
Abstract
Pathological links between neurodegenerative disease and cancer are emerging. LRRK2 overactivity contributes to Parkinson’s disease, whereas our previous analyses of public cancer patient data revealed that decreased LRRK2 expression is associated with lung adenocarcinoma (LUAD). The clinical and functional relevance of LRRK2 repression in LUAD is unknown. Here, we investigated associations between LRRK2 expression and clinicopathological variables in LUAD patient data and asked whether LRRK2 knockout promotes murine lung tumorigenesis. In patients, reduced LRRK2 was significantly associated with ongoing smoking and worse survival, as well as signatures of less differentiated LUAD, altered surfactant metabolism and immunosuppression. We identified shared transcriptional signals between LRRK2-low LUAD and postnatal alveolarization in mice, suggesting aberrant activation of a developmental program of alveolar growth and differentiation in these tumors. In a carcinogen-induced murine lung cancer model, multiplex IHC confirmed that LRRK2 was expressed in alveolar type II (AT2) cells, a main LUAD cell-of-origin, while its loss perturbed AT2 cell morphology. LRRK2 knockout in this model significantly increased tumor initiation and size, demonstrating that loss of LRRK2, a key Parkinson’s gene, promotes lung tumorigenesis.
Similar content being viewed by others

Introduction
Lung adenocarcinoma (LUAD), one of the deadliest cancers worldwide1, is a molecularly heterogeneous disease that originates in the distal lung, affecting smokers and non-smokers. Analyses of tumor evolution suggest that LUAD progression and patient outcome are highly influenced by subclonal heterogeneity2, making functional characterization of molecular alterations captured by large-scale, multi-omics efforts like The Cancer Genome Atlas (TCGA) a continued priority. In a previous analysis of TCGA LUAD RNA-seq data, we identified a marked reduction in expression of Parkinson’s susceptibility gene Leucine Rich Repeat Kinase 2 (LRRK2), in tumors versus matched normal lung3. Pathogenic links between cancer and Parkinson’s disease (PD) are emerging: while cancer cells can proliferate indefinitely, diseased neurons die prematurely, placing cancer and neurodegenerative disease at opposite ends of a spectrum of aberrant cell survival4. Overactive LRRK2 is a common genetic cause of PD5, which when inhibited or knocked out perturbs pulmonary surfactant homeostasis in alveolar type II (AT2) cells6—a main cell of origin in LUAD7. LRRK2 inhibition is poised to be the first disease-modifying therapy in PD, with a small-molecule inhibitor in early phase clinical trials8. While pre-clinical studies of systemic LRRK2 inhibition in primates have revealed a reversible lung phenotype, which does not affect pulmonary function tests with short-term treatment9, potential irreversible effects beyond respiration remain to be defined—particularly in the setting of long-term LRRK2 inhibition.
Large-scale epidemiological surveys of PD patients have revealed intriguing links between brain and lung health. PD patients, with either hereditary or idiopathic disease, are less likely to develop smoking-related cancers, with a dramatically reduced incidence rate of lung cancer (0.40) compared to the general population10, while ongoing cigarette smoking halves the risk of developing PD compared to neversmokers11. Mechanisms governing these associations have yet to be elucidated, including whether altered LRRK2 plays a mediating role. Several epidemiological studies report that LRRK2 G2019S carriers are at increased risk for various non-lung cancers12, while others contradict these results13,14; therefore, the cancer epidemiology of mutant LRRK2 remains unclear. In addition, as LRRK2 mutation carriers have been the focus of most LRRK2-related epidemiological studies of cancer incidence, an overactive LRRK2 phenotype was presupposed; consequently, to our knowledge, a role for LRRK2 loss in cancer pathogenesis has yet to be considered, including for LUAD.
