
Abstract
Drosera rotundifolia has been traditionally used for the treatment of respiratory diseases in phytotherapy and homeopathy. The mechanisms of action recognized so far are linked to the known effects of specific components, such as flavonoids, but are not completely understood. In this study, the biological functions of D. rotundifolia were explored in vitro following the treatment of bronchial epithelial cells, which are the potential targets of the pharmacological effects of the herbal medicine. To do so, the whole plant ethanolic extract was 1000-fold diluted in water (D. rotundifolia 3×) and added to a 16HBE human cell line culture for 3 h or 6 h. The effects on gene expression of the treatments and corresponding controls were then investigated by RNA sequencing. The differentially expressed genes were validated through RT-qPCR, and the enriched biological functions involved in the effects of treatment were investigated. D. rotundifolia 3× did not impair cell viability and was shown to be a stimulant of cell functions by regulating the expression of dozens of genes after 3 h, and the effects were amplified after 6 h of treatment. The main differentially expressed genes encoded ligands of epithelial growth factor receptor, proteins involved in xenobiotic detoxification and cytokines, suggesting that D. rotundifolia 3× could stimulate self-repair systems, which are impaired in airway diseases. Furthermore, D. rotundifolia 3× acts on a complex and multifaceted set of genes and may potentially affect different layers of the bronchial mucosa.
Similar content being viewed by others


Introduction
Pathologies of the airways are among the most common diseases. Cough is a common complaint in patients of all ages, with air pollutants contributing to airway challenge. In children, cough is the second most common symptom of respiratory disease after runny nose, with a 46–56% prevalence depending on the age of the child. The use of pharmacological treatment without side effects could be useful not only to reduce cough symptoms but also to prevent the toxic effects of pollution. Low doses of plant-derived drugs are largely used for cold and cough symptoms in adults and children1,2,3,4,5,6,7. Often, these are used with the hope of resolving ailments not successfully cured by conventional drugs as a complementary treatment to reduce the consumption of anti-inflammatory drugs or steroids that may have adverse effects and to relieve certain symptoms and improve the quality of life. However, there is presently no definitive explanation for the possible biological activity of such preparations at the cellular level.
European sundew species (Drosera rotundifolia L., Drosera intermedia Hayne, and Drosera anglica Huds) have been used as traditional medicines in the therapy of respiratory tract infections. Drosera Herba, which is comprised chiefly of Drosera rotundifolia, has been commonly used for its spasmolytic properties in the treatment of convulsive or whooping cough since the seventeenth century. The general action on the respiratory system of D. rotundifolia was described in the homeopathic Materia Medica8 and includes profuse expectoration and a spasmodic, paroxysmal, dry, and irritative cough, similar to whooping cough, and the plant is often included as a component in homeopathic complexes used for upper respiratory tract ailments. The efficacy of such remedies for the relief of symptoms or the improvement of quality of life was analysed in clinical studies, which reported positive results5,9,10,11,12.
Ethanol extracts of Drosera species contain high concentrations of flavonoids (hyperoside, isoquercitrin, quercetin, and myricetin-3-O-galactoside) together with phenolic acids (ellagic acid)13. Flavonoids have been reported to have anti-inflammatory14, antioxidant and antimicrobial properties, which are efficacious in the treatment of respiratory diseases15. Traces of naphthoquinones were also found in some preparations16.
Bronchial epithelial cells are specialized cells strictly connected to a tissue that form a barrier that protects against external agents of damage. Epithelial membrane integrity could be injured by pathogenic events that damage basal cellular activities (cell cycle, viability, energy balance and oxidative stress) or impair specific activities, such as the maintenance of the electrolytic balance (functioning of membrane ion channels)17,18,19. With appropriate stimulation, the bronchial epithelium can contribute to innate immunity by secreting immune-stimulatory and modulatory mediators, including cytokines, chemokines, growth factors, and lipid mediators that recruit and activate effector cells and antigen-presenting cells20,21,22. Differentiated 16HBE cells simulate many of the activities of the bronchial epithelium and are considered a good model for basic studies.
The aim of this study was to evaluate the effect of a low homeopathic concentration of D. rotundifolia ethanolic extract on the transcriptome of the human bronchial epithelial cell line 16HBE. Transcriptomics, which is an important tool of functional genomics research, can be used to study overall gene expression and function, which could be the first step in revealing specific biological processes and molecular mechanisms that are involved in the occurrence of the disease. For this study, a D. rotundifolia third decimal (3×) dilution was used (1000 times more diluted than the whole ethanolic extract). The 3× terminology corresponds to the traditional homeopathic nomenclature according to the Anglo-American convention23. The goal of the study was to discover the potential gene targets of D. rotundifolia 3× after 3 and 6 h of incubation by means of the RNA-seq technique. After validating the main genes using RT-qPCR, the main biological functions involved in D. rotundifolia 3× action were identified.
Results
Analysis of polyphenols in the D. rotundifolia ethanolic extract
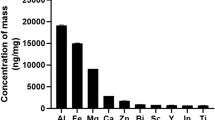
The concentration of polyphenols in the D. rotundifolia ethanolic extract was estimated to be 7.1 × 10−3 mol/L in terms of the gallic acid equivalents. This concentration corresponds to approximately 1.2 mg/mL of total polyphenols, which in turn corresponds to a concentration of 12 µg/mL in the D. rotundifolia 3× test sample and 1.2 µg/mL (7.1 × 10−6 mol/L) in the final cell culture. The D. rotundifolia ethanolic extract was also qualitatively analysed by mass spectrometry. In the HPLC 6.590 min retention time peak, the mass spectrum showed peaks corresponding to ellagic acid (m/z = 300.997905) and isoquercitrin (m/z = 463.087935). In addition, at the 8.004 min retention time peak, quercetin (m/z = 301.035282) was detected. Regarding the presence of hyperoside, which has the same molecular mass as isoquercitrin, there is a chance that the isoquercitrin peak represented a mixture of both compounds. The results confirmed the presence of important bioactive components in D. rotundifolia ethanolic extract13.
