
Abstract
The performance and productivity of livestock have consistently improved by natural and artificial selection over the centuries. Both these selections are expected to leave patterns on the genome and lead to changes in allele frequencies, but natural selection has played the major role among indigenous populations. Detecting selective sweeps in livestock may assist in understanding the processes involved in domestication, genome evolution and discovery of genomic regions associated with economically important traits. We investigated population genetic diversity and selection signals in this study using SNP genotype data of 14 indigenous sheep breeds from Middle East and South Asia, including six breeds from Iran, namely Iranian Balochi, Afshari, Moghani, Qezel, Zel, and Lori-Bakhtiari, three breeds from Afghanistan, namely Afghan Balochi, Arabi, and Gadik, three breeds from India, namely Indian Garole, Changthangi, and Deccani, and two breeds from Bangladesh, namely Bangladeshi Garole and Bangladesh East. The SNP genotype data were generated by the Illumina OvineSNP50 Genotyping BeadChip array. To detect genetic diversity and population structure, we used principal component analysis (PCA), admixture, phylogenetic analyses, and Runs of homozygosity. We applied four complementary statistical tests, FST (fixation index), xp-EHH (cross-population extended haplotype homozygosity), Rsb (extended haplotype homozygosity between-populations), and FLK (the extension of the Lewontin and Krakauer) to detect selective sweeps. Our results not only confirm the previous studies but also provide a suite of novel candidate genes involved in different traits in sheep. On average, FST, xp-EHH, Rsb, and FLK detected 128, 207, 222, and 252 genomic regions as candidates for selective sweeps, respectively. Furthermore, nine overlapping candidate genes were detected by these four tests, especially TNIK, DOCK1, USH2A, and TYW1B which associate with resistance to diseases and climate adaptation. Knowledge of candidate genomic regions in sheep populations may facilitate the identification and potential exploitation of the underlying genes in sheep breeding.
Similar content being viewed by others

Introduction
Genetic diversity in livestock is important for improving productivity and addressing future challenges, including food security and mitigating climate change1. Sheep were probably first domesticated in the Fertile Crescent region approximately 10,000 years ago2. Asia has about 40 percent of the world’s sheep breeds and diverse agro-ecological conditions have led to the development of more than 80 native sheep breeds in different geographical districts of Iran, Afghanistan, India, and Bangladesh3,4. Sheep play an important role in the livelihood of many rural and nomadic families in these countries4. The number of sheep average from 2010 to 2018 were in India 64 million, Iran 42.7 million, Afghanistan 13.6 million, and Bangladesh 2 million.
Study of population structure gives information on anthropogenic activities and historical processes that have influenced recent gene pools and the genetic relationships among breeds (Ju et al. 2019). Population structure among breeds can be studied using principal component analysis (PCA), admixture and phylogenetic analyses. A range of demographic forces and evolutionary trends affects linkage disequilibrium (LD) patterns on the genome5. The LD patterns provide good historical information on the population demography.
Natural and artificial selections leave patterns on the genome that result in differences in allele frequencies among populations6. If the selection pressure is high at the level of an individual locus, the frequency of the selected variant increases. In addition, selection will change the diversity pattern around the selected variant through genetic hitchhiking, known as a selective sweep7. As a result, different genetic variations and various haplotype structures are fixed over time within separated subpopulations, leading to a wide range of farm animal breeds and distinct genetic populations8. Selective sweeps detected in livestock breeds can add to new information about their population history.
Several methods have been developed to scan genome-wide selective sweeps9. Most of the methods are based on: (1) increases in derived allele frequency and decreases in genetic variation near a selective sweep (hitchhiking) within a population, (2) haplotype length and structure measured by extended haplotype homozygosity (EHH) or EHH-derived statistics, and (3) the differentiation of genetic populations measured by FST or related statistics10.
To capture any signal in the genome, depending on the number of populations, temporal context scale, and type of selection signatures more than one method is often needed6. Therefore, we implemented four complementary statistical tests, FST, FLK (the extension of the Lewontin and Krakauer), xp-EHH (cross-population extended haplotype homozygosity), and Rsb (extended haplotype homozygosity between-populations). We studied selection signature in 14 indigenous sheep breeds from Iran, Afghanistan, India and Bangladesh, the four neighboring countries located in the Middle East and South Asia having more than 80 indigenous sheep breeds adapted to diverse ecological conditions. The selection signatures can illuminate selection patterns at the genome level of these indigenous sheep breeds, from adaptation to local environment and selection by breeders to improve production.
Materials and methods
Populations and genotypic data
We employed 50 K SNP genotype data on 453 individuals from 14 indigenous sheep breeds located in Iran, Afghanistan, India, and Bangladesh. Unpublished genotype data from three indigenous Iranian sheep breed, Iranian Balochi (IBL), Lori-Bakhtiari (LOR), Zel (ZEL) were used along with publicly available genotype data on another three Iranian sheep breeds, namely Afshari (AFS), Moghani (MOG), and Qezel (QEZ). We included data on three unpublished genotype data of Afghan sheep breeds, Arabi (ARB), Afghan Balochi (BLO), and Gadik (GDK). From South Asia, we included three Indian sheep breeds, Changthangi (CHA), Indian Garole (GAR), Deccani (IDC), and two Bengal sheep breeds, Bangladeshi Garole (BGA) and Bangladesh East (BGE)11. Information on these 14 breeds is summarized in Table 1.
Genotype quality control
OvineSNP50 BeadChip (Illumina, San Diego, CA, USA) was used to genotype animals. The SNP location information was taken from the Illumina Oar_v4 assembly, retrieved from SNPChiMp v.312.
The genotype data from different breeds were merged using PLINK13. We excluded the SNPs located on sex chromosomes and those with unknown chromosomal position. The quality control was performed using PLINK13. SNPs that were genotyped in less than 90% of the animals, had a minor allele frequency (MAF) lower than 1%, or departed from Hardy–Weinberg proportions at a P-value < 10–3 were discarded. Furthermore, individuals with more than 10% missing genotypes were removed from the data set. After quality control, we used Beagle V5.0 software to impute sporadic missing genotypes14. The fcGENE v1.7 software was used to convert the PLINK formatted files to Beagle format and vice versa15.
Genetic diversity and population structure
Individual genetic distances for the 14 sheep breeds were represented by a neighbor-joining tree and displayed using VCF-kit v0.1.616 and FigTree.v1.4.417.
We performed a PCA to investigate the population structure and to check whether samples for a breed came from a homogeneous population. PCA was done for the 14 sheep breeds using the smartpca program, which is part of EIGENSOFT 7.2.118.
Linkage disequilibrium (mean of r2) among SNPs was estimated for the breeds using PopLDdecay v1.01 software, and a Perl script was applied to visualize the results19.
Admixture analysis
For admixture analysis, quality filtered genotype data were pruned using PLINK based on LD. In a sliding window of 50 SNPs, LD pruning was carried out, moving the window in steps of 5 SNPs at a time, and removing all SNPs within each window exceeding the 1.7 variance inflation factor (VIF) threshold (–indep 50 5 1.7). VIF is known as 1/(1 − r2), with r2 being the correlation of the squared inter-variant allele count20.
We analyzed ancestry using ADMIXTURE v1.3.0 to infer breed origins and quantify the populations' admixture21. For a priori defined ancestry component (K), individual ancestry proportions were calculated with ADMIXTURE v1.3.0, which was an assumption of the number of ancestral populations20. Using 14-fold cross-validation for K values ranging from 2 to 14, admixture analysis was performed. To identify the most likely number of ancestral populations, the lowest 14-fold cross-validation error was applied. Finally, the admixture graphs were visualized using the R package BITE22.
