
Abstract
Juvenile idiopathic arthritis (JIA) is one of the most common chronic diseases in children. While clinical outcomes for patients with juvenile JIA have improved, the underlying biology of the disease and mechanisms underlying therapeutic response/non-response are poorly understood. We have shown that active JIA is associated with distinct transcriptional abnormalities, and that the attainment of remission is associated with reorganization of transcriptional networks. In this study, we used a multi-omics approach to identify mechanisms driving the transcriptional abnormalities in peripheral blood CD4+ T cells of children with active JIA. We demonstrate that active JIA is associated with alterations in CD4+ T cell chromatin, as assessed by ATACseq studies. However, 3D chromatin architecture, assessed by HiChIP and simultaneous mapping of CTCF anchors of chromatin loops, reveals that normal 3D chromatin architecture is largely preserved. Overlapping CTCF binding, ATACseq, and RNAseq data with known JIA genetic risk loci demonstrated the presence of genetic influences on the observed transcriptional abnormalities and identified candidate target genes. These studies demonstrate the utility of multi-omics approaches for unraveling important questions regarding the pathobiology of autoimmune diseases.
Similar content being viewed by others
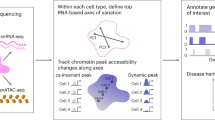


Introduction
Juvenile idiopathic arthritis (JIA) is a broad term that describes a clinically heterogeneous group of diseases characterized by chronic synovial hypertrophy and inflammation, with onset before 16 years of age1. JIA therefore represents an exclusion diagnosis that encompasses all forms of chronic childhood arthritis of an unknown cause. The reported prevalence of the disease is between 16 and 150 per 100,000 in the developed world, making it the most common rheumatic disease of childhood and one of the most common chronic illnesses in children2.
The causes of JIA are poorly understood, but both genetic and environmental factors are thought to play a role. A genetic component has been inferred from twin and affected sibling studies where concordance between monozygotic twins is between 25 and 40%3,4, and from numerous genome-wide association studies that demonstrate more than 40 regions associated with JIA 5,6,7,8,9. Like many complex diseases, many of the identified genetic variants that are associated with JIA are found outside of protein coding regions 8,10. It is possible that these variants alter the function of genomic regulatory elements, such as enhancers and promoters, which control gene expression and may contribute to disease initiation and progression. Our group has identified numerous transcriptional differences between JIA patients with active disease, those in clinical remission, and healthy controls in whole blood samples, peripheral blood mononuclear cells and neutrophils 11,12,13,14,15. In order to understand JIA pathogenesis, we believe that it is necessary to understand the events that lead to the transcriptomic alterations observed in children with JIA.
The control of transcription is a tightly choreographed process involving numerous cis- and trans- acting factors. Modifications to histone proteins may facilitate the recruitment of nucleosome remodeling complexes, which then evict nucleosomes and lead to remodeled chromatin16. Accessible chromatin can then be bound by sequence-specific transcription factors, which recruit co-factors, and mediate the three dimensional looping of DNA to bring distal regulatory elements into close physical contact with proximal promoters 16,17,18.
The CCCTC-binding factor (CTCF) is a sequence specific DNA binding factor, known for its role in establishing and maintaining long-range, three-dimensional chromatin interactions19. It was first categorized as an insulating factor, capable of preventing the interaction between enhancers and promoters in reporter constructs. More recently, CTCF has been shown to help establish topologically associated domains (TADs) and is involved in mediating promoter-enhancer interactions 20. Moreover, differential CTCF binding and the resulting differences in three-dimensional chromatin architecture have been implicated in disease pathogenesis for asthma and chemically induced cancers21,22,23.
Recent work has added an additional layer of complexity to transcriptional control. Transcriptional activation appears to occur within nuclear condensates, which contain high concentrations of transcription factors, co-factors and the general transcriptional machinery24,25,26,27,28. Moreover, single cell chromatin conformation capture studies have shown that one promoter or enhancer may interact simultaneously with multiple other promoters and enhancers29,30. These data suggest that multiple regulatory elements physically interact with each other in a hub-like complex, surrounded by high concentrations of transcription factors and transcriptional machinery 31.
Results
Widespread transcriptional changes in active disease and restoration with remission
We performed RNA-seq on a cohort of 32 patients and healthy controls. This was a cross-sectional study that included 12 patients with active designated as having “active disease, treated” (ADT), 10 patients, who met the definition of clinical remission on medication (CRM) 32,33,34, and 10 healthy controls (HC) (See “Methods”). We identified 19,691 genes that were expressed in any group (See methods). We performed differential gene expression analysis on every pair of groups, identifying 693 genes that were differentially expressed in peripheral blood CD4+ T cells between ADT and HC (Supp. Table 1), 533 genes that demonstrated expression differences between ADT and CRM (Supp. Table 2), and 52 genes that were expressed differently between HC and CRM (Fig. 1A, Supp. Table 3). Of interest was the small number of genes that reached a threshold for differential expression between HC and CRM. We have previously noted that the attainment of clinical remission did not result in normalization of neutrophil transcriptomes 11,12,13,14,15. In the CD4+ T-cells examined here, there was considerable normalization between healthy controls and patients in remission. The pattern of differential expression between ADT and CRM or between ADT and HC was very similar, with most of the genes identified as up-regulated in the ADT group.

Attainment of clinical remission in JIA normalizes gene expression in CD4+ T-cells. (A) Volcano plots for each of the pairwise comparisons of gene expression between groups. (B) Heatmap showing the expression of all genes that were considered differentially expressed between any two groups. Kmeans clustering was applied to separate genes into one of 5 clusters and hierarchal clustering was applied to cluster the groups.