Effect of D. rotundifolia 3× on 16HBE cell viability
The viability of the bronchial epithelial cells (16HBE) (Fig. 1) was not impaired after 3 h of exposure to D. rotundifolia 3× or the corresponding control (Ctrl), demonstrating the absence of cytotoxicity caused by the plant preparation at the dilutions used in the experiments. Furthermore, after 24 h of incubation with D. rotundifolia 3×, the cells showed a small (+ 6.9%) but significant (p = 0.019) increase in cell viability. No morphological changes or alteration of cell adherence to the bottom of the well could be observed by optical microscopic inspection (data not shown).

Viability of 16HBE cells incubated with D. rotundifolia 3 × or Ctrl for 3 h or 24 h. The colorimetric assay was based on the reduction of the WST reagent as described in the “Materials and method” section.
Changes in gene expression after D. rotundifolia 3× treatment
The effects of D. rotundifolia 3× treatment on the global gene expression of 16HBE cells were investigated after 3 h of incubation by comparison with the control. The experiments were reproduced in 4 different biological replicates, and the gene expression profile in response to D. rotundifolia 3× treatment was investigated by RNA-seq analysis.
The reads obtained by the sequencing were quantified as transcripts by Salmon, which showed an average mapping coverage of 92%. Differential gene expression analysis was then performed to identify the significant target genes of D. rotundifolia 3× treatment. The differential expression output obtained from DESeq2 analysis displayed a total of 69 statistically significant (padj < 0.05) differentially expressed genes (DEGs); 44 genes were upregulated (Table 1), and 25 genes were downregulated (Table 2). The lof2FoldChange ranged from + 0.7 (maximum upregulation, Table 1) to − 0.51 (maximum downregulation, Table 2).
Gene ontology classification of differentially expressed genes
To better understand the functions of the identified genes, functional analysis was performed by submitting the up- and downregulated gene list to gene ontology (GO) enrichment analysis using the Bioconductor/R package gprofiler2. The representative enriched biological processes and the genes associated with these processes are represented in Table 3. The DEGs were mainly enriched in biological processes, including general functions such as “regulation of response to stimulus”, which includes 34 of the 69 genes, and more specific biological activities, such as “vasculature development” (13 of 69 genes) and “positive regulation of vascular endothelial growth factor production” (5 of 69 genes). A group of 10 genes is associated with the function “epithelial cell proliferation”. This gene set includes the upregulated gene LGR5 (a GPCR receptor protein and member of the Wnt signalling pathway that controls cell proliferation), the downregulated gene IGFBP3 (encoding insulin-like growth factor-binding protein 3, which exerts anti-proliferative effects in many cell types) and 8 other genes with specific GO functions related to epithelial regulation (the upregulated epithelial growth factor-like ligands EREG and EPGN, the downregulated EGF-linked inhibitor ERRFI1), proteins involved in cell membrane repair (DYSF) or in tissue development (NOG) and two transcription factors (HIF1A and JUN). A group of DEGs is related to the production of cytokines, suggesting their possible role in immunity. Other processes that are significantly associated with the target genes are “regulation of cell death” and “positive regulation of the mitotic cell cycle”, suggesting their involvement in the recovery of epithelial tissue.
RT-qPCR validation of the D. rotundifolia 3× target genes
For mRNA expression validation, genes that are associated with enriched biological processes and identified by RNA-seq were investigated by RT-qPCR to validate the effects of D. rotundifolia 3× on mRNA expression (Table 4). Importantly, genes were selected mainly if they were associated with a biological function (Table 3), and then cut-off values were applied to the genes reported in Tables 1 and 2 as follows: log2FoldChange > 0.1 and < − 0.1, baseMean > 100 and transcripts per million (TPM) > 1 (data not shown). For many candidate genes, the analysis of their expression based on the experimental series of the RNA-seq study was corroborated by the inclusion of results from 4 new follow-up experiments (for a total of 8 independent samples).
As shown in Table 4, RT-qPCR confirmed the significant changes in the regulation of 10 genes identified by the RNA-seq analysis. The remaining investigated genes showed changes similar to those revealed by RNA-seq (see Tables 1, 2), which were not statistically significant. Notably, the differential expression changes in the relevant genes associated with the enriched biological processes, such as CY1B1, EREG, AREG, CTGF, and ERR1F1, were verified by RT-qPCR in new follow-up experiments performed with cells treated for 3 h with D. rotundifolia 3×.