Runs of homozygosity
Runs of homozygosity (ROHs) were studied for all 14 breeds. Using the R package “detectRUNS”23, ROHs were calculated. The sliding window method was applied to calculate ROH segments24. Conditions used to detect segments of ROH were: sliding window size (windowSize = 15 SNPs), minimum number of homozygous SNPs in a run (minSNP) = 20, threshold of windows overlapping, homozygous (threshold) = 0.05, minimum number of SNP per kbps (minDensity) = 1/103, maximum distance between two SNPs (maxGap) = 106 bps, and the minimum length of a homozygous run (minLengthBps) = 250,000 bps. By default settings defined in the detectRUNS package, the ROHs detected were divided into five categories. (0 to \(< { }\) 2 Mb, 2 to \(<\) 4 Mb, 4 to \(<\) 8 Mb, 8 to \(<\) 16 Mb and \(\ge\) 16 Mb). For each of the ROH length categories, the mean ROH sum per breed was determined by summing up all the ROHs per animal in that category and by averaging them per breed. The individual genomic inbreeding coefficient (FROH) was also determined as follows:
where \(\Sigma L_{ROH}\) is the total length of all ROHs observed in an individual’s genome and \(Lgenome\) is the sum of the length of the autosomes23.
Selection sweep, gene annotation, and functional analysis
Neighbor-joining tree and PCA analysis divided the sheep populations in three distinct categories, IR (contains AFS, MOG, QEZ, ZEL, LOR breeds), IN (contains BGA, BGE, GAR, CHA, IDC breeds), and AF (contains IBL, ARB, BLO, GDK breeds) Table 1. Therefore, we compared pairwise these three categories for selective sweeps analysis.
Selection sweep methods
We performed pairwise comparison for (a) IR vs. IN, (b) IR vs. AF, and (c) IN vs. AF to identify genomic regions under increasing differentiation using Fixation index (FST), FLK (the extension of the Lewontin and Krakauer), xp-EHH (cross-population extended haplotype homozygosity), and Rsb (extended haplotype homozygosity between-populations).
The FST analysis is a widely used approach to identify genetic differentiations between populations compared to the within-population polymorphic frequency25. We performed the FST to identify genomic regions under increasing differentiation using VCFtools v0.1.1526. For each comparison, the mean of FST value was computed in all 39,348 SNPs. Z transformation of the mean of FST values (Z(FST)) was performed using the “scale” command in R software.
The FLK test is an extension of the original Lewontin and Krakauer (LK) statistic27.
It calculates a population differentiation statistic, which includes a kinship matrix representing the relationship between populations28. This test accounts for population structure and differences in the effective population size by modeling the genetic divergence between populations as a result of drift and population division29.
For FLK analyses, p-values were computed as explained in the hapFLK software documentation30. For each comparison, the negative log p-value was calculated using the hapFLK R script30, and the candidate genomic regions under selection were plotted.
Extended haplotype homozygosity (EHH) detects selection signatures by comparing a high frequency and extended homozygosity based haplotype with other haplotypes at the selected locus31. Complete selective sweeps can be approached by using the cross-population EHH (xp-EHH) test, which compares each population regarding corresponding haplotypes to the other populations. The xp-EHH test compares the integrated EHH profiles between two populations at the same SNP31. The xp-EHH test has a high power to detect selection signatures in small sample sizes, and therefore grouping of genetically similar breeds may help in gaining power32,31.
Rsb test to identify selective sweeps is based on the same idea of estimation of EHH as xp-EHH test. However in contrast to xp-EHH test, it does not require phasing information28. Rsb compares the EHH patterns of the same allele between populations instead of comparing the EHH between alleles within one population, analogous to other statistics that are often focused on contrasting genetic variation between populations33.We used the xp-EHH and Rsb approaches33,34 to determine selected alleles with higher frequency than expected according to their haplotype length to obtain recent and generally segregating selective sweeps. The haplotypes were phased with Beagle14, and then xp-EHH and Rsb scores were calculated for each haplotype within a population. Haplotype frequencies were computed for 39,348 SNPs. For each locus, the xp-EHH and Rsb score were calculated using the rehh package35 in R and the candidate genomic regions under selection were obtained.
For each test, the genes that were considered as candidates were found within the intervals spanning the candidate genome regions and also overlapping candidate genes among the tests were captured using the Ovis Oar_v4 reference genome assembly in the Ensembl36. The candidate genes visualized using Venpainter tool (Lin et al. 2016).
Absolute correlation among four methods used to detect selection sweeps on: (a) IR vs. IN, (b) IR vs. AF, and (c) IN vs. AF sheep breeds were determined using R codes.
The biological enrichment and functional annotation of the genes under selective pressure were defined using Gene Ontology Consortium (http://geneontology.org).
Results
Populations and genotype data
After quality control and imputation of missing genotypes from 463 individuals genotype data for 39,531 SNPs from 14 sheep breeds Table 1, 453 individuals and 39,348 SNPs remained for analysis. In details 10 individuals removed due to missing genotype data (–mind), 180 SNPs removed due to missing genotype data (–geno), and 3 SNPs were removed due to minor allele frequency (–maf).
Population genetic structure and linkage disequilibrium
The Neighbor-joining phylogenetic tree analysis divided the 14 breeds into three main branches, IR, IN, and AF. The IR group included AFS, MOG, QEZ, ZEL, and LOR, in a main branch Fig. 1, blue color), which illustrated close relationships in the blue branch. These five breeds are from mountainous and forest areas with cold and temperate climates of Iran. The AF group has two distinct sub-branches, one for the three Afghan breeds (ARB, BLO, GDK), and the other own for the Iranian IBL breed Fig. 1, red color). The IBL sheep is from a hot dry climate in the south-eastern deserts of Iran, bordering Afghanistan and Pakistan and therefore IBL is geographically closer to Afghan breeds than the other Iranian breeds in this study. The ARB, BLO, and GDK breeds formed a dense sub-branch that indicates their close genetic relationship. The IN branch included BGA, BGE, GAR, IDC, and CHA Fig. 1, green color). In this branch, two Bengal breeds (BGA and BGE) and GAR formed a distinct cluster, and two other Indian sheep breeds were placed in two separate clusters. The GAR and BGA which are both named Garole breed live in West Bengal state of India and Bangladesh, respectively however, some breeding isolation between them occurred. Therefore, a close genetic relationship between these two breeds is expected.

Neighbor-joining phylogenetic tree for 14 sheep breeds based on autosomal SNPs. For breed abbreviations, see Table 1.
The LD patterns among the IR and IN groups indicated that the mean of correlation coefficient values (r2) in both groups dropped rapidly at approximately 10 Kb while the AF group showed a slower drop and its r2 values at 50 Kb was higher than the other groups (Supplementary Figure S3). The average r2 at 250 Kb for the IR, IN and AF breeds were 0.0351, 0.0230 and 0.0693, respectively. There was a big difference in r2 values more than 100 Kb between (IR and IN) and AF.