`We plotted all differentially expressed genes called between any pair of sample groups and used k-means clustering on the normalized expression values to group the genes into different gene clusters (Fig. 1B, Supp. Table 4). We also performed hierarchical clustering of the gene profiles of the individual samples. We find, as expected based on the differential analysis, that the CRM and HC groups clustered together and discretely from the ADT group. The gene clusters grouped into 5 distinct subgroups, representing different cellular functions (Supp. Table 5). Cluster 1, which was characterized by increased expression in ADT compared to HC, is involved in translation, indicating that in the active disease state, CD4+ T-cells are more active in their transcriptional and translational processes. Cluster 2, representing genes that were identified as showing lower expression in the ADT group compared the HC and CRM groups, were enriched for genes involved in signaling and response to stimuli. Cluster 3, which is characterized by higher expression in HC samples compared to either CRM or ADT samples, is enriched for genes involved in protein targeting and transport. Cluster 4, characterized by lower expression in HC compared to ADT and CRM, is enriched for genes involved in immune activation. Cluster 5 contained genes that showed a wide variety of expression patterns across the samples but show a general increase in expression in the ADT samples compared to the CRM and HC samples; this cluster is represented by genes involved in chromatin organization and silencing.
Widespread differences in chromatin accessibility in active disease and remission
Having identified differentially expressed genes between the three conditions, we next sought to understand how the regulatory networks were perturbed and contributed to the differences in transcription. We performed the assay for transposase-accessible chromatin with sequencing 35 (ATAC-seq) on 6 HC, 5 ADT samples and 5 CRM samples, all of which were included in the RNAseq samples studied for gene expression. We identified 108,596 consistent peaks in our HC samples, 223941consistent peaks in our ADT samples and 103,612 consistent peaks in our CRM samples (Fig. 2A; see “Methods”). Of these, 45,911 peaks were shared by all three groups, indicating a significant level of chromatin reorganization associated with disease status. This data supports the transcriptomic data, in that there are significant differences in the expression and regulation of genes between JIA patients and healthy children. We also found considerable differences in the genomic accessibility of CRM patients versus healthy controls, indicating a high level of chromatin reorganization in remission despite modest changes in gene expression.

Chromatin re-arrangement occurs in the absence of three-dimensional changes. (A) Overlap between the group-consistent accessible peaks identified in HC, ADT and CRM samples. (B) Volcano plot showing the differences in CTCF binding affinity between 5 HC samples and 5 ADT samples. Blue dots represent sites whose log10(fold-change) was at least 1 and whose FDR was less than or equal to 0.05.
Genetic variation in CTCF binding sites affects transcription of adjacent genes but does not seem crucial for the differences in expression data in active disease and healthy controls
CTCF is a critical genomic architectural protein that, with the aid of cofactors such as the cohesion complex, acts to mediate long-range chromatin interactions. These interactions are important for the establishment of regulatory domain boundaries, for mediating promoter-enhancer interactions, and the in the formation of insulator elements 20. There is also evidence that altered CTCF binding contributes to human disease 21,22,23. We hypothesized that differential CTCF binding, and the perturbed three-dimensional architecture that would result, may contribute to the transcriptional changes that we observed in the active disease. To test this hypothesis, we performed CTCF ChIP-seq on 10 patients not included in the first ATACseq and RNAseq studies. These included five ADT patients and 5 healthy controls. To directly investigate the three dimensional topology that is mediated by CTCF, we performed CTCF HiChIP on 6 participants, including 3 ADT patients and 3 healthy controls, of which one participant in each group was included in the CTCF ChIP-seq studies36. Using DiffBind37 to identify differential binding events, we identified 31 differentially bound CTCF sites using a threshold of FDR ≤ 0.05 and log10(fold-change) > = 1 (Fig. 2B, Supp. Table 7). Moreover, none of these sites were located in the LD blocks around JIA associated variants 9.
We further characterized two of the differentially bound CTCF sites, whose log10(counts per million) > 5 and whose log10(fold-change) was > 4 (Supp. Table 7). Using HiChIP, we characterized a statistically significant gain of binding event, located in the intergenic region between the promoters of LCOR and PIK3AP1 on chromosome 10 (Supp. Figure 1A), where CTCF binding occurred in some of the ADT group and not in the HC group. The site showed binding of CTCF in three out of the five ADT samples and none of the healthy controls (Supp. Figure 1B). The loops that were anchored on this site in the ADT patients interacted with several differentially expressed genes (Supp. Figure 1C). To determine whether genetic variations may be driving the binding differences observed between the conditions, we performed Sanger sequencing38,39 on all samples across the identified locus. We found that all three ADT samples that had CTCF binding at the locus had a C allele at position chr10:98549577, whereas every sample that did not show CTCF binding, including all 5 HC samples and 2 of the 5 ADT samples, had the reference T allele at that location (Supp. Figure 1B). The gain of binding allele has been previously annotated as rs7477274. These data indicate that CTCF binding at this site is driven primarily by genetic variation.
We next identified a loss of binding event located in the promoter region of RNF135 on chromosome 17 (Supp. Figure 2A). The loops that are anchored on this site in healthy samples interact with the promoter regions of the differentially expressed genes (Supp. Figure 2C) and non-differentially expressed genes. The three HC samples that have CTCF binding at this site have a G allele at position chr17: 29297397, annotated as rs7221217 (Supp. Figure 2B), which was confirmed through Sanger sequencing. Of the three samples with CTCF binding, two were homozygous and one was heterozygous (Supp. Figure 2B). The two HC samples without binding at this site, as well as all five ADT samples that have no CTCF binding, were all homozygous with the reference A allele (Supp. Figure 2B). This data indicates this loss of binding is mediated by genetic variation.
Overall, this data shows that differential CTCF binding and altered three-dimensional interactions occur and are associated with genetic variants and with differentially expressed genes. However, the rarity of the events and the fact that none occur in known JIA risk loci indicate it is unlikely to be a driving mechanism of transcriptional aberrations in in CD4+ T-cells in JIA.