Time course of the D. rotundifolia 3× effect
Some representative and very well-expressed genes were selected to investigate the kinetics of the effects of D. rotundifolia 3× during the time course of the cell treatment. A new series of four experiments was performed, in which the cells were incubated for 2 h, 6 h, and 24 h with the medicine or the control. Figure 2 displays the mRNA expression of AREG, CTGF, CXCL8, CYP1B1, EGR1, EREG, IL-1α and TIPARP in cells with and without D. rotundifolia 3× at the indicated times. The results of all four experiments are separately shown since one of the experiments resulted in higher levels of mRNA expression for a few genes (e.g., CXCL8, EREG and IL-1α). Despite the heterogeneity in the basal gene expression, the direction of the effect is clear-cut, and it is largely significant for at least one time point based on the statistics for the paired data. The experiments showed that the effect of D. rotundifolia 3× on most genes starts already at two hours, it is expressed in a higher degree after 6 h, while it decreases at 24 h. Exceptions to this trend are CYP1B1 and TIPARP, which showed the maximum activity at 2 h, which was decreased at 6 h and further decreased at 24 h.

Kinetics of AREG, CTGF, CXCL8, CYP1B1, EGR1, EREG, IL-1α and TIPARP mRNA expression in 16HBE cells incubated with D. rotundifolia 3x. 16HBE cells were cultured with D. rotundifolia 3× (Dro 3×) or Ctrl (Ctrl 3×) for up to 24 h to evaluate mRNA expression by RT-qPCR. Gene expression is depicted as the mean normalized expression (MNE) after GAPDH mRNA normalization (n = 4). Asterisks indicate a significant increase: *p < 0.05, **p < 0.01, ***p < 0.001, #p = 0.057 by two-way ANOVA followed by Bonferroni’s post-test.
Based on these results, a new RNA-seq analysis of the samples incubated for 6 h with D. rotundifolia 3× was performed to describe in detail and under the best conditions all of the effects of the plant on the gene transcription of bronchial cells.
RNA sequencing of 16HBE cells treated with D. rotundifolia 3× for 6 h
RNA-seq was performed using the same parameters used for the RNA-seq of 16HBE cells treated with D. rotundifolia 3× for 3 h as well as the bioinformatics analysis. All samples passed the quality control tests as described for the RNA-seq of the 3 h-treated samples.
The DESeq2 output generated a list of 495 DEGs in 16HBE cells after 6 h of treatment with D. rotundifolia 3× compared to treatment with the control (padj < 0.05), which contains a majority of upregulated genes (n = 334). Figure 3 depicts the DEGs in a Volcano plot, which highlights (blue dots) all the genes (n = 117) that had a statistically significant change (adjusted p-value < 0.05) and a log2FoldChange higher than + 0.4 or less than − 0.4. For this analysis, a log2FoldChange cut-off value was used since the number of DEGs was much higher compared to that identified by the RNA-seq performed for the 3 h treatment. Figure 3 shows that after 6 h of incubation, there were a high number of DEGs, especially upregulated genes, which is in accordance with what is shown in Fig. 2. From a qualitative point of view, a similarity is observed in the genes whose expression is increased at the two different incubation times. Such a similarity is reflected by the upregulation of some genes, such as CYP1B1, EPGN, EREG, SERPINB2, IL-1α, AREG, and PHDLA1, which were also upregulated by cells treated with D. rotundifolia 3× for 3 h (see Table 1). Regarding the downregulated genes (left side of Fig. 3), some new genes associated with an inhibitory effect are highlighted, including IGFBP3, PLAC8, MAN2B2, and BNIP3, which, except for IGFBP3, were identified only after 6 h of incubation and not after a shorter exposure (2 or 3 h). Moreover, genes that were slightly downregulated after 3 h of D. rotundifolia 3× treatment, such as ERRFI1, were not found to be regulated after 6 h of treatment.

Volcano plot of the differential expression analysis of 16HBE cells treated with D. rotundifolia 3× for 6 h. The scattered points represent genes: the x-axis is the log2foldchange for the D. rotundifolia 3×-treated 16HBE cells. The y-axis shows the log1p(− log10 adjusted p-value), which better highlights the differentially expressed genes in our experimental conditions. Blue dots represent genes that were significantly differentially expressed (adjusted p-value < 0.05) with a log2foldchange < − 0.4 or > 0.4 after 6 h treatment with D. rotundifolia 3×.
Functional analysis
The 117 differentially expressed genes were ranked by the adjusted p-value significance, and GO enrichment analysis was performed using gprofiler2 as described in the “Materials and methods” section. The enrichment results for the 6 h data set (Fig. 4) show a wide range of information concerning the biological processes (blue bars in Fig. 4) and molecular functions (red bars). Similar enriched biological processes were observed after 6 h and 3 h of D. rotundifolia 3× treatment, such as functions related to blood vessel development, regulation of epithelial cell proliferation and cytokine production. Moreover, there was an increase in biological processes related to inflammation and chemotaxis after 6 h of D. rotundifolia 3× treatment due to the upregulation of additional cytokines and chemokines. These data confirm that there is an increase in the effects and suggest new functions associated with D. rotundifolia 3× treatment of 16HBE cells after 6 h.

Representative gene ontology classifications of the differentially expressed genes in 16HBE cells treated with D. rotundifolia 3×. The differentially expressed genes were classified into two categories: biological processes and molecular functions. Gene ontology enrichment analysis was performed using the “gost” function from the gprofiler2 package according to the gene list ranked by the adjusted p-value. GO terms with p-values < 0.05 were considered significant.
To draw a functional picture of the enriched functions and their correlations, a network that associates the genes with the functions in the biological model of bronchial epithelial cells was constructed (Fig. 5). Functions are representative and were chosen based on our experimental model and enrichment p-value. The Log2FoldChange values from the DESeq2 output are depicted by the coloured nodes, which shows that the enrichment of these biological functions is mostly associated with the upregulated genes. This was expected since the number of upregulated genes observed after D. rotundifolia 3× treatment was almost 3 times higher than the number of downregulated genes. Moreover, genes that were upregulated by D. rotundifolia 3× are associated with many biological processes, while this was not the case for the downregulated genes.