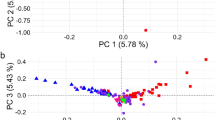
PCA results Fig. 2 also indicated close relationships within the IR, IN, and AF groups and supported separation into the three broad geographic groups that were identified by the neighbor-joining tree Fig. 1. Although the breeds clustered according to geographic origin, a gradient based on the geographic distance was less pronounced Fig. 2. In addition, the first principal component (PC1), explaining 13.3% of the total genetic variation among breeds, clearly separated the IR and IN breeds from the AF breeds, thus forming two clusters. Along with the PC1 projection spectra, both IBL and Afghan breeds formed the AF group but a large genetic variation are shown between them. Among the AF breeds, IBL is clearly distant from the other breeds and supported the phylogenetic results. The subclusters of MOG, GEZ, and AFS breeds overlapped, indicating a close relationship and possible admixture of these breeds from the same region in north-western Iran. The LOR breed clearly distant from the other IR breeds which show geographic distance between the LOR from the west and south-western of Iran and the other IR breeds from the north and north- western of Iran. The patterns of genetic variation observed for the AFS, MOG, and GEZ breeds suggested a recent admixture between these three Iranian breeds.

Principal components analysis (PC 1 and PC 3) of among 14 sheep breeds based on autosomal SNP markers. For breed abbreviations, see Table 1.
PC2, explaining 6.8% of the total genetic variation, separated the Afghan breeds from the other breeds, but it did not clearly show geographic distance between the IR and IN breeds (Supplementary Figure S1). PC3, explaining 3% of the total genetic variation, separated the IR from the IN and also showed close genetic relationship among two Bengal breeds and GAR, while genetic distances among IDC, CHA, and the other IN breeds Fig. 2. For a more clear assessment, we did PCA between the IR and IN breeds which were separated by PC1 (Supplementary Figure S2).
The occurrence and extent of breed admixture were examined by estimating individual ancestry proportions from quality filtered and LD pruned genotype data. During pruning the dataset 13,257 of 39,348 SNPs removed and 26,091 SNPs were remained. Admixture analyses were carried out with up to 14 ancestral components (K) Fig. 3. Cross-validation (CV) errors were calculated to identify the most likely number of ancestral populations. The lowest CV error was detected for K = 12 Fig. 3a. Although at K = 10, CV errors had stagnated after a decline, ancestry components up to K = 10 separate breeds, and so it was recognized as the optimal value of K Fig. 3b.The results of Admixture were in general agreement as PCA. Although the AF breeds, especially the IBL from the IR and IN breeds, were separated at the first ancestry components (K = 2) and also at K = 4, the IR breeds were separated from the IN breeds but a substantial IR ancestry is observed in the CHA breed at K = 4. In addition, based on geographic origin the breeds were divided as follows: K = 2: the AF breeds,K = 4: the IN breeds,K = 7: the IR breeds. At K = 10. The breed-specific ancestry components were clearly defined by all breeds except MOG and QEZ and the Afghan breeds (ARB, BLO, and GDK). However, our finding showed that increasing the number of K above 10 did not yield a consistent MOG and QEZ separation. Therefore, four ancestral components distinguished the five IR breeds where AFS, ZEL, and LOR were unambiguously recognized. Similar to PCA findings, the fourth component was shared between MOG and QEZ, confirming a close genetic relationship. There were no differences among the ARB, BLO, and GDK Afghan breeds from K = 2 to K = 14 which indicated close genetic relation sheep of them, confirming results from PCA and the neighbor-joining tree. The Bengal breeds (BGA and BGE) separated from K = 9, but despite expectation, BGA and GAR with the common name and root separated from K = 5. In general, compared with the other IN breeds, closer genetic relationships were seen between BGA, BGE, and GAR confirming PCA and the neighbor-joining tree analyses.

Prediction error (a) and circle admixture from K2 to K14 (b) plotted, respectively. For breed abbreviations, see Table 1.
ROHs
A total of 34,587 ROH segments were identified in the 14 studied breeds. The results are summarized in Supplementary Table S5. The number of ROH segments was highest in IBL (5,407) and GAR (4,605) breeds, and lowest in ARB (1,000) and GDK (1,018). The majority of ROH segments were shorter than 8 Mb (32,260), followed by intermediate ROH segments ranging from 8 to 16 Mb (1,520) and large ROH segments exceeding 16 Mb (807). Based on ROH, levels of FROH for each breed were estimated separately (Supplementary Figure S4). Genomic inbreeding ranged from 0.008 (in the AFS breed) to 0.5 (in the GAR breed).
Selective sweeps detection
Selective sweeps detection was performed using FST37, FLK27, Rsb33, and xp-EHH34. Based on the PCA and the neighbor-joining tree results, these four different tests were conducted for selective sweeps detection on the three pairwise comparisons: (a) IR and IN breeds, (b) IR and AF breeds, (c) IN and AF breeds. The Z-transformation of FST, Z(FST), values of 39,348 SNPs were estimated Fig. 4. For these three pairwise comparison, the maximum of Z(FST) values were 14.524 on chromosome 11 (IR vs. IN breeds), 4.744 on chromosome 24 (IR vs. AF breeds) and 4.556 located on chromosomes 24 (IN vs. AF breeds) Fig. 4. Based on the Z(FST), a total of 131 genes as top 1% candidates for selective sweeps were detected in (a) IR and IN breeds, 131 genes in (b) IR and AF breeds, and 121 genes in (c) IN and AF breeds (Supplementary Table S1). Among these candidate genes, several of them are known for association with economic traits, for example, SLC27A6, ANXA13, ADCY2, HDAC9, TTC8, and WDR70 association with milk traits. HERC2, FTO, TP73, GRM3, KCNIP4, GRM7, and UBR2 are related to body weight and growth traits. TMEM132B, TMEM232, and SLC8A3 affect fertility traits. ADAMTS6, ADAMTS20, GALNT6, ATP2C1, TMPRSS3, PCDH15, MAGI2, TRPC4, DOCK1, DOCK4, DOCK10, MAPK10, ADAM7, PPA2, CHD3, ITGA4, NBEAL1, NFATC1, and ZNF609 involve in the immune system and environment adaptation.

The distribution of absolute Z(FST) values on 26 sheep autosomes: (a) IR vs. IN breeds (the horizontal blue line, Z(FST) ≥ 3.93), (b) IR vs. AF breeds (the horizontal blue line, Z(FST) ≥ 3.18), (c) IN vs. AF breeds (the horizontal blue line, Z(FST) ≥ 3.08). The data points above the horizontal line (blue line) are top 1% Z(FST) values. FST fixation index. For breed abbreviations, see Table 1.
The xp-EHH scores were calculated for haplotype frequencies Fig. 5. The top 1% of xp-EHH, considered as selective sweeps, identified 164 genes for (a) IR and IN breeds,236 genes for (b) IR and AF breeds; and 221 genes for (c) IR and AF breeds (Supplementary Table S2). Many candidate genes found by the xp-EHH method are related to economic traits, such as, OXT, HSPB1, TBX6, GNA12, BMP7, MYH10, TRHDE, IL27, IL4R, and IL21R involved in heat stress; ATP2A1, ATP2B1, LRP12, CD19, MAPK3, PLCE1, VPS16, PTPRA, ADAM2, MYO18A, PCDH17, BBS9, NFATC2IP, RNF26, RNF139, ZNF572, ZNF655, and ZNF789 associated with immune system and environment adaptation; and MEF2C, TRHDE, FAM222B, FAM177A1, and SSC4D influenced body weight and growth traits.

Genomic distribution of standardized cross-population extended haplotype homozygosity (xp-EHH) scores on 26 sheep autosomes pairwise: (a) IR and IN breeds, (b) IR and AF breeds, (c) IN and AF breeds. For breed abbreviations, see Table 1.
