TG and apoB in all dyslipidaemic individuals26. Like the TC/HDL-C, the ApoB/ApoA-I ratio has been linked to cardiovascular risk30. Indeed, a previous study identified the ApoB/ApoA-I ratio as a potential biomarker for FH12. The TG/ApoB ratio was selected both in the first and second ranked models, the later delivering the highest specificity and PPV. This model further includes two ‘Basic’ biochemical parameters (TC and TC/HDL-C) and LDL1 from Lipoprint analysis (see methods). Of note, LDL1 is the most commonly selected biochemical parameter across all top 10 models, suggesting it holds relevant information for the specific identification of FH+ individuals.
The parameters used by the best two models are in agreement with the biology behind FH. Supplementary Fig. 2 shows data for these parameters. TC and LDL-C have higher values for FH+ compared to FH− subjects. This is unsurprising, because FH+ subjects present single-gene mutations that disrupt the clearance of LDL particles by the liver31. The TG/ApoB ratio is lower for FH+ compared to FH− subjects. This is understandable, given both the lower clearance of ApoB in FH+ subjects as well as a higher expected TG in FH− subjects. Hypercholesterolaemia in FH− subjects is likely to have environmental influence, such as cholesterol and TG-rich diets. This should lead to a production of more and ‘bigger’ VLDL particles, containing more TG32. It has been shown that fatty acids can also modulate lipoprotein lipolysis and clearance33. Therefore, the observed TG/ApoB ratio difference is biologically understandable. The TC/HDL-C and ApoB/ApoA-I ratios are higher for FH+ compared to FH− subjects. Higher TG availability in FH− subjects leads to more lipolysis of VLDL through LPL. The cholesterol released is transported back to the liver as HDL, raising HDL-C and ApoA-I concentrations. This mechanism is plausible because LPL gain-of-function and loss-of-function polymorphisms lead to higher and lower HDL-C respectively34. We consistently find higher LDL1 concentration for FH+ versus FH− subjects. This is in accordance with the findings of Teng et al.35. Explaining the observed high LDL1 requires distinguishing between lipolysis through lipoprotein lipase (LPL) and hepatic lipase (HL). The mechanistic modelling study by van Schalkwijk et al.36 suggests that lipolysis outside the liver by LPL mostly affects larger ApoB-containing lipoproteins such as VLDL, while HL mostly targets smaller IDL through LDL particles. Given the impaired binding of ApoB-containing particles to LDLR on the liver, FH+ subjects can be expected to have a lower HL lipolysis and liver clearance than FH− subjects. The lower HL lipolysis explains the accumulation of LDL1 particles. Therefore, even though other LDL subfractions will increase due to a longer circulation time, accumulation of the larger LDL1 particles is especially marked. In addition, several studies have associated altered HL activity or expression, namely in association to genetic polymorphisms, to more severe FH phenotypes37,38. All parameters identified in this study to discriminate between FH+ and FH− subjects are therefore biologically plausible.
Overall, our results suggest that modelling, together with the inclusion of novel lipid parameters, can support an improved classification of FH+ and FH− individuals, with a significant impact on the yield of genetic screening programs and corresponding costs.
Our top models can already be used by clinicians to obtain more precise estimates of the likelihood that their patients are FH+ in comparison to SB criteria. The PPVs and NPVs described in Supplementary Table S4 should be taken into consideration when interpreting results. All the required information for their application is provided in GitHub (see link in Results). The availability of larger patient datasets will be crucial to identify which of the new, non-standard parameters used by our models will be worth incorporating into clinical practice.
References
M. W. Freeman, Lipid Metabolism and Coronary Artery Disease, in Principles of Molecular Medicine, 2nd edn., M. S. Runge and C. Patterson, Eds. Humana Press, Inc., 2006, pp. 130–137.
Wilson, P. W. F. et al. Prediction of coronary heart disease using risk factor categories. Circulation 97(18), 1837–1847 (1998).
Benito-Vicente, A. et al. Familial hypercholesterolemia: the most frequent cholesterol metabolism disorder caused disease. Int. J. Mol. Sci. 19(11), 3426 (2018).