This study explores the association of LRRK2 transcriptional repression with established clinical, genomic and transcriptomic phenotypes assembled for TCGA LUAD patient samples, identifies a significant overlap between transcriptional changes seen in LRRK2-low LUAD and those documented in normal postnatal lung development in mice, and, significantly, presents the first in vivo evidence that LRRK2 loss promotes carcinogen-induced lung tumorigenesis.
Results and discussion
LRRK2 reduction occurs in high-risk non-terminal respiratory unit (non-TRU) type lung adenocarcinoma (LUAD)
In a previous molecular screen of autophagy-associated genes across 11 TCGA cancer types, we identified a striking reduction of LRRK2 expression in LUAD tumors (n = 517) versus matched normal lung tissue (n = 59)3. To determine the potential clinical relevance of altered LRRK2 levels in LUAD, we examined the association of LRRK2 expression levels with clinicopathological variables. While a median split of tumor samples based on LRRK2 expression showed a significant difference in survival between LRRK2-low versus -high patients (Log-rank test P = 0.033; Supplementary Fig S1), we chose to apply a supervised (and potentially more biologically meaningful) method to refine the LRRK2 cutpoint, by deriving LRRK2-low and -high cohorts via optimization of a two-group cohort correlation with overall survival (OS) data, which was achieved by identifying a cutpoint that minimized the P-value defined by Kaplan–Meier OS analysis and Log-rank test (X-tile cohort separation algorithm15 with Monte Carlo cross-validation P < 0.05, n = 10,000 repetitions; Fig. 1A). We found that 204 of 517 (39.5%) TCGA LUAD tumors expressed a dramatic reduction of LRRK2 expression, which stratified both overall survival (OS; Log rank test (60 months) P = 0.0024) and disease specific survival (DSS; Log rank test (60 months) P = 0.025) (Fig. 1B). Survival data was obtained from the TCGA Pan-Cancer Clinical Data Resource16, which was curated for high quality outcome data. Differences in sample numbers between OS and DSS analyses (Fig. 1B) occur, as only patients (from Fig. 1A) with reliable survival follow-up per outcome type were included, as determined by Liu et al.16 (Supplementary Table S1). LRRK2-low patients survived more poorly, with a hazard ratio of 1.42 (Cox proportional hazards regression adjusted for pathological stage; CI 1.06–1.91; Wald test P = 0.019; Log likelihood test P < 0.0001). Further, patients expressing the lowest amount of tumoral LRRK2 (n = 50) had an increased OS hazard ratio of 2.42 (CI 1.19–4.90; Wald test P = 0.014; Log likelihood test P < 0.0001) and an increased DSS hazard ratio of 3.03 (CI 1.20–7.68; Wald test P = 0.019; Log likelihood test P < 0.0001), compared to patients expressing the highest amount of tumoral LRRK2 (n = 49).

LRRK2-low lung adenocarcinoma is associated with poor patient survival, non-TRU expression-based molecular subtypes and worse predicted tumor differentiation. (A) Plot of LRRK2 mRNA levels in LUAD tumors (dichotomized into LRRK2-low and -high expression groups) and adjacent normal lung tissue (RSEM values), from TCGA LUAD patients. (B) Kaplan–Meier plot of LUAD patient OS or DSS stratified by LRRK2 expression status. (C) Pairwise Fisher’s exact test for enrichment of expression subtype frequency within LRRK2 expression groups: TRU-like versus non-TRU type LUAD. (D) (Left) Standardized LRRK2 tumoral expression per sample (RSEM values), annotated for lower risk TRU-like or higher risk non-TRU type tumors or (Right) grouped by the combined status for LRRK2 expression and expression subtype (median centred boxplot of RSEM values). (E) Pairwise Fisher’s exact test for enrichment of smoking history within LRRK2 expression groups: TRU-like versus non-TRU type LUAD. (F) Correlation of LRRK2 expression with a previously published gene expression-based score representing LUAD tumor differentiation status (Spearman’s correlation coefficient − 0.59 with Holm’s adjP < 0.0001; positive scores represent increasingly poor differentiation). (G) Stratification of the tumoral gene expression of established markers for alveolar and bronchiolar epithelial cell types, by the combined LRRK2 and expression subtype status of LUAD tumors (median-centred boxplot of standardized RSEM values; Dunn’s test BH adjP < 0.05).