The Rsb scores were calculated for haplotype frequencies Fig. 6. The top 1% of Rsb, considered as selective sweeps, identified 185 genes for (a) IR and IN breeds, 249 genes for (b) IR and AF breeds, and 233 genes for c) IR and AF breeds (Supplementary Table S3). Many candidate genes specially associated with immune response and heat stress were identified by Rsb test, such as, ATP2B1, ATP2C1, ATP6V1H, BMPR1B, PLCE1, LRP1B, CXCL1, CD19, DOCK1, DOCK4, PTPRA, MAPK3, UNC5C, ANKRD2, BBS9, NAFTC2IP, RNF139, and ZNF695 in immune system and environment adaptation, and IFT22, EIF2A, HSPB1, TBX6, TBX21, GNA12, BMP7, IL16, IL27, IL4R, and IL21R in heat stress. Furthermore, HOXD1, HEXD2, and MTX2 affect the horn traits,and PRLP, TBC1D10B, TMEM151A, TMEM65, TMEM225B, BMPRIB, and BMP7 genes associated with fertility traits were detected as candidate genes using Rsb.

Genomic distribution of standardized haplotype differentiation (Rsb) scores on 26 sheep autosomes pairwise: (a) IR and IN breeds, (b) IR and AF breeds, (c) IN and AF breeds. For breed abbreviations, see Table 1.
The -log(p-value) values of 39,348 SNPs for the FLK test are presented in Fig. 7. Based on the -log(p-value), a total of 244 genes as top 1% candidates for selective signals were detected in (a) IR and IN breeds,265 genes in (b) IR and AF breeds; and 247genes in (c) IN and AF breeds (Supplementary Table S1). Several candidate genes identified using FLK test are related to economic traits, for example, FABP3, SLC27A6, ACP7, ANXA13, HEATR5B, ADCY2, BRD4, BRD8, HDAC9, TTC8, TTC23, WDR7, WDR31, WDR70, and POU6F1 related to milk traits as well as HERC2, FAM169A, FTO, TP73, GRM2, GRM3, and UBR2 for body weight and growth traits. Several candidate genes related to immune system and climate adaptation were detected by FLK, such as, ADAMTS6, ADAMTS20, ARHGAP26 GALNT6, GALNT13, GALNT18, ATP2C1, ADAM19, MAPK10, MAGI2 ADAM33, LRP1B, CXCL14, TMPRSS3, TRPC4, NBEAL1, CD34, COL12A1, PCDH15, DOCK1, DOCK4, DOCK10, UNC5B, BBS9, CDH6, CHD3, IRF6, ITGA, LRP1B, NAFATC1, RNF26, ZNF609, and ZNF692.

Genomic distribution of single marker statistic (FLK) scores on 26 sheep autosomes pairwise: (a) IR and IN breeds, (b) IR and AF breeds, (c) IN and AF breeds. For breed abbreviations, see Table 1.
The FST and FLK tests with average 128 and 252 genes showed the minimum and maximum captured genes among these four tests. Furthermore, five, six and three concordant genomic regions for (a) IR and IN breeds, (b) IR and AF breeds, (c) IN and AF breeds were identified by FST, xp-EHH, Rsb, and FLK tests as candidates for selection signals, respectively Fig. 8. These overlapping candidate genes for (a) IR and IN breeds include the following genes,Scm-like with four MBT domains protein 1 (SFMBT1) on chromosome 19, plays a role during spermatogenesis. Dedicator of cytokinesis protein 1(DOCK1) on chromosome 22, has an essential role in embryonic development and involved immune response, Neural EGFL like 2 (NELL2) on chromosome three, involved in involved in pubertal development. NCK-interacting protein kinase (TNIK) on chromosome one, the protein encoded by this gene plays important role in embryonic development, especially during the early embryo to blastocyst stages, participates in the regulation of the inflammatory response against infections.

Venn diagram showing the unique and shared candidate genes for FST, Rsb, xp-EHH, and FLK tests on: (a) IR vs. IN, (b) IR vs. AF, and (c) IN vs. AF sheep breeds. For breed abbreviations, see Table 1.
The overlapping candidate genes for (b) IR and AF breeds include, Echinoderm microtubule-associated protein-like 5 (EML5) on chromosome seven, may change the assembly dynamics of microtubules to make microtubules are slightly longer but more dynamic and it is possible that EML5 plays a role during neuronal development in the regulation of cytoskeletal rearrangements, IQ domain-containing protein E (IQCE) on chromosome 24, involved in body development, TRNA-YW Synthesizing Protein 1 Homolog B (TYW1B) on chromosome 24, influenced on the wybutosine biosynthesis pathway. Usherin (USH2A) on chromosome 12, may be involved in the function of synapses and plays an important role in the development and maintenance of cells in the inner ear and retina. SPO11 initiator of meiotic double-stranded breaks (SPO11) on chromosome 13, involved in the production of double-strand breaks (DSB) of DNA and it is specifically involved in the growth of the testis, maintenance of the male germ line, and maturation of sperm. Three overlapping candidate genes for c) IN and AF breeds were detected; the IQCE, TYW1B, and an unknown gene with Ensemble number (ENSOARG00000025902) which all of these three genes were detected before (IR vs. AF).
We also detected overlapping candidate genes for IR vs. IN, IR vs. AF, and IN vs. AF data on the FST, xp-EHH, Rsb, and FLK tests Fig. 9. For the FST test PPA2, involved in the immune system, and KCNIP4 plays important role in heart performance and it is related to skeletal muscle growth and also immune response. SYT1, associated with feeding behavior traits such as residual feed intake and TMEFF2, involved in a wide range of traits such as,immune response, milk production and sperm morphology, were detected as overlapping candidate genes for the Rsb test. For the FLK test, PPA2, EML5 genes, which have been found in the previous tests, MGAT5, associate with dry matter intake and NEB, involved in environment adaptation, were detected as overlapping candidate genes on the three different data. We did not find any overlapping candidate genes on the all data by the xp-EHH test.

Venn diagram showing the unique and shared candidate genes for IR vs. IN, IR vs. AF, and IN vs. AF data on: (a) FST, (b) xp-EHH, (c) Rsb, and (d) FLK tests. For breed abbreviations, see Table 1.
Biological enrichment analysis of significant biological processes for candidate genes under positive selective pressure revealed 26 Gene Ontology (GO) terms Table 2. These GO terms reflected protein function and biosynthetic processes, including TNIK and DOCK1 genes associated with cytoskeleton organization (GO:0007010) and six other GOs related to the TNIK gene. Four other GOs were associated with DOCK1. The SFMBT1 gene associated with negative regulation of transcription (GO:0035556). Seven GOs were associated with the spo11. Four GOs were related to the USH2A gene. Finally, tRNA processing (GO:0008033) was associated with the TYW1B gene.
Absolute correlation coefficients among these four tests on (a) IR vs. IN, (b) IR vs. AF, and (c) IN vs. AF sheep breeds showed the maximum correlation between FST and FLK on the all comparisons (average: 0.861) and the minimum correlation between FLK and Rsb on IR vs. IN (0.107) and FST and Rsb on IR vs. AF and IN vs. AF data (average: 0.021) Fig. 10.

Absolute correlation among different methods used to detect selective sweeps on: (a) IR vs. IN, (b) IR vs. AF, and (c) IN vs. AF sheep breeds. For breed abbreviations, see Table 1.