Functional network of enriched biological processes and associated genes. The network was constructed using the selected significant GO terms shown in Fig. 4, which were uploaded into Cytoscape software as described in “Materials and method” section. Diamonds indicate biological processes and circles indicate associated genes.
Discussion
In this study, functional genomic research was performed to explore the effects of a medicinal plant traditionally used for respiratory diseases on its potential target cells in vitro, i.e., bronchial epithelial cells. This study shows that a diluted herbal extract used in a traditional homeopathic pharmacopoeia5,9,10,11,12 can induce a change in mRNA expression that can be measured reliably and reproducibly. Low doses of traditional plants and homeopathic dilutions are now being investigated by means of molecular biology techniques for differential gene expression analysis and bioinformatics24,25,26,27,28.
Screening in bronchial cells with RNA-seq analysis showed that low amounts of D. rotundifolia at a 3× dilution, which represents a dose commonly used in homeopathic and phytotherapeutic syrups, changed the expression of dozens of genes after 3 h, and this effect was amplified after 6 h of treatment. Validation with RT-qPCR confirmed the differential expression of the genes of interest and showed that after 6 h of D. rotundifolia 3× treatment, there was an increase in mRNA expression. Since RT-qPCR sensitivity is higher than that of RNA-seq, such results support the findings of the RNA-seq analysis, suggesting that treatment with D. rotundifolia × can be followed-up by the investigation of the expression of these genes.
In our experiments, D. rotundifolia 3× did not impair cell viability or adherence, suggesting that this dilution is safe when tested directly in cell culture. The same dilution exerted a stimulatory effect on the expression of several genes, including those of inflammatory cytokines. Furthermore, the transcriptomic analysis and RT-qPCR confirmation suggest that, at the dilution used, D. rotundifolia 3× works mainly as a stimulant and not as an inhibitor of cell functions. This result is different from the data reported by others in HMC-1 human mast cells29, suggesting the anti-inflammatory effects of the plant. This apparent discrepancy could be due to the difference in the biological model, since mast cells are typical inflammatory cells and were stimulated before treatment with D. rotundifolia, while our epithelial cells were treated without previous inflammatory stimuli. However, even more importantly, different doses may be utilized: Fukushima et al.29 obtained an inhibitory effect by using nondiluted fractions of the whole extract of D. rotundifolia, while in the present study, the cells were treated with a 3× dilution (1000 times) of the ethanolic extract.
Paper et al.30 reported that extracts of D. rotundifolia and other species, tested in hen’s egg model, show efficacy as anti-inflammatory, antispasmodic and antiangiogenic agents. In contrast, the data of our study suggest that treatment with D. rotundifolia 3× can trigger a mild inflammatory response in 16HBE cells. The latter effect can be attributed to the increase in the mRNA expression of pro-inflammatory cytokines such as IL-1α, IL-1β and IL-6 and chemokines such as CXCL1, CXCL2 and CXCL8. Discrepancies in the data from Paper et al.30 could be due to the different models or, again, to the doses applied, since we used a homeopathic dilution of the plant extract, which represents a biological stimulus and does not inhibit key cell functions24, as conventional anti-inflammatory agents do. Moreover, it is well known that a mild inflammatory response could be beneficial to the immune system by increasing the apoptosis and clearance of inflammatory cells31,32. This concept is fully in agreement with homeopathic theory and tradition, in which low doses of pathogenic substances and/or minimally stressful stimuli trigger endogenous responses of “vital energy”, which eventually lead to healing at the cell, tissue or systemic levels33,34,35. The presence of CXCL1, CXCL2 and CXCL8, which are mostly chemoattractants for neutrophils, suggests that treatment with D. rotundifolia 3× can help to establish a ready-state immune barrier against pathogens in case of bronchial epithelial tissue damage36.
Interestingly, D. rotundifolia 3× treatment increased the expression of specific epidermal growth factors, such as AREG, EREG and EPGN, by 16HBE cells. The upregulation of these factors suggests that D. rotundifolia 3× plays a positive role in the regulation of cell survival, cell proliferation and wound healing. Moreover, all these factors bind to epidermal growth factor receptor (EGFR) to accomplish their functions. In this context, EGFR is widely expressed on the cells present in the bronchial environment, suggesting that the possible release of these growth factors affects not only the proliferation of bronchial epithelial cells but also that of endothelial cells, fibroblasts and vascular cells. In the case of damage to bronchial epithelial tissue, D. rotundifolia 3× could contribute to faster healing of the bronchial microenvironment.
CYP1B1 was the most highly expressed gene after treatment with D. rotundifolia 3× at both time points shown in this study. CYP1B1 is probably activated as a response to treatment with D. rotundifolia 3x, since CYP1B1 belongs to the cytochrome family and is involved in the metabolism of xenobiotics. Moreover, as shown in Fig. 5, CYP1B1 is connected to the regulation of angiogenesis and the positive regulation of cytokine production because of its role as an antioxidant and NF-kB regulator37,38, respectively.