Identifying interaction domains with CTCF ChIP-seq and HiChIP
Our profiling of the CD4+ T cell genome has yielded several features not previously recognized. We observed widespread transcriptional differences in CD4+ T cells in the comparison between active JIA and healthy controls, widespread changes in chromatin accessibility between all three conditions. We did not observe widespread changes in CTCF binding and, therefore, in the three dimensional structure of the genome. We thus hypothesize that due to the lack of three dimensional interaction differences, the variability in gene expression patterns must be a function of the variability in the activity of regulatory elements. However, due to the small expression changes observed between healthy controls and patients in remission and the widespread changes in accessibility between those groups, we further hypothesize that only a small fraction of the regulatory variability accounts for the differences in expression. We therefore focused next on previously identified JIA risk loci and employed a novel method based on emerging models of gene regulation to identify the regulatory elements and genes driving the observed genomic differences.
Recent studies, as noted previously, have suggested that transcription occurs within “regulatory hubs”, containing high concentrations of transcriptional machinery, and multiple genomic regions that physically interact with each other24,25,26,27,28,29,30,31. Additional studies have shown that genomic regulatory regions could simultaneously interact with numerous other elements and gene regions29,30,31. . Our data identify multiple regions connected to one another through chains of CTCF-mediated loops (Fig. 3). These observations challenge the paradigm that transcriptomic regulation is mediated exclusively by differential three-dimensional contacts31. Instead, numerous regulatory elements and gene promoters interact together in regulatory hubs, with intra-hub regulation occurring through a mechanism other than simple physical proximity. With this concept in mind, we attempted to understand transcriptomic regulation by modeling the expression of a gene as a function of the accessibility of all regulatory elements that physically interacted with the gene promoter and used feature selection to identify elements contributing to variability in the gene’s expression (see methods).

Identifying regulatory hubs. Snapshot showing the regulatory hub anchored on JIA risk variants rs6946509, rs7808122, rs10280937 and rs73300638. Top row shows LD blocks of the risk variants, followed by averaged ATAC-seq, RNA-seq, CTCF ChIP-seq signal across the three sample groups, as well as the HiChIP loops for the healthy control and active disease groups, followed by differentially expressed genes and all RefSeq genes.
Genome-wide association studies (GWAS) and genetic fine mapping studies have identified at least 46 genetic loci that are associated or suggestively associated with JIA7,40. Linkage disequilibrium blocks, genomic regions with highly correlated patterns of genetic heritability, are enriched for histone marks associated with regulatory activity 9. We found that 33 out of 46 of LD blocks around previously identified JIA genetic risk loci7,9,40 had regulatory elements and CTCF loop anchors located within them (Fig. 3). This indicates that some of the genetic risk for JIA may act by affecting CD4+ T-cell function.
Starting with the 33 JIA risk loci that contained CTCF loop anchors, we identified the regulatory hubs of which they were a part (Fig. 3) and predicted which regulatory elements controlled the gene’s expression (see “Methods”). We then considered any regulatory element that was within the JIA associated LD block as a putative casual element, and that any genes that were predicted to be controlled by that element as putative target genes for the JIA risk locus (Table 1). The total set of predicted target genes showed strong enrichment for gene ontology (GO) terms such as antigen presentation and T cell regulation and proliferation, while differentially expressed predicted target genes showed enrichment for cell signaling and gene regulation pathways (Supp.Tables 2–3). This enrichment for immune system pathways supports our predictions for JIA target genes. We believe that these putative targets and casual elements represent a starting point for understanding the genomic pathogenesis of JIA.
Discussion
In this study, we observed widespread changes in gene expression profiles and chromatin accessibility in CD4+ T cells of patients with active JIA when we compared them to those of healthy controls. These differences reveal several important features of JIA. The first is that the transcriptional abnormalities that are observed in other cell types, such as neutrophils 11,14, PBMCs41 and whole blood samples 15 are present within the CD4+ T cell compartment. It is likely that the abnormalities reflect differences within similar cell populations between disease states, as opposed to differences in the proportions of naïve, memory or regulatory T cell subtypes, as evidenced by recent work showing little change in cell subtype ratios between patients with JIA and controls42,43. Second, the transcriptional differences reflect activation of peripheral blood immune components, which implicates a global immune dysfunction, as opposed to an isolated process involving the joints or resident lymphocytes within the synovium44.
We have shown that the CD4+ T cells of children with JIA display a three dimensional genomic architecture that is similar to that in healthy children. Alterations in CTCF binding, and the corresponding changes in loop structures, are observed and are associated with genetic variants, but are rare events that are not associated with any known JIA genetic risk locus. These findings, combined with the widespread changes in chromatin accessibility that we observed, point to a mechanism where disrupted regulatory elements may drive gene expression changes, rather than a rewiring of the three dimensional genome.
These features pose a critical question to our understanding of JIA: does this activation of peripheral immune cells simply reflect the brisk inflammatory milieu of the synovial tissues or is it an intrinsic immune dysfunction? Recently, researchers have observed an enhanced IFNγ signaling phenotype in the CD4+ T cells of active JIA patients 43, suggesting that an intrinsic dysfunction is occurring. We provide further evidence of an intrinsic dysfunction by showing that more than half of JIA associated genetic risk LD blocks contain T-cell specific regulatory elements that physically interact with other regulatory elements and gene promoters. Leveraging this feature allowed for the prediction of numerous putative target genes, which may have pathogenic roles in JIA.
One of the surprising findings from this work was the relative normalization in expression patterns that we observed in CD4+ T cells from patients who have achieved clinical remission while on MTX and etanercept. This stands in contrast to what we have observed in other cell populations, especially neutrophils 41. This leads us to an interesting hypothesis concerning CD4+ T-cells in JIA and their role in the disease. We hypothesize that CD4+ T-cells are “primed” for a hyper-inflammatory response, due to the effect of genetic variations on the expression of our predicted target genes. When CD4+ T-cells interact with another cell type or with molecules secreted by other cells, they initiate an inflammatory response, which progresses unchecked to an elevated level. We hypothesize that pharmacological intervention acts by disrupting the extracellular signaling, returning the CD4+ T-cells to a resting, but “primed” state, while other cell types remain altered.