Gene expression phenotyping of multiple LUAD cohorts over the last decade17,18,19,20 have confirmed that non-terminal respiratory unit (non-TRU) type LUAD is associated with worse patient outcomes. To situate LRRK2 transcriptional repression with respect to established expression-based molecular subtyping of LUAD17,19,21, we determined whether LRRK2-low tumors were enriched for higher-risk non-TRU type tumors or lower-risk terminal respiratory (TRU)-like tumors. Stratification of tumors by both LRRK2 status and expression subtype revealed that most LRRK2-low tumors were of the higher-risk non-TRU type (Frequency (F) = 0.90; n = 517 patients; pairwise Fisher’s exact test, Benjamini Hochberg adjusted P (BH adjP) < 0.0001; Fig. 1C,D). Together these results associate reduced LRRK2 expression with poor survival outcomes and the high risk non-TRU subtype of lung cancer.
LRRK2-low non-TRU type tumors are associated with ongoing smoking, subtype-specific genome instability and less differentiated tumors
Non-TRU type tumors are associated with smoking, KRAS, TP53 and STK11/LKB1 mutations, and peripheral lung architecture destruction, while TRU-like tumors have established associations with never smoking, EGFR mutation, and preservation of peripheral lung architecture17,22. Non-TRU tumors can be further broken down into proximal-inflammatory (PI) tumors, enriched for TP53 and NF1 co-mutation, high grade solid-type morphology, and higher expression of immunotherapy targets, or proximal-proliferative (PP) tumors, enriched for KRAS and STK11/LKB1 co-alteration and low immune-related gene expression17,23,24. To identify disease covariates associated with a change in LRRK2 status, potentially independent of expression subtype, we performed an enrichment analysis for various clinical and molecular covariates in LRRK2-low (n = 183) versus -high (n = 146) non-TRU type tumors only.
Similar to non-TRU type LUAD in general, LRRK2-low non-TRU type tumors were associated with smoking history; however, patients with LRRK2-low tumors represented 55.5% of all ‘Current Smokers’ (pairwise Fisher’s exact test, LRRK2-low versus -high non-TRU BH adjP = 0.043; Fig. 1E; Supplementary Table S1), suggesting that marked LRRK2 reduction was associated with ongoing smoking. Pathological mechanisms triggered by cigarette smoking include genome instability25, global gene expression changes26, alteration of epithelial differentiation pathways26, and small airway tissue remodeling27. At the molecular level, reduced LRRK2 was associated with subtype-specific genome instability: LRRK2-low PP tumors carried higher genome-wide somatic copy number load than LRRK2-high PP tumors (pairwise Dunn’s test BH adjP = 0.042), while LRRK2-low PI tumors were more likely to have highly amplified genes (n = 77) from chromosome bands 5p12, 5p13 or 5p15 (pairwise Fisher’s exact test BH adjusted P < 0.05) (Supplementary Fig. S2 and Supplementary Tables S2, Suppl Table S3). Interestingly, somatic mutation prevalence in genes reported to be significantly mutated in LUAD21 (n = 52 genes) remained equivalent between tumor groups (Supplementary Tables S2), suggesting LRRK2 transcriptional repression was not a consequence of the oncogenic activation of particular LUAD driver genes.