Discussion
The present study investigates the genetic diversity and selective sweeps of 14 sheep breeds from Iran, Afghanistan, India, and Bangladesh. The selective sweeps were studied using the FST, FLK, xp-EHH and Rsb statistical methods on the three cluster of breeds (IR, IN, and AF). Our goal in the current study was to search the genomes of these indigenous sheep breeds to highlight genetic variants that can be used in developing next-generation productive breeds, better suited to diverse Iran environments, in a comparative scale with Indian, Bengal, and Afghan sheep breeds. Furthermore, the other goal was using four comparable selective sweeps tests to cover all the regions of the genomes and capture maximum candidate genes, as well as review their biological function. The results showed that these breeds' genomes contain multiple regions under selection. These regions contain well-known economic trait-related candidate genes. This could help sheep breeders to: (1) improve adaptation in extant breeds; (2) develop native breeds that are better adapted to local agro-climatic conditions; (3) launch future research work on the genomes of Iranian, Afghan, Indian, and Bengal sheep, and highlight essential genetic variants or haplotypes that can be used in the production of higher productivity and efficiency next-generation breeds, better adapted to various Iranian environments, on a comparative scale with Afghan, Indian, and Bengal sheep breeds.
Genetic relatedness and geographic origin
We demonstrated that the IR sheep breeds are genetically distinct from the breeds of IN and AF. Based on their geographic origins, the studied sheep breeds are well clustered. We categorized the IR, IN and AF breeds into three phylogeographic clades. Close connections between breeds originating in the same geographical region have been found. In fact, phylogenetic analysis showed a close genetic relationship among the IR breeds. These breeds are from cold and temperate climates of Iran. On the other hand, the IN breeds showed a closer relationship among BGA, BGE, and GAR breeds from the eastern region of India and Bangladesh. In contrast, IDC from the western peninsular region and CHA from northern Himalayan part of India formed two distinct sub clusters.
Furthermore, the AF cluster showed an IBL sub-cluster and a compact sub-cluster of three Afghan breeds, indicating a closer relationship among Afghan breeds and their genetic distance from IBL.
These findings are consistent with previous research on sheep38,39,3,40, which showed that individuals were separated by global population structure patterns according to their geographical origin.
In accordance with previous findings2,41,38 PCA results demonstrated that the genetic variation was associated with the separation among sheep breeds from different parts of the world. This was further supported by neighbour-joining tree analysis revealing that the population was split according to geographic origin (IR, IN, and AF). Population structure analyses of the IR, IN, and AF breeds clearly reflected the geographic distribution at PC1 and the separation of northern from southern breeds at PC3.
Admixture and phylogenetic patterns
In accordance with the previous analyses, admixture results confirmed that the first few ancestral breed components (K = 2 to K = 5) were related to the geographic origins. High levels of breed admixture were detected among the Iranian (IR and IBL) breeds, and also among the IN breeds. A significant IR ancestry is observed in the CHA breed at K = 4, which probably due to the same climate between the CHA from Kashmir and the IR breeds from the northwest of Iran Table1, and also due to historical ties and neighborliness between Iran and India, especially in the Kashmir region. It is possible that the IR breeds and CHA have common ancestors. However, low levels of admixture events among the breeds originating from the different geographical regions were detected. For example, although the GAR and BGA from India and Bangladesh have a common breed name (Garole), they separated at k = 5 ancestral breed components, while BGA and BGE which are known as two different breeds in Bangladesh showed more relationship and they have been separated at k = 9 which confirm the effect of geographic origin in breeds separation. Admixture results confirmed genetic divergence identified through the neighbor-joining and PCA.
Inference based on population neighbour-joining trees based on genome-wide allele frequencies clustered the breeds into three monophyletic clades according to the geographical origin. The deepest population split among the AF breeds separated IBL from the other AF breeds. Among the IN breeds, IDC and CHA showed deeper population splits, in line with geographic clades detected by the PCA and admixture analysis. These results support the previous findings2,41,38.
Runs of homozygosity
The history of inbreeding within a population can be estimated from the length distribution of ROH segments42. We estimated FROH to study genomic inbreeding. The average FROH levels estimated for the breeds was (0.09), which was almost the same as the previous study3. In the studied breeds, the range of FROH recorded 0.008 to 0.5, was higher than the previous maxima but the observed minima were consistent with previous findings. Mastrangelo et al.43 reported the range of FROH from 0.016 to 0.099 in domestic sheep breeds.
The IBL and IN breeds had the largest number of ROHs, and therefore showed highest FROH levels, indicating the relationship between the number of ROH segments and FROH levels3. The results of ROH segment also showed that more than 93% of the ROH segments were shorter than 8 Mb, which indicated older events of inbreeding and a board effective population size of sheep flocks43. The IN breeds displayed a higher FROH variance than the IR and AF breeds which could indicate more effective population size variation in the IN breeds. Inbreeding and extended ROH segments can be increased by small population size and intense selection, thereby continuing to express the deleterious alleles44.
Genome-wide selective sweeps
The ability of specific genomic regions to detect selective sweeps depends on the selection of analytical tools appropriate to the biological situation but no single method can detect selective sweeps that are both starting and nearly completed. However, combining several tests increases significantly the power to recognize the region selected6,7. Therefore, we used FST, FLK, xp-EHH, and Rsb test statistics to detect genome-wide selective sweeps in (a) IR and IN breeds, (b) IR and AF breeds, (c) IN and AF populations. FST was first implemented to measure the degree of genetic differentiation between populations based on variations in allele frequency45. The genomic variation information is provided by FST at a locus between the populations compared to within the populations. Therefore, the FST is an evidence of selection: high FST values indicate positive local adaptation46. The older selection events between populations are expected to be identified by FST47,48. The xp-EHH test is an extension of EHH31, that incorporates information on the relationship between an allele's frequency and LD measurements with neighboring alleles. Therefore, this test may provide maximal statistical power and low ascertainment bias sensitivity33. The Rsb test is population comparison test to identify selective sweeps33. The test is based on the same idea as the XP-EHH, identifies loci similar to the XP-EHH test under selection, but can be implemented with unphased data28. Generally, the xp-EHH and Rsb tests are used to detect recent positive selection within population and between-populations, respectively9. The FLK (extended Lewontin and Krakauer test) test is based on the assumption that two new populations are formed by the splitting of a population,calculates a statistic of population differentiation, which incorporates a matrix of kinship describing the relationship between populations27,28. For each SNP, the FLK test calculates a global FST, but allele frequencies are first rescaled using a matrix of population kinship. This matrix, which is estimated from the genome-wide data observed, measures the amount of genetic drift that can be predicted along all branches of the population tree under neutral evolution27. Therefore, the integration of these four complementary statistical tests provides a valuable tool for detecting, with greater confidence, positive selection of genomic regions.
For FST and FLK, only the top 1% Z(FST) values and the top 1% -log(p-value) were considered, respectively to be representing selective sweeps as recommended in previous studies29,40.
Analyses of selective sweeps were reported for several international sheep populations from several countries, including China49, Europe41,50, Russia51, Egypt52, Brazil53, and New Zealand54. Furthermore, several studies of selective sweeps on sheep carried out using different tests, including the FLK and hapFLK2,55, hapFLK, FLK, FST, and hapFST10, REHH and xp-EHH56, FST and hapFLK49, FST and iHS52, FST, Rsb, and iHS53.
This study, using FST, xp-EHH, Rsb, and FLK, detected on average 128, 207, 222, and 252 genomic regions as candidates for selective sweeps, respectively. Although the selected candidate regions are narrow and are distributed across different chromosomes, however for FST and FLK tests, chromosome 1 showed a low value for IR vs. AF and IN vs. AF comparisons which may indicate the genome of two populations are the same in this region and many common genes were expected to be fixed in both populations48, Figs. 4 and 7. Several of these genes encode economically important traits. For example, genes that have directly or indirectly influence traits for adaptation to hot arid conditions and heat tolerance (TRHDE, IL4R,IL21R, and SLC4A4), which are reported as candidate genes involved in heat tolerance on sheep57. The heat shock protein B1 (HSPB1) gene which expresses both at mRNA and protein levels under heat stress on poultry58, reported in sheep59, and cattle60. All of these candidate genes were detected in IR vs. AF, and vs. AF clusters, where the AF breeds are common (Supplementary Tables S1, S2, and S3). This indicates that the AF breeds, which are from a hot dry climate, have undergone selection for heat tolerance.