We recognize that the present study has several limitations. The first is the fact that this is a cross-sectional study and therefore it is not known whether the differences in chromatin organization that we observed change through the course of disease progression or through treatment response. Another limitation of this study is the small number of patients and the fact that the same assays were not performed on every subject. These limitations largely reflect the constraints of undertaking translational studies in a pediatric population. Given the limited sample volumes that can be obtained from children, investigators are limited in the number of studies that can be performed on any individual patient sample. We are sanguine, however, that continued technologic advances will allow us to perform more complex studies on larger groups of patients in the near future. However, the validity of our approach is shown in the fact that that datasets from one group of participants corroborated the inferences made with an independent set of participants.
Methods
Patients and patient samples
Patients were recruited from the pediatric rheumatology clinic at the Women & Children’s Hospital of Buffalo. All children fit criteria for polyarticular-onset, rheumatoid factor-negative JIA as established by the International League Against Rheumatism (ILAR) 45. Patients included 18 girls and 4 boys who ranged in age from 4 to 16 years. Children with JIA were classified as having active disease on therapy (ADT) or clinical remission on medication (CRM) as determined by standard criteria 32,33,34. Children with JIA were all taking combinations of methotrexate and the TNF inhibitor, etanercept. Samples were obtained from children classified as ADT within 6 weeks after initiating therapy. All children classified as ADT had at least 4 warm, swollen joints. Samples obtained from children with CRM were obtained at the first clinic visit at which CRM status was confirmed, typically 12–15 months after the initial diagnosis.
Healthy control (HC) children (n = 10) were recruited from the Hodge General Pediatrics Clinic of the University at Buffalo Jacobs School of Medicine and Biomedical Sciences and ranged in age from 5 to 16 years and included two boys and eight girls. Children were excluded if they had fever ≥ 38 °C within the previous 48 h, had another autoimmune disease (e.g., type 1 diabetes), or were taking either systemic glucocorticoids or antibiotics for any reason.
University at Buffalo IRB approval was obtained for this study, and informed consent documents executed with the parents of all patients and controls. For children over the age of 7 years, age-appropriate assent documents were also executed. All research procedures were carried out in compliance with the IRB-approved protocol.
Blood was drawn into CPT tubes (Beckton-Dickinson # BD362761) and brought immediately to the pediatric rheumatology research laboratory. Sample processing was begun within an hour of obtaining the sample.
CD4+ T cells
CD4+ T cells were purified from whole blood by negative selection using the StemSep™ Human CD4+ T Cell Enrichment Kit (STEMCELL Technologies Inc., Vancouver, Canada) as previously described 46.
RNA purification and sequencing
Total RNA was extracted using TRIzol™ reagent and was further purified using RNeasy MiniElute Cleanup kit, including a DNase digest as previously described 46. RNA was quantified spectrophotometrically (Nanodrop, Thermo Scientific, Wilmington, DE) and assessed for quality by capillary gel electrophoresis (Agilent 2100 Bioanalyzer; Agilent Technologies, Inc., Palo Alto, CA). cDNA libraries were prepared for each sample using the Illumina TruSeq RNA Sample Preparation Kit following the manufacture’s recommended procedures. Libraries were sequenced using 100 base pair (bp) paired-end reads on the Illumina HiSeq 2500 platform. Library construction and RNASeq were performed at the University at Buffalo Genomics and Bioinformatics Core.
ATAC-seq on CD4+ T cells
The assay for transposase-accessible chromatin sequencing (ATAC-seq) was carried out according to Buenrostro’s protocol 47. To prepare nuclei, 50,000 CD4+ T cells were spun at 500 × g for 5 min, followed by a wash using 50 μL of cold 1 × PBS and centrifugation at 500 × g for 5 min. Cells were lysed using cold lysis buffer (10 mM Tris–Cl, pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.1% IGEPAL CA-630). Immediately after lysis, nuclei were spun at 500 × g for 10 min using a refrigerated centrifuge. Immediately following the nuclei prep, the pellet was resuspended in the transposase reaction mix (25 μL 2 × TD buffer, 2.5 μL Transposase (Illumina) and 22.5 μL of nuclease free water). The transposition reaction was carried out for 30 min at 37 °C. Directly following transposition the sample was purified using a Qiagen Minelute kit. Following purification, library fragments were amplified using 1 × NEBnext PCR master mix and 1.25 μM of PCR primer1 and Barcoded PCR Primer 2, using the following PCR conditions: 72 °C for 5 min, 98 °C for 30 s, followed by thermocycling at 98 °C for 10 s, 63 °C for 30 s and 72 °C for 1 min. To reduce GC and size bias in our PCR we monitored the PCR reaction using qPCR in order to stop amplification prior to saturation. To do this, the full libraries were amplified for 5 cycles, after 5 cycles , 5 μl of PCR reaction was added 10 μl of the PCR cocktail with Sybr Green at a final concentration of 0.6x. This reaction was run for 20 cycles, to determine the additional number of cycles needed for the remaining 45 μl reaction. To calculate the additional number of cycles needed, we plotted linear Rn versus cycle and to determine the cycle number that corresponded to one-third of the maximum fluorescent intensity. The libraries were purified using AMPure XP beads yielding a final library 17.5 μl. Sequencing was performed using 50 bp paired-end sequencing on the Illumina HiSeq 2500 platform at the University at Buffalo Genomics and Bioinformatics Core.