We further tested whether LRRK2 expression level was associated with an empirically derived, gene expression-based ‘tumor differentiation’ score, calculated for TCGA LUAD cohort by Chen et al. (Supplementary Table S1)18. The authors developed a gene signature from an alternate cohort of LUAD patients22, which had been histopathologically categorized by growth pattern into either less or more differentiated tumors. Less differentiated tumors (called ‘bronchial-derived’) exhibited invasive features with architectural destruction, while more differentiated tumors (called ‘bronchioloalveolar’) exhibited preservation of the lung architecture. Morphological indications of LUAD invasiveness are used to inform clinical prognosis28. Stratification of this tumor differentiation score by LRRK2 and expression subtype status indicated that non-TRU type tumors were less differentiated (Dunn’s test, BH adjP < 0.0001)17,19, with LRRK2-low non-TRU tumors predicted to be the least differentiated (Spearman’s correlation coefficient − 0.59 with Holm’s adjP < 0.0001; Fig. 1F). Further, tumoral expression of established markers for key distal lung epithelial cell types (e.g., alveolar type (AT)1, AT2 and club cells)29,30 were significantly reduced in LRRK2-low non-TRU type tumors (Fig. 1G; Supplementary Fig. S3). To evaluate the cell type specific expression of LRRK2 in lung, we took advantage of a recent publicly available single cell RNA-seq dataset (GSE13190731) that included 42,996 sequenced cells from the normal lung of 11 patients. Using cell type annotation reported by the authors, we determined that the vast majority of LRRK2 expression in human lung occurs in AT2 cells and not in other known lung epithelial cells (Supplementary Fig. S4). Taken together, these results suggest that LRRK2 reduction in LUAD likely occurs in AT2 cells, is associated with ongoing smoking, and may be associated with disease-related changes to peripheral lung architecture.
Surfactant metabolism genes are tightly co-expressed with LRRK2 in LUAD
To profile the transcriptional landscape associated with a change in LRRK2 expression, we compared the results of differential expression analyses for two biological contexts in LUAD with a marked decrease of LRRK2 expression: in tumor (n = 517) versus normal lung (n = 59), and in tumors expressing the lowest (n = 50) versus highest deciles (n = 49) of LRRK2 expression. A comparison of differentially expressed genes (DEGs) identified in each context revealed a considerable overlap of misexpressed genes (Supplementary Table S4), suggesting that altered cell processes associated with the transformation of normal lung dominated the transcriptional changes found in LRRK2-low tumors. Figure 2 depicts the hierarchical agglomerative clustering of the expression of a selection of DEGs, common to both contexts, plotted for the complete TCGA LUAD cohort and ordered by tissue, LRRK2 expression status and expression subtype (n = 385 DE genes with absolute median RSEM value fold change > 2; Fig. 2A). To tease out DEGs associated with reduced tumoral LRRK2 in an expression subtype-independent manner, we also stratified the expression of DEGs by LRRK2 expression status in non-TRU subtype tumors only (Supplementary Table S4).

The transcriptional landscape of LRRK2 repression in lung adenocarcinoma patients. (A) Heatmap depicting the hierarchical agglomerative clustering of genes differentially expressed in common between two biological contexts in TCGA LUAD tumors, each with a marked reduction of LRRK2 expression: (1) across all tumors versus normal lung and (2) in the lowest LRRK2 expressing tumors versus the highest LRRK2 expressing tumors. Columns: n = 385 DEGs with absolute fold change of median RSEM value > 2; Rows: n = 59 normal lung and n = 517 LUAD tumors, ordered by tissue, LRRK2 expression status and expression subtype. (B) Exemplar DEGs identified in Cluster 1, enriched for genes that act in mitotic cell cycle (Metascape algorithm; q < 0.05), stratified by tumor group (multicolour boxplots of exemplar gene expression; Dunn’s test BH adjP > 0.05). Tumor groups represent the combined sample status for LRRK2 expression and non-TRU subtype. (C) Exemplar DEGs identified in Cluster 2, associated with tumor purity (consensus purity estimate or CPE; Spearman’s correlation coefficient ≥ 0.5 with Holm’s corrected P < 0.0001) and enriched for immune response genes (Metascape algorithm; q < 0.05), stratified by tumor group (multicolour boxplots of exemplar gene expression; Dunn’s test BH adjP < 0.01). (D) Exemplar DEGs identified in Cluster 3, enriched for genes that act in surfactant metabolism (Metascape algorithm; q < 0.05), stratified by tumor group (multicolour boxplots of exemplar gene expression; Dunn’s test BH adjP < 0.001) or plotted against LRRK2 expression (multicolour scatterplots; Spearman’s correlation coefficient ≥ 0.6 with Holm’s corrected P < 0.0001; standardized RSEM Transcripts Per Million or TPM).