This study has some limitations since some of the findings are based on bioinformatics analysis, and mRNA expression and functional experiments are required to verify the mechanisms of action and effects of the treatment with D. rotundifolia 3×. In addition, it is important to observe that after 6 h of D. rotundifolia 3× treatment, there was a number of differentially expressed genes with log2FoldChange values ranging from + 0.4 to − 0.4, which were not included in the analysis for this manuscript but could be considered for a future study. Indeed, the effects on gene regulation represent only a first functional step of the action of the plant that we have highlighted, and our hypotheses will have to be confirmed with adequate studies in laboratory animals to investigate the therapeutic potential and the mechanisms of action of this plant.
In conclusion, the data highlight the complex and multifaceted action of the plant on the different layers of the bronchial mucosa, as summarized in Fig. 6. The mRNA expression changes in 16HBE cells treated with D. rotundifolia 3× suggest its direct action on the epithelial cell, which protects its integrity with respect to toxic substances (CYP1B1) and stimulates its reparative capacity (AREG, EREG and EPGN). In addition, epithelial cells transmit molecular signals that activate mild inflammation (pro-inflammatory cytokines) and recruit innate defence- and angiogenesis-associated cells.

Illustrative graphic summarizing the potential effects of D. rotundifolia 3× on bronchial epithelial cells. The presence of D. rotundifolia 3× induces the expression of epidermal growth factors (AREG, EREG and EPGN), leading to the growth/repair of bronchial epithelial cells and other cells in the bronchial environment. Those genes, which bind to EGFR to accomplish their functions, are regulated by ERFFI1, which is in turn downregulated by D. rotundifolia 3× in the first 3 h. CYP1B1 is highly expressed and is related to the metabolism of xenobiotics and could be involved in the regulation of angiogenesis. Furthermore, inflammatory cytokines (IL-1α and IL-6) and chemokines (CXCL1, CXCL2 and CXCL8) are induced by D. rotundifolia 3× and can trigger mild inflammation, increasing chemotaxis and angiogenesis, helping the system in fighting infections.
Materials and methods
Preparation of D. rotundifolia samples
The original round leaf sundew plant, identified with the botanical name Drosera rotundifolia, was commercially purchased (Monteagle Herbs, ON, Canada; Document of Authenticity #42808). To the best of our knowledge, no issues related to the Convention on the Trade in Endangered Species of Wild Fauna and Flora have been raised. Drosera rotundifolia (ethanolic extract) was prepared by Standard Homeopathic Company, CA, USA. The extract contained 1 g dried whole plant macerated in 10 mL of 45% EtOH, which corresponds to D. rotundifolia 1×. Before the experiments, D. rotundifolia 1× ethanolic extract was serially diluted 1:10 (0.5 mL + 4.5 mL) twice in sterile pyrogen-free water (B-Braun, Melsungen, Germany) in a 14-mL clean glass tube, which immediately vigorously shaken (succussed) with a Dyna A mechanical shaker that delivered 20 strokes/s for 7.5 s with an 11-mm travel distance. The final solution corresponds to a 3× dilution in 0.45% ethanol solution. The control solution was prepared starting from 45% ethanol (AppliChem, Darmstadt, Germany) fresh solution and succussed as previously described. This was considered the 1× control solution. Two serial decimal dilutions/succussions in ultrapure water were applied as reported above to obtain the control 3× dilution (Ctrl). No filtration was applied at any step. These solutions were used in the culture media at a 1:10 ratio (0.1 mL test solution in 0.9 mL culture medium). Therefore, the final dilution of D. rotundifolia was 1000 times greater than that of the ethanolic extract and the ethanol concentration was 0.045%.
Phenolic quantification
The total phenolic content of the D. rotundifolia ethanolic extract was determined by the Folin–Ciocalteu assay39. Briefly, 50 μL of extract at an appropriate dilution was mixed with 155 μL of Folin–Ciocalteu reagent diluted 1:10 v/v with water. After 1 min, 40 μL of 20% Na2CO3 solution was added, and the samples were incubated for 30 min in the dark at 37 °C. The absorbance of each sample was measured at 765 nm. Gallic acid was used as a standard for the calibration curve, and the phenolic content was expressed as gallic acid equivalents. Each determination was repeated three times, and the results are expressed as the mean ± SD.
Mass spectrometry
A series 1260 HPLC (Agilent Technologies, Waldbronn, Germany) in tandem with a Q-TOF mass spectrometer was used for the present study. The separation was performed on a Zorbax Eclipse XDB (2.1 × 150 mm, 5 µm particle size, Agilent Technologies) with gradient elution. Formic acid (0.1%) was solvent A, and methanol was solvent B; the flow rate was 500 µL/min. An injection volume of 5 µL was used in all experiments. Samples were eluted with a linear gradient from 20 to 95% of solvent B. The MS analysis was carried out using a model 6540 Accurate-Mass Q-TOF LC/MS (Agilent Technologies, Palo Alto, CA, USA) online with HPLC. QTOF-MS was implemented with an electrospray ion source with Agilent Jet Stream technology operating in negative ionization mode. Data acquisition was performed in full scan mode in the mass range of 100–1000 m/z40. The standards used were quercetin, ellagic acid (Sigma–Aldrich, Saint Louis, MO, USA), isoquercitrin and hyperoside (HWi, Ruelzheim, Germany).