ChIP-seq for CTCF
The ChIP assay was carried out using a kit from Diagenode (Denville, NJ). Briefly, Purified CD4+ T cells were fixed in 1% formaldehyde (J.T.Baker) in PBS for 10 min at room temperature, followed by 5 min blocking in 125 mM glycine. Cells were rinsed two times in ice-cold PBS and pelleted (500 g, 10 min, 4 °C). All fixed cell pellets were stored at − 80 °C. Approximately four million cells were resuspended in 0.5 ml of nuclei extraction buffer (5 mM PIPES pH 8; 85 mM KCl; 0.5% Igepal CA-630) supplemented with Protease Inhibitor Cocktail (Cell Signaling Technologies). Cell suspensions were sonicated using Bioruptor Plus (Diagenode) at low power with four cycles (15 s on and 30 s off). Samples were centrifuged (1000 g, 5 min, 4 °C) and washed once in 1 ml of the nuclei extraction buffer. Isolated nuclei were resuspended in the shearing buffer supplemented with Protease Inhibitor Cocktail and sonicated using Bioruptor Plus at high power with 25 cycles (30 s on and 30 s off; water temperature 4 °C) to reach a fragment size distribution of 100–700 bp. ChIP reactions were carried out in a final volume of 320 μl (70 ul washed Protein A-coated magnetic beads with antibody and 250 ul of sheared chromatin). 5 μl of sheared chromatin is stored at 4 °C as input sample. The antibody against CTCF was acquired from Diagenode (Cat No C15410210, Lot No A2359-00234p). After immunoprecipitation (IP) overnight at 4 °C, the beads were washed sequentially with different wash buffer, followed by incubation with elution buffer. Finally, the decross-linked DNA was resuspended in 25 ul buffer. Then DNA-sequencing was conducted using the Illumina HiSeq 2500 at the University at Buffalo Genomics and Bioinformatics Core.
HiChIP for CTCF
CD4+ T cells were cross-linked using 1% formaldehyde for ten minutes at room temperature and then quenched by 125 mM glycine for five minutes. Four million cross-linked cells per sample were used in HiChIP assays as described in Mumbach et al., 2016 with some modifications 36. Briefly, nuclei were isolated and subjected to in situ digestion using MboI for 4 h at 37 °C. The cutting sites were then filled and labeled with dCTP, dGTP, dTTP and biotin-dATP. In situ proximity ligation was performed at room temperature overnight. After fragmentation of the DNA by sonication for two minutes, chromatin was immunoprecipitated using CTCF antibody (Cell Signaling, #3418S). DNA was eluted from the beads and purified by Zymo DNA Clean & Concentrator Column. Streptavidin M-280 Dynabeads were used to capture biotin-labeled DNA fragments. The sequencing libraries were generated on the streptavidin beads using the Nextera DNA Library Prep Kit.
Sanger sequencing
DNA from whole blood was purified using DNeasy Blood & Tissue Kit (Qiagen, USA). DNA sequencing was performed in the Roswell Park Comprehension Cancer Institute.
Data processing: aligning high-throughput sequencing reads to the reference genome
All RNA-seq, ATAC-seq and ChIP-seq read files, in FASTQ format, were aligned to the hg19 reference genome using bowtie248. All of the files were run through bowtie2 using default parameters. RNA-seq and ATAC-seq data were run using paired-end mapping options (-1 and -2 options), while the CTCF ChIP-seq data was run using the unpaired options (-U option). HiChIP raw reads (fastq files) were aligned to hg19 human reference genome using HiC-Pro49, which was augmented with positions of Mbo1 restriction fragment cut sites for identifying alignment end sites. The resulting SAM files, outputted by HiC-Pro or bowtie2, were converted into BAM file formats using samtools 50.
Data processing: RNA-seq analysis
After the data was aligned to the reference genome and converted into BAM format, the RNA-seq data was used to create a count table for differential analysis. For every gene, the number of RNA-seq reads for each sample mapping to that gene were counted using HT-Seq 51. HT-Seq was run using default parameters, using the GENCODE 52 version-19 gene annotations. This count table was then used as the input into the edgeR package for differential gene expression analysis 53.
Differential gene expression was identified using edgeR’s common dispersion protocol. A gene was considered expressed if at least five samples had a counts-per-million (CPM) value of at least 1. 14,357 genes were considered expressed under these conditions, in any of the three groups. edgeR was then run on the remaining list of expressed genes, first by calculating the normalization factors (calcNormFactors()), followed by calculating the common and then tag-wise dispersions (estimateCommonDisp(), estimateTagwiseDisp()), followed by an exact test and FDR correction (extactTest(), decideTestsDGE(,adjust.method = ”BH”,p.value = 0.05) to determine genes that were differentially expressed.
The volcano plots for differential gene expression were made by plotting the fold-change and p-value for every expressed gene, with genes whose p-value was ≤ 0.05 and whose log fold change was ≥ 1 colored blue and those genes not matching these criteria colored red. The heat-map was created by identifying all genes that were considered differentially expressed in any of the three pairwise comparisons (4783 genes were considered), and clustering them based on their log CPM values into five clusters with k-means clustering. The individual samples were clustered with hierarchal clustering. Genes belonging to the five individual clusters were used for GO term analysis using GOrilla 54. GOrilla was run using the two gene set option, with the cluster genes as the input and the total list of expressed genes as the background set.
Data processing: ChIP-seq and HiChIP analysis
After the ChIP-seq data was aligned to the reference genome and converted to BAM format, those BAM files were used as the input into MACS2 55 for identifying CTCF binding sites. Each IP file had a matching input file that was used as inputs into MACS2. The q-value threshold for reporting peaks from MACS2 was set to 0.05 (option –q 0.05), which is the default value. Once peaks were called, those peaks were used as inputs into the DiffBind software37. For each sample, DiffBind takes the ChIP BAM files, the control input BAM files and the peaks from MACS2 as inputs. In total, DiffBind identified 61 CTCF binding sites that were considered differentially bound (FDR ≤ 0.1). The volcano plot showing the fold-change vs p-values (Fig. 2B) showed a possible artifact, evidenced by the diagonal line of points on the right side of the figure. Due to this, none of those sites were included in any further analysis and were not characterized with additional assays.
HiC-Pro alignments, in BAM format, were input into HiChipper56 using mostly default parameters. Mbo1 restriction fragment cut sites and CTCF peaks generated above, that were present in at least one sample, were input as anchor locating guides. Reads contributing to anchors were required to have a mapping quality of MAPQ ≥ 20.
Once the HiChIP loops had been identified, several post processing steps were conducted. We first sought to retain only those loops whose anchors were at valid, group-specific binding sites. To identify these we first took all ADT or HC CTCF peaks from MACS2 and determined consensus peaks for each group by finding sites that were considered peaks in at least 60% of samples in the group. Then every HiChIP loop, in each sample, was checked to see if both of its anchors were located at a consensus peak. If the loop was not anchored on both ends at a consensus peak, it was removed from further analysis.