LRRK2 status failed to be associated with the differential expression of most Cluster 1 cell cycle-related genes in non-TRU subtype tumors only (e.g., GO:000734632; Fig. 2B), but was associated with a marked decrease of a number of Cluster 2 immune-related genes (e.g., GO:001922132; Fig. 2C) and many Cluster 3 genes, enriched for genes that function in surfactant metabolism (e.g., Reactome R-HSA-568382633; Fig. 2D). Moreover, Cluster 3 genes, including all surfactant genes (SFTPA1, SFTPA2, SFTPB, SFTPC, SFTPD) and various lamellar body-related genes (ABCA3 and LAMP3), were strongly co-expressed with LRRK2 (absolute Spearman’s correlation coefficient > 0.6 with Holm’s adjP < 0.0001), suggesting the tight transcriptional co-regulation of LRRK2 with pathways known to be important for normal AT2 cell differentiation34 and function35. Therefore, differential expression results show that increased proliferation was associated with non-TRU type LUAD, regardless of LRRK2 expression status, while LRRK2 reduction was strongly associated with decreased surfactant gene expression, regardless of expression subtype. Indeed, applying principal component (PC) analysis, as an unbiased technique to reduce the dimensionality of the tumor versus normal transcriptional landscape of TCGA LUAD cohort, revealed that the top gene contributor to PC1—the component that explained the most variance—was SFTPC, while LRRK2 was quantified as one of the top 3% of gene contributors to PC1 (Supplementary Fig. S5; gene loadings and sample scores in Supplementary Table S6).
While TCGA LUAD patient data were not available to assess changes to LRRK2 and surfactant gene expression at the protein level, surfactant protein deficiency is known to be associated with smoking and lung disease in patients36,37, including cancer38,39. Given that LRRK2 knockout (KO) in rodents, or inhibition in primates, perturbs the morphology of lamellar bodies that package, store and secrete surfactant6,40, a pathological cycle may be triggered whereby ongoing smoking contributes to the repression of LRRK2 and the surfactant pathway, while decreased LRRK2 further disrupts surfactant biogenesis and/or function. Pulmonary surfactant acts as a protective mechanical barrier, with surfactant proteins A and D directly modulating alveolar innate immunity41. Therefore, reduced LRRK2 may lead to altered alveolar immunity that could undermine tumor immune surveillance42. Of note, LRRK2-low PP tumors from the TCGA LUAD cohort were predicted to have significantly lower mRNA levels of colony-stimulating factor-1 (CSF1) macrophage regulation genes, monocyte and macrophage marker genes, as well as major histocompatibility complex II (MHC-II) pathway genes (Fig. 2C). Further, Cluster 2 immune-related genes—but not LRRK2 (Spearman’s correlation coefficient = − 0.14 with Holm’s adjP < 0.0001)—were strongly negatively associated with tumor purity (consensus purity estimate43; Spearman’s correlation coefficient ≤ − 0.5 with Holm’s corrected P < 0.0001) (Supplementary Table S4), predicting that their expression depended to a large extent on non-tumor cells. To corroborate this interpretation, we mined the above mentioned recent public dataset of single cell RNA-seq from normal human lung (GSE13190731). Using the cell type annotation reported by the authors, we confirmed that the myeloid-related genes displayed in Fig. 2C were expressed in this new dataset mainly in myeloid cells, while the MHC-II gene HLA-DPA1 was expressed in some epithelial cells, as well as in various white blood cells (Supplementary Fig. S6). Therefore, we predict that the changes in myeloid-related gene expression we observed in LRRK2-low versus -high PP tumors (Fig. 2C) represented changes in tumor infiltration by myeloid cells, whereas the changes in expression of MHC-II genes, such as HLA-DPA1, may have occurred in both tumor cells and various immune cells.