Many of the candidate genes identified in this study are effective in genetic resistance to disease, immune response and climate adaptation, which indicates differential selections among the studied breeds. Since genetic resistance against diseases and harsh environmental conditions are important characteristics of indigenous animal breeds, the identification of a large number of genes in this study points toward the associated genes have been under selection pressure over time due to the natural selection of immune response traits61,62. For example, we detected the DOCK family (DOCK1, DOCK4, DOCK10)63,64, ZNF family (ZNF572, ZNF655, ZNF609, ZNF692, and ZNF789)65,66, ATP family (ATP2A1, ATP2B1, and ATP2C1)67, TMEFF268, CXCL169, PCDH1569, and (COL12A1, COL15A1, COL27A1)69, VPS16 and PTPRA70, PLCE171, ATAMTS2072, TPRC473, candidate genes involved in the immune response and climate adaptation (Supplementary Tables S1, S2, and S3). Almost all these genes were detected in all three clusters, a (IR and IN), b (IR and AF), and c (IN and AF), which may indicate genetic resistance and high immune response against diseases and harsh environmental conditions in these native breeds.
We detected some genes involved in production traits and indirectly related to climate adaptation, such as, FABP3. Calvo et al.74 showed linkage disequilibrium between FABP3 gene and quantitative trait loci (QTL) for milk fat content. Other related milk traits candidate genes included the LRP1B and CNTN4 which previously reported on sheep75 and cattle76. The ITPR2 and SLC27A6 are also two examples of important candidate genes detected by Li et al.75 on sheep and both have been proposed to be candidate genes for milk and fat production in cattle and indirectly involved in climate adaptation77,78. All of these genes were found in b (IR and AF), and c (IN and AF) clusters, indicating AF breeds may be under selection pressure related to milk traits but it needs further research to conclude. (Supplementary Tables S1, S2, and S3).
We found several candidate genes involved in body weight and growth traits specially post-weaning gain in all population clusters, such as the TRHD, UBR2, GRM2, GRM3 which also related to climate adaptation indirectly79,80.
Furthermore, 11 overlapping candidate genome regions were detected for FST, Rsb, xp-EHH, and FLK tests on: (a) IR vs. IN, (b) IR vs. AF, and (c) IN vs. AF sheep breeds Fig. 8. The number of overlapping unique candidate genes are consistent with the previous results using FST, Rsb, and iHS tests81, FST, xp-EHH, and iHS tests48, ROH, FST, and xp-EHH82.
Four of the genes (DOCK1, TYW1B, USH2A, and TNIK) play important roles in resistance against diseases and climate adaptation. DOCK1 located on chromosome 22 is involved in immune response63,64. TNIK on chromosome 1 plays several functions in embryonic development, especially during the early embryo to blastocyst stages, participates in the regulation of the inflammatory response against infections83, TYW1B on chromosome 24 influences artery disease and blood pressure in human84, USH2A on chromosome 12 may be involved in the function of synapses and plays an important role in the development and maintenance of cells in the inner ear and retina85.
In total, seven unique candidate genes were detected for IR vs. IN, IR vs. AF, and IN vs. AF comparisons by FST, Rsb, and FLK analysis, but no overlapping candidate gene was found for the xp-EHH method Fig. 9.
PPA2 on chromosome 6 is associated with immune response and disease resistance in cattle86. KCNIP4 gene on chromosome 6 is directly involved in processes related to muscle growth and fat deposit and indirectly climate adaptation in sheep87 and was reported in cattle involved in bovine growth and calcium metabolism88. SYT1 gene on chromosome 3 is associated with feeding behavior traits related to local adaptation89, and TMEFF2 gene on chromosome 2 is involved in a wide range of traits such as, immune response, milk production and, sperm morphology68,90. For the FLK test, PPA2, EML5, MGAT5, and NEB genes were detected, which PPA2 and EML5 have been found in the previous tests Fig. 9. NEB gene on chromosome 2 is involved in environmental adaptation. Among 1262 selected genomic regions reported by Yudin and Larkin91, only NEB gene was a shared candidate gene among cattle, sheep, mammoth, polar bear, and whale genomes.
GO classifications of the candidate genes were performed to enable a better understanding of their molecular functions. Based on the GO biological process (BP) for a significant threshold (p ≤ 0.05), we implemented the GO on 11 overlapping candidate genes. Only six genes (TNIK, DOCK1, SFMBT1, SPO11, USH2A, and TYW1B) associated with the 26 GO terms were identified. In total 11 GOs were related to TNIK, USH2A, TYW1B, and DOCK1, which are associated with local adaptation (resistance against diseases).
In confirmation of our results, Nie et al.92, reported different GO terms associated with the TNIK gene in human92. Four other GOs associated with DOCK1. DOCK family genes have several biological functions63,64.
Absolute correlation among the FST, FLK, xp-EHH, and Rsb tests were calculated Fig. 10. The xp-EHH, and Rsb are based on the frequency of extended haplotypes between two populations33,34, whereas FST and FLK are based on allele frequencies6,37. So as expected, maximum correlations were observed between FST and FLK, as well as between xp-EHH and Rsb. On the other hand, we detected minimum correlations between haplotype based tests (xp-EHH, and Rsb) and allele based tests (FST and FLK). These findings are consistent with the previous reports8,28.
Conclusions
Our results showed the population structure and selective candidate genomic regions of the 14 indigenous sheep breeds from Middle East and South Asia. This information would be valuable in future study on genetic basis for local adaptation of indigenous breeds. In FST, FLK, xp-EHH, and Rsb complementary statistical tests, some candidate genomic regions under selective pressure were detected in indigenous sheep breeds and these candidate genomic regions may facilitate identification of the underlying genes and possible exploitation in future sheep breeding.
Data availability
Genotype data from the sheep breeds (Afshari, Moghani, Qezel, Bangladeshi Garole, Bangladesh East, Indian Garole, Changthangi, and Deccani) are available through the Sheep HapMap project11. The ZEL, Lori-Bakhtiari, Iranian Balochi, Arabi, Afghan Balochi, and Gadik breeds data are part of the Iranian national genetic evaluations of economic traits conducted at the Animal Breeding Center of Iran. Any request for data should be addressed to the corresponding author.
References
Groeneveld, L. et al. Genetic diversity in farm animals—a review. Anim. Genet. 41, 6–31 (2010).
Alberto, F. J. et al. Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 9, 1–9 (2018).
Eydivandi, S., Sahana, G., Momen, M., Moradi, M. & Schönherz, A. Genetic diversity in Iranian indigenous sheep vis-à-vis selected exogenous sheep breeds and wild mouflon. Anim. Genet. 51, 772–787 (2020).
FAOSTAT F. Crop statistics (2018).
Ardlie, K. G., Kruglyak, L. & Seielstad, M. Patterns of linkage disequilibrium in the human genome. Nat. Rev. Genet. 3, 299–309 (2002).
Hohenlohe, P. A., Phillips, P. C. & Cresko, W. A. Using population genomics to detect selection in natural populations: Key concepts and methodological considerations. Int. J. Plant Sci. 171, 1059–1071 (2010).
Vatsiou, A. I., Bazin, E. & Gaggiotti, O. E. Detection of selective sweeps in structured populations: a comparison of recent methods. Mol. Ecol. 25, 89–103 (2016).