Data processing: ATAC-seq analysis
The aligned BAM files for the ATAC-seq data were first subjected to preprocessing steps to generate input files for peak calling. The unsorted BAM files were first sorted and then indexed using samtools (samtools sort followed by samtools index) 50. Bedtools 57 was used to convert the sorted and indexed BAM files into a bed file (bedtools bamtobed) and these bed files were then used to create a genome wide bigwig file of read coverage, first with bedtools and then with UCSC’s bedGraphToBigWig tools (bedtools genomecov –bga followed by bedGraphToBigWig). The sorted BAM files, the BAM index files and these BigWig files were used as inputs into the ATAC-seq peak calling algorithm, HMMRATAC 58. Additionally, a genome size file was downloaded from UCSC’s genome browser as was a list of blacklisted sites for the hg19 genome annotation (wgEncodeDacMapabilityConsensusExcludable.bed with the –e option in HMMRATAC) 59,60. HMMRATAC was run with the upper and lower range settings for finding candidate peaks to build the model, set to 20 and 10 respectively (-u 20 and –l 10 options). For each sample, HMMRATAC outputted a peak file, with the locations of every accessible region, the top 100,000 of which were kept for further analysis. Consistent peaks were considered to be genomic regions that were identified as a peak in at least 60% of the group’s samples. A total of 108,596 consistent HC peaks, 223,941 consistent ADT peaks and 103,612 consistent CRM peaks were identified. GO term enrichment for any group of peaks was determined by using GREAT61, with default settings.
Data processing: identifying linkage disequilibrium blocks for JIA risk loci
Zhu et al. had previously identified the linkage disequilibrium blocks for 35 JIA risk associated SNPs 9. Additional risk loci have been identified since and we aimed to incorporate those into our analysis7,40. Because the method used to identify the LD blocks from Zhu et al. is no longer maintained, we processed all JIA risk SNPs using SNiPA, using the same settings for each SNP 62. Using the “Block Annotation” tool from the SNiPA suite, we ran each SNP using the GRCh37 genome assembly, the 1000 Genomes, Phase 3 v5 variant set, set the population to European and the genome annotation to Ensembl 87. Each block used an r2 of 0.8 to identify linkage disequilibrium.
Data processing: identifying interaction domains around JIA risk loci
ATAC-seq, ChIP-seq and RNA-seq data was converted from raw BAM files into BigWig files and then merged together and averaged. HiChIP loops were converted into a paired end BED format and filtered to keep only those loops with at least 2 reads supporting it. All of this data, along with a BED file of JIA risk associated LD blocks from Zhu et al. 9, was then uploaded onto the WashU genome browser 63 for visualization. Starting with each LD block, we determined whether the LD region contained a CTCF loop anchor. If not, then the LD block was considered to be inactive in CD4+ T cells and was excluded from further analysis. If the LD blocks that contained one or more CTCF loop anchors, we then determined all genomic regions that physically interacted with the block. We then determined all regions that interacted with those anchors, and so forth, until we had identified all regions that could come into close physically proximity to the LD block. An example of a positive region is shown in Fig. 3.
Data processing: determining enhancer-TSS links within regulatory hubs
Having defined all regulatory hubs that are anchored on a JIA risk associated locus, we sought to determine which enhancers within a hub controlled the expression of which genes. Using HT-seq 51, we counted the number of RNA-seq reads that mapped to each gene for each sample and the number of ATAC-seq reads that mapped to each identified peak for each matched sample, i.e. those samples where there was both RNA-seq and ATAC-seq data. We then converted those read counts into log(Counts per million) using edgeR. Then, for each regulatory hub, we constructed a matrix of the log(CPM) for RNA-seq reads for each gene in the hub and log(CPM) for ATAC-seq reads for each peak in the hub. We then perform feature selection with Boruta64, modeling the expression of every gene (Y) as a function of the expression level of all other genes in the hub (T) as well as the accessibility (A) of all ATAC-seq peaks (P) within the hub.
Our modeling approach is similar to recent studies that have used the level of accessibility of regulatory elements to predict enhancer-promoter interactions65,66. Because of the stochastic nature of the Boruta algorithm, we ran it 50 times per gene and only retained features that were considered significant in at least 95% of the runs. Finally, we focused on those genes whose features were located within the JIA risk associated LD blocks.
References
Ravelli, A. & Martini, A. Juvenile idiopathic arthritis. Lancet (London, England) 369, 767–778. https://doi.org/10.1016/s0140-6736(07)60363-8 (2007).
Singsen, B. H. Rheumatic diseases of childhood. Rheum. Dis. Clin. N. Am. 16, 581–599 (1990).
Ansell, B. M., Bywaters, E. G. & Lawrence, J. S. Familial aggregation and twin studies in Still’s disease. Juvenile chronic polyarthritis. Rheumatology 2, 37–61 (1969).
Prahalad, S. et al. Twins concordant for juvenile rheumatoid arthritis. Arthritis Rheum. 43, 2611–2612. https://doi.org/10.1002/1529-0131(200011)43:11%3c2611::aid-anr33%3e3.0.co;2-t (2000).
Ozen, S. et al. A pilot study for genome wide association study (GWAS) in patients with juvenile idiopathic arthritis (JIA) and their parents. Pediatr. Rheumatol. 9, P152 (2011).
Thompson, S. D. et al. Genome-wide association analysis of juvenile idiopathic arthritis identifies a new susceptibility locus at chromosomal region 3q13. Arthritis Rheum. 64, 2781–2791. https://doi.org/10.1002/art.34429 (2012).
Hinks, A. et al. Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat. Genet. 45, 664–669. https://doi.org/10.1038/ng.2614 (2013).
Wong, L., Jiang, K., Chen, Y. & Jarvis, J. N. Genetic insights into juvenile idiopathic arthritis derived from deep whole genome sequencing. Sci. Rep. 7, 2657. https://doi.org/10.1038/s41598-017-02966-9 (2017).