Berberich, A. J. & Hegele, R. A. The complex molecular genetics of familial hypercholesterolaemia. Nat. Rev. Cardiol. 16(1), 9–20 (2019).
Akioyamen, L. E. et al. Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open 7(9), e016461 (2017).
Nordestgaard, B. G. et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 34(45), 3478–3490 (2013).
Marks, D., Thorogood, M., Neil, H. A. W. & Humphries, S. E. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 168(1), 1–14 (2003).
Rodenburg, J. et al. Familial hypercholesterolemia in childhood: diagnostics, therapeutical options and risk stratification. Curr. Opin. Lipidol. 15(4), 405–411 (2004).
Sturm, A. C. et al. Clinical genetic testing for familial hypercholesterolemia. J. Am. Coll. Cardiol. 72(6), 662–680 (2018).
Dron, J. S. & Hegele, R. A. Polygenic influences on dyslipidemias. Curr. Opin. Lipidol. 29(2), 133–143 (2018).
Medeiros, A. M., Alves, A. C. & Bourbon, M. Mutational analysis of a cohort with clinical diagnosis of familial hypercholesterolemia: considerations for genetic diagnosis improvement. Genet. Med. 18(4), 316–324 (2016).
Medeiros, A. M., Alves, A. C., Aguiar, P. & Bourbon, M. Cardiovascular risk assessment of dyslipidemic children: analysis of biomarkers to identify monogenic dyslipidemia. J. Lipid Res. 55(5), 947–955 (2014).
Medeiros, A. M., Alves, A. C., Francisco, V. & Bourbon, M. Update of the Portuguese familial hypercholesterolaemia study. Atherosclerosis 212(2), 553–558 (2010).
Scientific Steering Committee on behalf of the Simon Broome Register Group, Risk of fatal coronary heart disease in familial hypercholesterolaemia, BMJ, 303(6807), 893–896, 1991.
Chora, J. R., Medeiros, A. M., Alves, A. C. & Bourbon, M. Analysis of publicly available LDLR, APOB, and PCSK9 variants associated with familial hypercholesterolemia: application of ACMG guidelines and implications for familial hypercholesterolemia diagnosis. Genet. Med. 20(6), 591–598 (2018).
Hoefner, D. M. et al. Development of a rapid, quantitative method for LDL subfractionation with use of the Quantimetrix Lipoprint LDL System. Clin. Chem. 47(2), 266–274 (2001).
N. Clouet-Foraison, F. Gaie-Levrel, P. Gillery, & V. Delatour, Advanced lipoprotein testing for cardiovascular diseases risk assessment: a review of the novel approaches in lipoprotein profiling, Clin. Chem. Lab Med. 55(10) 2017.
Hirayama, S. & Miida, T. Small dense LDL: An emerging risk factor for cardiovascular disease. Clin. Chim. Acta 414, 215–224 (2012).
M. K. C. from Jed Wing et al., caret: Classification and Regression Training. 2018.
Li, B., Sharma, A., Meng, J., Purushwalkam, S. & Gowen, E. Applying machine learning to identify autistic adults using imitation: an exploratory study. PLoS ONE 12(8), e0182652 (2017).
Salvador, R. et al. Evaluation of machine learning algorithms and structural features for optimal MRI-based diagnostic prediction in psychosis. PLoS ONE 12(4), e0175683 (2017).
Gao, L., Ye, M. & Wu, C. Cancer classification based on support vector machine optimized by particle swarm optimization and artificial bee colony. Molecules 22(12), 2086 (2017).
Li, L.-G., Yin, X. & Zhang, T. Tracking antibiotic resistance gene pollution from different sources using machine-learning classification. Microbiome 6(1), 93 (2018).
Eissa, M. A., Mihalopoulos, N. L., Holubkov, R., Dai, S. & Labarthe, D. R. Changes in fasting lipids during puberty. J Pediatr 170, 199–205 (2016).