Ma, Y. et al. Properties of different selection signature statistics and a new strategy for combining them. Heredity 115, 426–436 (2015).
Oleksyk, T. K., Smith, M. W. & O’Brien, S. J. Genome-wide scans for footprints of natural selection. Philos. Trans. R. Soc. B Biol. Sci. 365, 185–205 (2010).
Fariello, M. I., Boitard, S., Naya, H., SanCristobal, M. & Servin, B. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics 193, 929–941 (2013).
Sempéré, G. et al. WIDDE: A Web-Interfaced next generation database for genetic diversity exploration, with a first application in cattle. BMC Genom. 16, 1–8 (2015).
Nicolazzi, E. L. et al. SNPchiMp: A database to disentangle the SNPchip jungle in bovine livestock. BMC Genom. 15, 123 (2014).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Browning, S. R. & Browning, B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097 (2007).
Roshyara, N.R. Documentation for Genotype Format Converter: fcGENE (2014).
Cook, D. E. & Andersen, E. C. VCF-kit: assorted utilities for the variant call format. Bioinformatics 33, 1581–1582 (2017).
Rambaut, A. FigTree–molecular evolution, phylogenetics and epidemiology (2007). Available Accessed 14 (2015).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Zhang, C., Dong, S.-S., Xu, J.-Y., He, W.-M. & Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788 (2019).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Johnston, S. E., Bérénos, C., Slate, J. & Pemberton, J. M. A genomic region containing RNF212 and CPLX1 is associated with sexually-dimorphic recombination rate variation in Soay sheep (Ovis aries). BioRxiv, 024869 (2015).
Milanesi, M., Capomaccio, S., Vajana, E., Bomba, L., Garcia, J.F., Ajmone-Marsan, P. & Colli, L. BITE: An R package for biodiversity analyses. bioRxiv, 181610 (2017).
Vogelzang, C. Effects of sample size and single-vs. multiple-breed analyses on characterizing runs of homozygosity in dairy cattle (2018).
Bjelland, D., Weigel, K., Vukasinovic, N. & Nkrumah, J. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. J. Dairy Sci. 96, 4697–4706 (2013).
Chang, L.-Y., Toghiani, S., Ling, A., Aggrey, S. E. & Rekaya, R. High density marker panels, SNPs prioritizing and accuracy of genomic selection. BMC Genet. 19, 4 (2018).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Bonhomme, M. et al. Detecting selection in population trees: The Lewontin and Krakauer test extended. Genetics 186, 241–262 (2010).
Weigand, H. & Leese, F. Detecting signatures of positive selection in non-model species using genomic data. Zool. J. Linnean Soc. 184, 528–583 (2018).
Bertolini, F. et al. Signatures of selection and environmental adaptation across the goat genome post-domestication. Genet. Select. Evol. 50, 1–24 (2018).
Fariello, M. I., Boitard, S., Naya, H., SanCristobal, M. & Servin, B. Using haplotype differentiation among hierarchically structured populations for the detection of selection signatures. arXiv preprint. arXiv:1210.7583 (2012).
Sabeti, P. C. et al. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918 (2007).
Pickrell, J. K. et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 19, 826–837 (2009).
Tang, K., Thornton, K. R. & Stoneking, M. A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 5, 1 (2007).
Sabeti, P. C. et al. Positive natural selection in the human lineage. Science 312, 1614–1620 (2006).
Gautier, M. & Vitalis, R. rehh: An R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 28, 1176–1177 (2012).
Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A. & Girón C.G.J.N. Ensembl 2018. 46, D754–D761 (2017).
Reynolds, J., Weir, B. S. & Cockerham, C. C. Estimation of the coancestry coefficient: Basis for a short-term genetic distance. Genetics 105, 767–779 (1983).
Ciani, E. et al. Merino and Merino-derived sheep breeds: A genome-wide intercontinental study. Genet. Select. Evol. 47, 64 (2015).
Deniskova, T. et al. Population structure and genetic diversity of sheep breeds in the Kyrgyz Republic. Front. Genet. 10, 1311 (2019).
Kijas, J. W. et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 10, 1 (2012).
Barbato, M. et al. Genomic signatures of adaptive introgression from European mouflon into domestic sheep. Sci. Rep. 7, 1–13 (2017).
Leroy, G. et al. Next-generation metrics for monitoring genetic erosion within populations of conservation concern. Evol. Appl. 11, 1066–1083 (2018).
Mastrangelo, S. et al. Runs of homozygosity reveal genome-wide autozygosity in Italian sheep breeds. Anim. Genet. 49, 71–81 (2018).
Muchadeyi, F., Malesa, M., Soma, P. & Dzomba, E. Runs of homozygosity in Swakara pelt producing sheep: implications on sub-vital performance. In: Proc. Assoc. Advmt. Anim. Breed. Genet, pp. 310–313 (2015).
Wright, S. The genetical structure of populations. Ann. Eugen. 15, 323–354 (1949).
Kullo, I. J. & Ding, K. Patterns of population differentiation of candidate genes for cardiovascular disease. BMC Genet. 8, 48 (2007).
Ma, Y., Zhang, H., Zhang, Q. & Ding, X. Identification of selection footprints on the X chromosome in pig. PLoS ONE 9, e94911 (2014).
Maiorano, A. M. et al. Assessing genetic architecture and signatures of selection of dual purpose Gir cattle populations using genomic information. PLoS ONE 13, e0200694 (2018).
Yuan, Z. et al. Selection signature analysis reveals genes associated with tail type in Chinese indigenous sheep. Anim. Genet. 48, 55–66 (2017).
Purfield, D. C., McParland, S., Wall, E. & Berry, D. P. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 12, e0176780 (2017).
Yurchenko, A. A. et al. High-density genotyping reveals signatures of selection related to acclimation and economically important traits in 15 local sheep breeds from Russia. BMC Genom. 20, 294 (2019).
Kim, E.-S. et al. Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. Heredity 116, 255–264 (2016).
de Simoni Gouveia, J. J. et al. Genome-wide search for signatures of selection in three major Brazilian locally adapted sheep breeds. Livestock Sci. 197, 36–45 (2017).
McRae, K. M., McEwan, J. C., Dodds, K. G. & Gemmell, N. J. Signatures of selection in sheep bred for resistance or susceptibility to gastrointestinal nematodes. BMC Genom. 15, 637 (2014).
Fariello, M.-I. et al. Selection signatures in worldwide sheep populations. PLoS ONE 9, e103813 (2014).
Zhao, F. P. et al. A genome scan of recent positive selection signatures in three sheep populations. J. Integr. Agric. 15, 162–174 (2016).
Berihulay, H., Abied, A., He, X., Jiang, L. & Ma, Y. Adaptation mechanisms of small ruminants to environmental heat stress. Animals 9, 75 (2019).
Sharma, A., Kumar, B. S., Dash, S., Singh, S. & Verma, R. Heat shock protein B1 expression is associated with age at sexual maturity in Rhode Island Red and Punjab Red layers under heat stress. Int. J. Biometeorol. 1, 1–11 (2020).
Armstrong, E. et al. Novel genetic polymorphisms associated with carcass traits in grazing Texel sheep. Meat Sci. 145, 202–208 (2018).
Srikanth, K., Kwon, A., Lee, E. & Chung, H. Characterization of genes and pathways that respond to heat stress in Holstein calves through transcriptome analysis. Cell Stress Chaperones 22, 29–42 (2017).
Mwai, O., Hanotte, O., Kwon, Y.-J. & Cho, S. African indigenous cattle: Unique genetic resources in a rapidly changing world. Asian Austral. J. Anim. Sci. 28, 911 (2015).