Zhu, L. et al. Chromatin landscapes and genetic risk for juvenile idiopathic arthritis. Arthritis Res. Ther. 19, 57. https://doi.org/10.1186/s13075-017-1260-x (2017).
Jiang, K. et al. Disease-associated single-nucleotide polymorphisms from noncoding regions in juvenile idiopathic arthritis are located within or adjacent to functional genomic elements of human neutrophils and CD4+ T cells. Arthritis Rheumatol. (Hoboken, N.J.) 67, 1966–1977, https://doi.org/10.1002/art.39135 (2015).
Jarvis, J. N. et al. Evidence for chronic, peripheral activation of neutrophils in polyarticular juvenile rheumatoid arthritis. Arthritis Res. Ther. 8, R154. https://doi.org/10.1186/ar2048 (2006).
Knowlton, N. et al. The meaning of clinical remission in polyarticular juvenile idiopathic arthritis: Gene expression profiling in peripheral blood mononuclear cells identifies distinct disease states. Arthritis Rheum. 60, 892–900. https://doi.org/10.1002/art.24298 (2009).
Jiang, K. et al. Whole blood gene expression profiling predicts therapeutic response at six months in patients with polyarticular juvenile idiopathic arthritis. Arthritis Rheumatol. (Hoboken, N.J.) 66, 1363–1371, https://doi.org/10.1002/art.38341 (2014).
Jiang, K., Sun, X., Chen, Y., Shen, Y. & Jarvis, J. N. RNA sequencing from human neutrophils reveals distinct transcriptional differences associated with chronic inflammatory states. BMC Med. Genomics 8, 55. https://doi.org/10.1186/s12920-015-0128-7 (2015).
Jiang, K. et al. Whole blood expression profiling from the TREAT trial: Insights for the pathogenesis of polyarticular juvenile idiopathic arthritis. Arthritis Res. Ther. 18, 157. https://doi.org/10.1186/s13075-016-1059-1 (2016).
Venters, B. J. & Pugh, B. F. How eukaryotic genes are transcribed. Crit. Rev. Biochem. Mol. Biol. 44, 117–141. https://doi.org/10.1080/10409230902858785 (2009).
Dekker, J., Marti-Renom, M. A. & Mirny, L. A. Exploring the three-dimensional organization of genomes: Interpreting chromatin interaction data. Nat. Rev. Genet. 14, 390–403. https://doi.org/10.1038/nrg3454 (2013).
Tsompana, M. & Buck, M. J. Chromatin accessibility: A window into the genome. Epigenet.Chromatin 7, 33. https://doi.org/10.1186/1756-8935-7-33 (2014).
Ghirlando, R. & Felsenfeld, G. CTCF: Making the right connections. Genes Dev. 30, 881–891. https://doi.org/10.1101/gad.277863.116 (2016).
Merkenschlager, M. & Odom, D. T. CTCF and cohesin: Linking gene regulatory elements with their targets. Cell 152, 1285–1297. https://doi.org/10.1016/j.cell.2013.02.029 (2013).
Schmiedel, B. J. et al. 17q21 asthma-risk variants switch CTCF binding and regulate IL-2 production by T cells. Nat. Commun. 7, 13426. https://doi.org/10.1038/ncomms13426 (2016).
Norton, H. K. & Phillips-Cremins, J. E. Crossed wires: 3D genome misfolding in human disease. J. Cell Biol. 216, 3441–3452. https://doi.org/10.1083/jcb.201611001 (2017).
Rea, M., Gripshover, T. & Fondufe-Mittendorf, Y. Selective inhibition of CTCF binding by iAs directs TET-mediated reprogramming of 5-hydroxymethylation patterns in iAs-transformed cells. Toxicol. Appl. Pharmacol. 338, 124–133. https://doi.org/10.1016/j.taap.2017.11.015 (2018).
Boehning, M. et al. RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat. Struct. Mol. Biol. 25, 833–840. https://doi.org/10.1038/s41594-018-0112-y (2018).
Boija, A. et al. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175, 1842-1855.e1816. https://doi.org/10.1016/j.cell.2018.10.042 (2018).
Cho, W. K. et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415. https://doi.org/10.1126/science.aar4199 (2018).
Chong, S. et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science https://doi.org/10.1126/science.aar2555 (2018).
Sabari, B. R. et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science https://doi.org/10.1126/science.aar3958 (2018).
Allahyar, A. et al. Enhancer hubs and loop collisions identified from single-allele topologies. Nat. Genet. 50, 1151–1160. https://doi.org/10.1038/s41588-018-0161-5 (2018).
Oudelaar, A. M., Davies, J. O. J., Downes, D. J., Higgs, D. R. & Hughes, J. R. Robust detection of chromosomal interactions from small numbers of cells using low-input Capture-C. Nucleic Acids Res. 45, e184. https://doi.org/10.1093/nar/gkx1194 (2017).
Oudelaar, A. M. et al. A revised model for promoter competition based on multi-way chromatin interactions. bioRxiv 612275, https://doi.org/10.1101/612275 (2019).
Wallace, C. A. The use of methotrexate in childhood rheumatic diseases. Arthritis Rheum. 41, 381–391. https://doi.org/10.1002/1529-0131(199803)41:3%3c381::aid-art2%3e3.0.co;2-3 (1998).
Wallace, C. A., Bleyer, W. A., Sherry, D. D., Salmonson, K. L. & Wedgwood, R. J. Toxicity and serum levels of methotrexate in children with juvenile rheumatoid arthritis. Arthritis Rheum. 32, 677–681 (1989).
Wallace, C. A. et al. Trial of early aggressive therapy in polyarticular juvenile idiopathic arthritis. Arthritis Rheum. 64, 2012–2021. https://doi.org/10.1002/art.34343 (2012).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. https://doi.org/10.1038/nmeth.2688 (2013).
Mumbach, M. R. et al. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922. https://doi.org/10.1038/nmeth.3999 (2016).
Stark R, B. G. DiffBind: Differential binding analysis of ChIP-Seq peak data. Bioconductor. http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf (2011).
Sanger, F. & Coulson, A. R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 94, 441–448 (1975).