Cell treatment
16HBE human bronchial epithelial cells (line 16HBE14o-, kindly provided by Dr Gruenert, University of California, San Francisco) were grown for 1 week in EMEM medium supplemented with 10% FBS (including 2 mM UltraGlutamine, 100 U/mL penicillin and 100 µg/mL streptomycin) from cryogenically frozen samples (2 × 106 cells/vial) prepared from the batch culture (P14, 14th culture passage from the original culture). Fibronectin/collagen/BSA-coated flasks were used41. The culture medium was replaced every 3 days. Prior to the treatment, on day 1, cells were seeded in 24-well plates (uncoated) at a density of 0.4 × 106 cells/well in EMEM medium with 2% FBS (including 2 mM UltraGlutamine, 100 U/mL penicillin and 100 µg/mL streptomycin). The plates were incubated until complete adhesion of the cells occurred (16–20 h). On day 2, the culture medium was replaced by 0.9 mL of fresh EMEM with 2% FBS. D. rotundifolia 3× or Ctrl was added (0.1 mL/well). The plates were incubated for 2 h, 3 h, 6 h or 24 h depending on each experimental setting. The final volume was 1 mL/well. The Ctrl cell cultures were carried out in parallel with those treated with D. rotundifolia for the indicated times in the same culture plates. After the desired incubation period, treated and Ctrl 16HBE cells were collected and subjected to RNA extraction.
Evaluation of cell viability
Cell viability was checked by the cell proliferation reagent WST-1 (Gibco ThermoFisher Scientific). The WST assay evaluates the cell metabolic activities (NADH reductase) by measuring the chemical modification of the WST tetrazolium salt. 16HBE cells were seeded at a density of 50,000 cells/well in 96-well plates and treated for 3 or 24 h with the D. rotundifolia 3× or the Ctrl solutions, which were added to the cell culture at a 1/10 volume ratio. After treatment, 1:10 (v/v) prewarmed WST-1 solution was added to the cells, and the plate was incubated for 90 min. The absorbance (OD) of the samples was measured using a Victor3 multilabel reader (PerkinElmer, Shelton, CT, USA) at 450 nm. Cell viability data were evaluated by t-test statistics (D. rotundifolia 3× vs Ctrl).
RNA extraction
Total RNA from cultured 16HBE cells was isolated using the RNeasy mini Kit (Qiagen, Venlo, Limburg, Netherlands) according to the manufacturer’s instructions. An on-column DNase digestion with the RNase-free DNase kit (Qiagen) was also performed during total RNA isolation to completely remove any possible contaminating DNA. RNA quality and quantity were determined using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA)42.
RNA sequencing (RNA-seq)
RNA samples were processed in the Genomic and Transcriptomic Unit at the Technologic Platform Centre (University of Verona, Italy). Total RNA extracted from 16HBE cells was assessed for quality (integrity) using an RNA 6000 Nano Kit (Agilent, Wokingham, UK). The samples with RNA integrity numbers > 9 were considered adequate for library preparation. RNA aliquots (2.5 μg) were used to isolate poly(A) mRNA for the preparation of a directional Illumina RNA-Seq library using the Illumina TruSeq Stranded mRNA Library Prep Kit (Illumina Inc., San Diego, CA, USA). The quality of the library before the generation of the clusters was checked through the visualization of the DNA fragments in the miniaturized electrophoresis system Tape Station (Agilent D1000 Screen Tape system). The average insert sizes were in the range of 300–370 bp. The libraries were also quantified by qPCR using the KAPA Library Quantification kit (Kapa Biosystems Inc., Woburn, MA, USA). The libraries (differentially labelled) were pooled in equal amounts before sequencing. Sequencing was performed with an Illumina NextSeq 500 (Illumina, CA, USA). The samples were sequenced with a single-end protocol (75 base pairs) with a sequence depth of approximately 30 M reads for the sample. After sequencing, the FastQC High Throughput Sequence QC Report (Version 0.11.8) was used to assess the quality of the sequencing based on the FASTQ files.
Transcript quantification was performed with the high-performance computing server at the Computational Unit of the Technologic Platform Centre (University of Verona, Italy). Transcript quantification was determined from the FASTQ reads using the mapping-based mode of Salmon (Version 0.13.1)43 with the following parameters: “-i index -l SR -r name -validateMappings -gcBias”. The reference sequence and annotation files were downloaded from the GENCODE repository (Human Release 32, GRCh38.p13) at the following website: https://www.gencodegenes.org/human/. The output of Salmon was then imported into the RStudio environment (R Version 3.5.3; R studio version 1.1.463) using Bioconductor/R package tximport (Version 1.10.10)44, which converted data from the transcript level to the gene level during the importing process. Genes with less than 10 reads were discarded from the posterior analysis. Differential expression analysis was performed using the Bioconductor/R package DESeq2 (Version 1.22.2)45. After differential analysis, the lfcShrink function in DESeq2 was applied to shrink the log2FoldChanges. Significance values were based on a Wald significance test, and differences in gene expression with an adjusted p-value < 0.05 (corrected by the Benjamini–Hochberg method) were considered significant. For the RNA-seq analysis of the experiments, in which the cells were treated for 6 h with D. rotundifolia 3×, a log2FoldChange cut-off value (< 0.4 and > 0.4) was applied. To associate a function with the differentially expressed genes, functional enrichment analysis was performed using the Bioconductor/R package gprofiler2 (Version 0.1.7)46. The differentially expressed genes were ranked by the adjusted p-value significance, and gene ontology (GO) analysis was queried using the “gost” function in gprofiler2 in “ordered” mode and “g_SCS” as correction/statistical method. The list of significant GO terms was used for the construction of the network using Cytoscape software (Version 3.7.2)47.