While the mechanisms that link surfactant dysfunction to lung tumorigenesis remain unknown, robust associations exist between smoking history, surfactant defects, immunosuppression and lung cancer. Low LRRK2 non-TRU tumors may constitute a LUAD tumor subset that reflects increased surfactant dysfunction and altered tumor immunity.
LRRK2-low LUAD shares patterns of differential gene expression with postnatal developing lung, during alveolarization
A literature search of genes misexpressed in LRRK2-low tumors identified a recent study of lung development in mice44, in which, just as in TCGA LUAD cohort, cell cycle genes and surfactant metabolism genes were reported to be inversely or co-expressed with LRRK2 (respectively), during postnatal alveolarization. Alveolarization refers to the postnatal differentiation of alveoli from prenatal saccules. To assess whether DEGs in LRRK2-low tumors overlapped significantly with genes found to be differentially expressed in developing mouse lung, we converted the human DEGs to their mouse gene orthologs45,46 (Supplementary Table S5), and employed a Monte Carlo permutation simulation (n = 100,000 repetitions; Bonferroni corrected empirical P-value) to generate an empirical distribution of the overlap that would occur by chance47. Table 1 summarizes the significance of overlap between DEGs in LRRK2-low LUAD tumors and genes differentially expressed between adjacent developmental stages in mice. The observed intersection of DEGs in LRRK2-low tumors and a particular developmental time point—between two stages of gene expression-defined secondary alveolar septation (i.e. postnatal days P0-P3 (called ALV1) versus P4-P7 (called ALV2)44)—was substantially greater than expected by chance: 4.5 times the expected number of differentially increased genes (n = 208 genes) and 2.8 times the expected number of differentially decreased genes (n = 252 genes) overlapped between studies. Gene set enrichment analysis revealed that shared increased DEGs function in cell cycle pathways (e.g., R-HSA-1640170), while shared decreased DEGs function in surfactant metabolism (e.g., R-HSA-5683826) (33,48; Supplementary Table S5).
Morphological characterizations of postnatal lung development in rodents have documented an exponential increase in alveoli number that occurs between postnatal days P3 and P1449,50,51, where new growth of septal membrane outpaces overall lung growth on days P4 to P752. Alveoli have also been shown to increase dramatically in human lung during the first 2 years of life, followed by a slow but continuous increase into adolescence53. It is possible that postnatal alveolar development mechanisms remain available in adult lung and could be co-opted by lung cancer cells—perhaps during alveolar regeneration following repetitive lung injury54, or by smoking-related oxidative stress that mimics the transition from a hypoxic in utero environment to the 21% oxygen of ambient air55. Based on shared patterns of differential gene expression, we propose that aberrant activation of a developmental program of alveolar growth and differentiation contributes to the hyperproliferation and surfactant defects predicted in our study, for LRRK2-low non-TRU type LUAD.