Landis, J. R. & Koch, G. G. The measurement of observer agreement for categorical data. Biometrics 33(1), 159–174 (1977).
Mach, F. et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur. Heart J. 00, 1–78 (2019).
De Castro-Orós, I., Pocoví, M. & Civeira, F. The fine line between familial and polygenic hypercholesterolemia. Clin. Lipidol. 8(3), 303–306 (2013).
Ajufo, E. & Cuchel, M. Improving the yield of genetic testing in familial hypercholesterolaemia. Eur. Heart J. 38, 574–576 (2016).
Khera, A. V. et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J. Am. Coll. Cardiol. 67(22), 2578–2589 (2016).
Nordestgaard, B. G. et al. Quantifying atherogenic lipoproteins for lipid-lowering strategies: Consensus-based recommendations from EAS and EFLM. Atherosclerosis 294, 46–61 (2020).
Lagace, T. A. PCSK9 and LDLR degradation. Curr. Opin. Lipidol. 25(5), 387–393 (2014).
Beck, C. Assembly and Secretion of Atherogenic Lipoproteins (Göteborg University Sahlgrenska Academy, Göteborg, 2008).
van Schalkwijk, D. B. et al. Dietary medium chain fatty acid supplementation leads to reduced VLDL lipolysis and uptake rates in comparison to linoleic acid supplementation. PLoS ONE 9(7), e100376 (2014).
Sagoo, G. S. et al. Seven lipoprotein lipase gene polymorphisms, lipid fractions, and coronary disease: a huge association review and meta-analysis. Am. J. Epidemiol. 168(11), 1233–1246 (2008).
Teng, B., Sniderman, A. D., Soutar, A. K. & Thompson, G. R. Metabolic basis of hyperapobetalipoproteinemia. Turnover of apolipoprotein B in low density lipoprotein and its precursors and subfractions compared with normal and familial hypercholesterolemia. J. Clin. Invest. 77(3), 663–672 (1986).
van Schalkwijk, D. B. et al. Improved cholesterol phenotype analysis by a model relating lipoprotein life cycle processes to particle size. J. Lipid Res. 50(12), 2398–2411 (2009).
Guay, S.-P., Brisson, D., Lamarche, B., Gaudet, D. & Bouchard, L. Epipolymorphisms within lipoprotein genes contribute independently to plasma lipid levels in familial hypercholesterolemia. Epigenetics 9(5), 718–729 (2014).
Brunzell, J. D., Zambon, A. & Deeb, S. S. The effect of hepatic lipase on coronary artery disease in humans is influenced by the underlying lipoprotein phenotype. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1821(3), 365–372 (2012).
Acknowledgements
We are thankful to Francisco R. Pinto for his suggestions regarding the modelling approach and strategies for correction of overfitting. This work was supported by UIDB/04046/2020 Research Unit grant from FCT, Portugal (to BioISI). MC is recipient of a fellowship from the BioSys Ph.D. programme PD65-2012 (Ref PD/BD/114387/2016) from FCT (Portugal).
Author information
Authors and Affiliations
Contributions
M.C. implemented the dataset characterization and modelling approach, produced and discussed results, prepared all manuscript figures and data, and wrote the manuscript; E.K. contributed to the development of the modelling approach and to the interpretation of results; D.S. contributed to the development of the modelling approach, the interpretation of results and the writing of the manuscript; M.B. is responsible for the PFHS and the production of the respective biochemical and genetic data, she defined the problem to be addressed in this study and contributed to the discussion of the results and writing of the manuscript; M.G.C. defined the research approach, supervised the research work, analysed and interpreted the results, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Correia, M., Kagenaar, E., van Schalkwijk, D.B. et al. Machine learning modelling of blood lipid biomarkers in familial hypercholesterolaemia versus polygenic/environmental dyslipidaemia. Sci Rep 11, 3801 (2021). https://doi.org/10.1038/s41598-021-83392-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-83392-w
This article is cited by
-
International Atherosclerosis Society guidance for implementing best practice in the care of familial hypercholesterolaemia
Nature Reviews Cardiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