Reverse transcription quantitative real-time PCR (RT-qPCR)
Total RNA extracted from 16HBE cells was reverse transcribed into cDNA using the PrimeScript RT reagent Kit (Takara Bio, Kusatsu, Japan), while qPCR was carried out using TB Green Premix Ex Taq (Tli RNase H Plus) (Takara Bio)42. The sequences of the gene-specific primers (Thermo Fisher Scientific, Waltham, Massachusetts, USA) used in this study are listed in Table 5. qPCR was performed using a Viia7 Real-time PCR system (Thermo Fisher Scientific). Data were calculated by Q-Gene software (http://www.gene-quantification.de/download.html) and expressed as the mean normalized expression (MNE) units after GAPDH normalization42. Statistical evaluation was performed by two-way ANOVA followed by Bonferroni’s post hoc test. Values of p < 0.05 were considered statistically significant.
Ethics approval and consent to participate
Not applicable, as the study did not involve patients, volunteers or animals.
Data availability
The raw and processed RNA-seq data are available in the public GEO database (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE144215.
References
Sevar, R. Audit of outcome in 455 consecutive patients treated with homeopathic medicines. Homeopathy 94, 215–221 (2005).
Haidvogl, M. et al. Homeopathic and conventional treatment for acute respiratory and ear complaints: A comparative study on outcome in the primary care setting. BMC Complement. Altern. Med. 7, 7 (2007).
Saha, S. K., Roy, S. & Khuda-Bukhsh, A. R. Ultra-highly diluted plant extracts of Hydrastis canadensis and Marsdenia condurango induce epigenetic modifications and alter gene expression profiles in HeLa cells in vitro. J. Integr. Med. 13, 400–411 (2015).
Danno, K. et al. Management of the early symptoms of influenza-like illnesses and ear, nose and throat (ENT) disorders by pharmacists. Homeopathy 103, 239–249 (2014).
Zanasi, A. et al. Homeopathic medicine for acute cough in upper respiratory tract infections and acute bronchitis: A randomized, double-blind, placebo-controlled trial. Pulm. Pharmacol. Ther. 27, 102–108 (2014).
Jacobs, J. & Taylor, J. A. A randomized controlled trial of a homeopathic syrup in the treatment of cold symptoms in young children. Complement. Ther. Med. 29, 229–234. https://doi.org/10.1016/j.ctim.2016.10.013 (2016).
Frass, M. et al. Adjunctive homeopathic treatment in patients with severe sepsis: A randomized, double-blind, placebo-controlled trial in an intensive care unit. Homeopathy 94, 75–80 (2005).
Boericke, W. Pocket Manual of Homeopathic Materia Medica & Repertory (1997).
Allaert, F.-A., Villet, S., Vincent, S. & Sauve, L. Observational study on the dispensing of cough syrups to children with acute cough by community pharmacists in France. Minerva Pediatr. 70, 117–126 (2018).
Voß, H. W., Michalsen, A. & Brünjes, R. Efficacy and tolerability of a complex homeopathic drug in children suffering from dry cough-A double-blind, placebo-controlled, clinical trial. Drug Res. (Stuttg.) 68, 444–449 (2018).
Zanasi, A. et al. Does additional antimicrobial treatment have a better effect on URTI cough resolution than homeopathic symptomatic therapy alone? A real-life preliminary observational study in a pediatric population. Multidiscip. Respir. Med. 10, 25. https://doi.org/10.1186/s40248-015-0022-3 (2015).
Bordes, L. & Dorfman, P. Evaluation de l’activité antitussive du siropDrosetux: Etude en double aveugle versus placebo. Les Cah. d’otorhinolaringol. 21, 731–734 (1986).
Zehl, M. et al. Identification and quantification of flavonoids and ellagic acid derivatives in therapeutically important Drosera species by LC-DAD, LC-NMR, NMR, and LC-MS. Anal. Bioanal. Chem. 400, 2565–2576 (2011).
Paper, D. H., Karall, E., Kremser, M. & Krenn, L. Comparison of the antiinflammatory effects of Drosera rotundifolia and Drosera madagascariensis in the HET-CAM assay. Phyther. Res. 19, 323–326 (2005).
Mattioli, V. et al. Dietary flavonoids and respiratory diseases: A population-based multi-case-control study in Italian adults. Public Health Nutr. 23, 2548–2556. https://doi.org/10.1017/S1368980019003562 (2020).
Krenn, L. et al. In vitro antispasmodic and anti-inflammatory effects of Drosera rotundifolia. Arzneimittelforschung 54, 402–405 (2004).
Carsin, A. et al. Bronchial epithelium in children: A key player in asthma. Eur. Respir. Rev. 25, 158–169 (2016).
Heijink, I. H., Brandenburg, S. M., Postma, D. S. & van Oosterhout, A. J. M. Cigarette smoke impairs airway epithelial barrier function and cell-cell contact recovery. Eur. Respir. J. 39, 419–428 (2012).
Awatade, N. T. et al. Human primary epithelial cell models: Promising tools in the era of cystic fibrosis personalized medicine. Front. Pharmacol. 9, 1429 (2018).
Blume, C. & Davies, D. E. In vitro and ex vivo models of human asthma. Eur. J. Pharm. Biopharm. 84, 394–400 (2013).
Mayer, A. K., Bartz, H., Fey, F., Schmidt, L. M. & Dalpke, A. H. Airway epithelial cells modify immune responses by inducing an anti-inflammatory microenvironment. Eur. J. Immunol. 38, 1689–1699 (2008).