Scarpa, R. et al. Valuing indigenous cattle breeds in Kenya: An empirical comparison of stated and revealed preference value estimates. Ecol. Econ. 45, 409–426 (2003).
Kunimura, K., Uruno, T. & Fukui, Y. DOCK family proteins: key players in immune surveillance mechanisms. Int. Immunol. 32, 5–15 (2020).
Laurin, M. et al. The atypical Rac activator Dock180 (Dock1) regulates myoblast fusion in vivo. Proc. Natl. Acad. Sci. 105, 15446–15451 (2008).
Cassandri, M. et al. Zinc-finger proteins in health and disease. Cell Death Discov. 3, 1–12 (2017).
Feng, X. et al. Characterization of genome-wide segmental duplications reveals a common genomic feature of association with immunity among domestic animals. BMC Genom. 18, 293 (2017).
Benavides, M. V. et al. Identification of novel loci associated with gastrointestinal parasite resistance in a Red Maasai x Dorper backcross population. PLoS ONE 10, e0122797 (2015).
Richardson, I. W. et al. A genome-wide association study for genetic susceptibility to Mycobacterium bovis infection in dairy cattle identifies a susceptibility QTL on chromosome 23. Genet. Select. Evol. 48, 19 (2016).
Atlija, M., Arranz, J.-J., Martinez-Valladares, M. & Gutiérrez-Gil, B. Detection and replication of QTL underlying resistance to gastrointestinal nematodes in adult sheep using the ovine 50K SNP array. Genet. Select. Evol. 48, 4 (2016).
Liu, J. et al. Genetic signatures of high-altitude adaptation and geographic distribution in Tibetan sheep. Sci. Rep. 10, 1–13 (2020).
Lv, F.-H. et al. Adaptations to climate-mediated selective pressures in sheep. Mol. Biol. Evol. 31, 3324–3343 (2014).
Oget, C., Servin, B. & Palhiere, I. Genetic diversity analysis of French goat populations reveals selective sweeps involved in their differentiation. Anim. Genet. 50, 54–63 (2019).
Gossner, A., Wilkie, H., Joshi, A. & Hopkins, J. Exploring the abomasal lymph node transcriptome for genes associated with resistance to the sheep nematode Teladorsagia circumcincta. Vet. Res. 44, 68 (2013).
Calvo, J., Marcos, S., Jurado, J. & Serrano, M. Association of the heart fatty acid-binding protein (FABP3) gene with milk traits in Manchega breed sheep. Anim. Genet. 35, 347–349 (2004).
Li H., Wu X.L., Tait Jr R., Bauck S., Thomas D., Murphy T. & Rosa G. (2020) Genome‐wide association study of milk production traits in a crossbred dairy sheep population using three statistical models. Anim. Genet.
Marete, A. G. et al. A meta-analysis including pre-selected sequence variants associated with seven traits in three French dairy cattle populations. Front. Genet. 9, 522 (2018).
Chen, Z., Yao, Y., Ma, P., Wang, Q. & Pan, Y. Haplotype-based genome-wide association study identifies loci and candidate genes for milk yield in Holsteins. PLoS ONE 13, e0192695 (2018).
Nafikov, R. et al. Association of polymorphisms in solute carrier family 27, isoform A6 (SLC27A6) and fatty acid-binding protein-3 and fatty acid-binding protein-4 (FABP3 and FABP4) with fatty acid composition of bovine milk. J. Dairy Sci. 96, 6007–6021 (2013).
Gebreselassie, G., Berihulay, H., Jiang, L. & Ma, Y. Review on genomic regions and candidate genes associated with economically important production and reproduction traits in sheep (Ovies aries). Animals 10, 33 (2020).
Zhang, L. et al. Genome-wide association studies for growth and meat production traits in sheep. PLoS ONE 8, 1 (2013).
Cádiz, M. I. et al. Whole genome re-sequencing reveals recent signatures of selection in three strains of farmed Nile tilapia (Oreochromis niloticus). Sci. Rep. 10, 1–14 (2020).
Ablondi, M., Viklund, Å., Lindgren, G., Eriksson, S. & Mikko, S. Signatures of selection in the genome of Swedish warmblood horses selected for sport performance. BMC Genom. 20, 717 (2019).
Blanco, F. C., Soria, M. A., Klepp, L. I. & Bigi, F. ERAP1 and PDE8A are downregulated in cattle protected against bovine tuberculosis. J. Mol. Microbiol. Biotechnol. 27, 237–245 (2017).
Willer, C. J. et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45, 1274 (2013).
Kim, S.-J. et al. Cattle genome-wide analysis reveals genetic signatures in trypanotolerant N’Dama. BMC Genom. 18, 371 (2017).
Brym, P. & Kamiński, S. Microarray analysis of differential gene expression profiles in blood cells of naturally BLV-infected and uninfected Holstein-Friesian cows. Mol. Biol. Rep. 44, 109–127 (2017).
Pasandideh, M., Rahimi-Mianji, G. & Gholizadeh, M. A genome scan for quantitative trait loci affecting average daily gain and Kleiber ratio in Baluchi Sheep. J. Genet. 97, 493–503 (2018).
Smith, J. L. et al. Genome-wide association and genotype by environment interactions for growth traits in US Gelbvieh cattle. BMC Genom. 20, 926 (2019).
Pattaro, C. et al. A meta-analysis of genome-wide data from five European isolates reveals an association of COL22A1, SYT1, and GABRR2with serum creatinine level. BMC Med. Genet. 11, 41 (2010).
Zhang, F. et al. Genetic architecture of quantitative traits in beef cattle revealed by genome wide association studies of imputed whole genome sequence variants: I: feed efficiency and component traits. BMC Genom. 21, 36 (2020).
Yudin, N. & Larkin, D. Shared signatures of selection related to adaptation and acclimation in local cattle and sheep breeds from Russia. Russ. J. Genet. 55, 1008–1014 (2019).
Nie, F.-Y. et al. Methylome-wide association study of first-episode schizophrenia reveals a hypermethylated CpG site in the promoter region of the TNIK susceptibility gene. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1, 110081 (2020).
Browning, S. R. & Weir, B. S. Population structure with localized haplotype clusters. Genetics 185, 1337–1344 (2010).
Ceballos, F. C., Joshi, P. K., Clark, D. W., Ramsay, M. & Wilson, J. F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 19, 220 (2018).
Consortium B.H. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 324, 528–532 (2009).
Faostat F. Available online: http://www.fao.org/faostat/en/#data. QC (accessed on January 2018) (2017).
Luu, K., Bazin, E. & Blum, M. G. pcadapt: an R package to perform genome scans for selection based on principal component analysis. Mol. Ecol. Resour. 17, 67–77 (2017).
Randhawa, I. A. S., Khatkar, M. S., Thomson, P. C. & Raadsma, H. W. Composite selection signals can localize the trait specific genomic regions in multi-breed populations of cattle and sheep. BMC Genet. 15, 34 (2014).
Author information
Authors and Affiliations
Contributions
S.E. revised the main manuscript text and did the new analyses, G.S. helped to revise the main manuscript, M.A.R. helped to do the new analysis and M.O.K. helped to complete Table 1.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eydivandi, S., Roudbar, M.A., Karimi, M.O. et al. Genomic scans for selective sweeps through haplotype homozygosity and allelic fixation in 14 indigenous sheep breeds from Middle East and South Asia. Sci Rep 11, 2834 (2021). https://doi.org/10.1038/s41598-021-82625-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-82625-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