Sanger, F., Nicklen, S. & Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A. 74, 5463–5467 (1977).
McIntosh, L. A. et al. Genome-wide association meta-analysis reveals novel juvenile idiopathic arthritis susceptibility loci. Arthritis Rheumatol. (Hoboken, N.J.) 69, 2222–2232, https://doi.org/10.1002/art.40216 (2017).
Jiang, K., Frank, M., Chen, Y., Osban, J. & Jarvis, J. N. Genomic characterization of remission in juvenile idiopathic arthritis. Arthritis Res. Ther. 15, R100. https://doi.org/10.1186/ar4280 (2013).
Wu, S. A., Yeh, K. W., Lee, W. I., Yao, T. C. & Huang, J. L. Persistent improper upregulation of Th17 and TReg cells in patients with juvenile idiopathic arthritis. J. Microbiol. Immunol. Infect. (Wei mian yu gan ran za zhi) 49, 402–408, https://doi.org/10.1016/j.jmii.2014.07.002 (2016).
Throm, A. A. et al. Identification of enhanced IFN-gamma signaling in polyarticular juvenile idiopathic arthritis with mass cytometry. JCI insight https://doi.org/10.1172/jci.insight.121544 (2018).
Park, C. O. & Kupper, T. S. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat. Med. 21, 688–697. https://doi.org/10.1038/nm.3883 (2015).
Petty, R. E. et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J. Rheumatol. 31, 390–392 (2004).
Wong, L. et al. Limits of peripheral blood mononuclear cells for gene expression-based biomarkers in juvenile idiopathic arthritis. Sci. Rep. 6, 29477, https://doi.org/10.1038/srep29477, https://www.nature.com/articles/srep29477-supplementary-information (2016).
Buenrostro, J. D., Wu, B., Chang, H. Y. & Greenleaf, W. J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21.29.21–29, https://doi.org/10.1002/0471142727.mb2129s109 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Servant, N. et al. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259. https://doi.org/10.1186/s13059-015-0831-x (2015).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. https://doi.org/10.1093/bioinformatics/btp352 (2009).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. https://doi.org/10.1093/bioinformatics/btu638 (2015).
Harrow, J. et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. https://doi.org/10.1101/gr.135350.111 (2012).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. https://doi.org/10.1093/bioinformatics/btp616 (2010).
Eden, E., Navon, R., Steinfeld, I., Lipson, D. & Yakhini, Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 10, 48. https://doi.org/10.1186/1471-2105-10-48 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. https://doi.org/10.1186/gb-2008-9-9-r137 (2008).
Lareau, C. & Aryee, M. hichipper: A preprocessing pipeline for assessing library quality and DNA loops from HiChIP data. bioRxiv http://biorxiv.org/content/early/2017/09/21/192302.abstract (2017).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. https://doi.org/10.1093/bioinformatics/btq033 (2010).
Tarbell, E. D. & Liu, T. HMMRATAC: A Hidden Markov ModeleR for ATAC-seq. Nucleic Acids Res. 47, e91. https://doi.org/10.1093/nar/gkz533 (2019).
Baugh, J. A. et al. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 3, 170–176 (2002).
An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, https://doi.org/10.1038/nature11247 (2012).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501. https://doi.org/10.1038/nbt.1630 (2010).
Arnold, M., Raffler, J., Pfeufer, A., Suhre, K. & Kastenmuller, G. SNiPA: An interactive, genetic variant-centered annotation browser. Bioinformatics (Oxford, England) 31, 1334–1336. https://doi.org/10.1093/bioinformatics/btu779 (2015).
Zhou, X. & Wang, T. Using the wash U epigenome browser to examine genome-wide sequencing data. Curr. Protoc. Bioinform. (editoral board, Andreas D. Baxevanis et al.) https://doi.org/10.1002/0471250953.bi1010s40 (2012).
Kursa, M. B. & Rudnicki, W. R. Feature selection with the boruta package. J. Stat. Softw. 36(13), 2010. https://doi.org/10.18637/jss.v036.i11 (2010).
Fulco, C. P. et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669. https://doi.org/10.1038/s41588-019-0538-0 (2019).
Trevino, A. E. et al. Chromatin accessibility dynamics in a model of human forebrain development. Science https://doi.org/10.1126/science.aay1645 (2020).
Acknowledgements
This work was supported by grants from a Delivering on Discovery grant from the Arthritis Foundation (#6490) as well as an Innovative Research Grant from the Rheumatology Research Foundation. It was also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR001412 to the University at Buffalo. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Contributions
E.T.—Performed the primary statistical analyses and was involved in data interpretation and preparation of the manuscript. K.J.—Prepared clinical samples and performed wet lab procedures for RNAseq and ChIPseq studies. Was involved in data interpretation and preparation of the manuscript. T.R.H.—Assisted in collection of clinical samples and assigning phenotype (disease activity). L.H.—Assisted in collecting samples from healthy children. S.W.—Assisted in collecting clinical specimens and assigning disease phenotype to patients. Y.F.—Performed HiChIP studies and assisted in their analysis. P.M.G.—Directed the performance of HiChIP studies and assisted in their analysis. Was involved in data interpretation and preparation of the manuscript. T.L.—Assisted in analysis of RNAseq, ChIPseq, and HiChIP studies. Was involved in data interpretation and preparation of the manuscript. J.N.J.—Designed the study, directed the wet lab procedures, and was involved in data interpretation and preparation of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tarbell, E., Jiang, K., Hennon, T.R. et al. CD4+ T cells from children with active juvenile idiopathic arthritis show altered chromatin features associated with transcriptional abnormalities. Sci Rep 11, 4011 (2021). https://doi.org/10.1038/s41598-021-82989-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-82989-5
This article is cited by
-
Systematic decoding of cis gene regulation defines context-dependent control of the multi-gene costimulatory receptor locus in human T cells
Nature Genetics (2024)
-
Broadening our understanding of genetic risk for scleroderma/systemic sclerosis by querying the chromatin architecture surrounding the risk haplotypes
BMC Medical Genomics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