Riechelmann, H. et al. Differential response of mono mac 6, BEAS-2B, and jurkat cells to indoor dust. Environ. Health Perspect. 115, 1325–1332 (2007).
Fisher, P. What is homeopathy? An introduction. Front. Biosci. (Elite Ed.) 4, 1669–1682 (2012).
Bellavite, P. et al. Cell sensitivity, non-linearity and inverse effects. Homeopathy 104, 139–160 (2015).
Marotti, I. et al. Transcriptome profiling of wheat seedlings following treatment with ultrahigh diluted arsenic trioxide. Evid.-based Complement. Altern. Med. 2014, 851263 (2014).
Marzotto, M. et al. Extreme sensitivity of gene expression in human SH-SY5Y neurocytes to ultra-low doses of Gelsemium sempervirens. BMC Complement. Altern. Med. 14, 104 (2014).
Bigagli, E., Luceri, C., Dei, A., Bernardini, S. & Dolara, P. Effects of extreme dilutions of Apis mellifica preparations on gene expression profiles of human cells. Dose-Response 14, 1–7 (2016).
Lagunin, A. A. et al. Combined network pharmacology and virtual reverse pharmacology approaches for identification of potential targets to treat vascular dementia. Sci. Rep. 10, 257 (2020).
Fukushima, K. et al. Drosera rotundifolia and Drosera tokaiensis suppress the activation of HMC-1 human mast cells. J. Ethnopharmacol. 125, 90–96 (2009).
Paper, D. H., Karall, E., Kremser, M. & Krenn, L. Comparison of the antiinflammatory effects of Drosera rotundifolia and Drosera madagascariensis in the HET-CAM assay. Phytother. Res. 19, 323–326 (2005).
Yang, Y., Jiang, G., Zhang, P. & Fan, J. Programmed cell death and its role in inflammation. Mil. Med. Res. 2, 12 (2015).
Marin, N. D., Dunlap, M. D., Kaushal, D. & Khader, S. A. Friend or foe: The protective and pathological roles of inducible bronchus-associated lymphoid tissue in pulmonary diseases. J. Immunol. 202, 2519–2526 (2019).
Bellavite, P., Ortolani, R., Pontarollo, F., Pitari, G. & Conforti, A. Immunology and homeopathy. 5. The rationale of the ‘ Simile’. Evid.-Based Complement. Altern. Med. 4, 149–163 (2007).
Bell, I. R. The complexity of the homeopathic healing response part 1: The role of the body as a complex adaptive system in simillimum-initiated recovery from disease. Homeopathy. https://doi.org/10.1055/s-0039-1694998 (2019).
Bell, I. R. The complexity of the homeopathic healing response part 2: The role of the homeopathic simillimum as a complex system in initiating recovery from disease. Homeopathy. https://doi.org/10.1055/s-0039-1694999 (2019).
Peiseler, M. & Kubes, P. More friend than foe: The emerging role of neutrophils in tissue repair. J. Clin. Investig. 129, 2629–2639 (2019).
Tang, Y. et al. CYP1B1 expression promotes the proangiogenic phenotype of endothelium through decreased intracellular oxidative stress and thrombospondin-2 expression. Blood 113, 744–754 (2009).
Palenski, T. L., Gurel, Z., Sorenson, C. M., Hankenson, K. D. & Sheibani, N. Cyp1B1 expression promotes angiogenesis by suppressing NF-κB activity. Am. J. Physiol. Physiol. 305, C1170–C1184 (2013).
Singleton, V. L. & Rossi, J. A. Colorimetry of total phenolics with phosphomolybdic-phosphotungstic acid reagents. Am. J. Enol. Vitic. 16, 144–158 (1965).
Gottardo, R. et al. Screening for synthetic cannabinoids in hair by using LC-QTOF MS: A new and powerful approach to study the penetration of these new psychoactive substances in the population. Med. Sci. Law 54, 22–27 (2014).
Lechner, J. F. & Laveck, M. A. A serum-free method for culturing normal human bronchial epithelial cells at clonal density. J. Tissue Cult. Methods 9, 43–48. https://doi.org/10.1007/BF01797773 (1985).
Arruda-Silva, F. et al. Human neutrophils produce CCL23 in response to various TLR-agonists and TNFα. Front. Cell. Infect. Microbiol. 7, 176 (2017).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences [version 2; peer review: 2 approved]. F1000Research 4, 1521 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Raudvere, U. et al. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Acknowledgements
The authors acknowledge Dr. Shahram Shahabi from Standard Homeopathic Company for providing the D. rotundifolia ethanolic extract and for valuable discussion regarding the manuscript. This work was supported by a grant from Standard Homeopathic Company, Los Angeles (USA) to the University of Verona, Department of Medicine. The funder contributed to the costs of materials, equipment and two post-doctoral scholarships. The funder did not play any role in the experiments, the analysis of results or data disclosure. The authors acknowledge the Centro Piattaforme Tecnologiche dell’Università degli Studi di Verona for the technical contribution in the RNA-seq and Mass Spectrometry experiments.
Author information
Authors and Affiliations
Contributions
F.A.S. and M.M. executed the experiments, analysed the results and wrote the manuscript. P.B. supervised the research program, the results and the writing of the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arruda-Silva, F., Bellavite, P. & Marzotto, M. Low-dose Drosera rotundifolia induces gene expression changes in 16HBE human bronchial epithelial cells. Sci Rep 11, 2356 (2021). https://doi.org/10.1038/s41598-021-81843-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-81843-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