LRRK2 knockout increases tumor initiation in a murine model of early lung cancer, induced by a carcinogen present in cigarette smoke
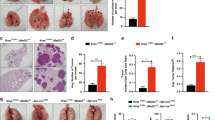
Given the association of reduced LRRK2 with current smoking and decreased patient survival, increased genome instability and less differentiated non-TRU tumor types, we hypothesized that LRRK2 has tumor suppressive effects in carcinogen-exposed lung tissue. To assess whether LRRK2 KO increases adenoma multiplicity and/or size, 6- to 8-week-old C57BL/6-Lrrk2tm1Mjfa null mice of both sexes, homozygous for a deletion in Exon 41 of the kinase domain, and wild type (WT) strain mates received 10 weekly intraperitoneal injections of urethane (1 g/kg diluted in PBS), and were sacrificed at 26 weeks post‐first injection56 (Fig. 3). WT mice produced an average of 2.53 ± 0.91 (mean ± SEM) visible adenomas (n = 16 mice; 95% CI 0.702–4.36 including outlier animal), while LRRK2 KO mice produced an average of 6.53 ± 0.75 adenomas per animal (n = 20 mice; 95% CI 5.02–8.03) (Welch’s two-sample t-test, t(30.94) = − 3.38, 95% CI − 6.41–1.58, P = 0.0020, d = 1.14) (Fig. 3A–C). LRRK2 KO adenomas were larger on average, per animal: 0.53 ± 0.06 mm in WT (95% CI 0.41–0.65) and 0.78 ± 0.02 mm in LRRK2 KO (95% CI 0.74–0.82) (n = 40 WT and 130 KO surface lesions; Welch's two-sample t-test, t(13.46) = − 3.93, 95% CI − 0.39–0.11, P = 0.0016, d = 1.72) (Fig. 3A–C). No sex-specific differences were found. IHC staining showed an equivalent percent of Ki-67 positive adenoma cells between genotypes, suggesting that LRRK2 KO may have facilitated tumor development at an earlier time point; granted, given the long latency of lesion development in this model, we cannot rule out that differences in proliferation rate existed closer to treatment.

LRRK2 knockout increases SFTPC-positive tumor initiation in a murine model of urethane-induced, early stage lung cancer. (A) Representative visible adenomas that developed in the lungs of both LRRK2 knockout (KO) mice and wild type (WT) strain mates, following urethane treatment. (B) Double blind analysis of adenoma counts and size (n = 16 WT and n = 20 LRRK2 KO animals, with n = 40 WT and n = 130 LRRK2 KO surface lesions). (C) Representative H&E stained sections of adenomas (20× magnification; transverse section; n = 16 WT and n = 20 LRRK2 KO animals, with n = 40 WT and n = 130 LRRK2 KO surface lesions). (Inset) Detailed view of representative vacuolated cells (arrows) found in LRRK2 KO lung only. (D) Representative images of multiplexed IHC staining for colocalization of alveolar type II (AT2)-specific cell marker SFTPC (Ferangi blue chromogen) and LRRK2 (3,3′-Diaminobenzidine (DAB) chromogen), in LRRK2 KO (top) and WT (bottom) mouse lung parenchyma (n = 7 WT mice with 14 lesions; n = 15 KO mice with 25 lesions). (E) Representative images of multiplexed IHC staining for SFTPC and LRRK2, in adenomas that developed in the lungs of both LRRK2 KO (top) and WT (bottom) control mice, following urethane treatment (n = 7 WT mice with 14 lesions; n = 15 KO mice with 25 lesions). (Inset) detailed view of staining at adenoma edge and adjacent lung parenchyma. (F) Representative images of multiplexed IHC staining for SFTPC and LRRK2, for adenomas that developed in the lungs of WT control mice following urethane treatment, showing spontaneous reduction of LRRK2 positivity. Representative of 4 of 14 lesions from n = 7 WT control mice. (Inset) AT2 cells in the adjacent lung parenchyma remained double positive for SFTPC and LRRK2.
To confirm LRRK2 expression in AT2 cells in our model, we applied multiplexed IHC staining for AT2 cell-specific marker SFTPC (Ferangi blue chromogen) with LRRK2 (3,3′-Diaminobenzidine chromogen) to determine colocalization (Fig. 3D). LRRK2 colocalized almost exclusively with SFTPC in WT mouse lung, in both adenomas and parenchyma (Fig. 3E), locating LRRK2′s main functional relevance in this model to AT2 cells, a predominant cell type of origin in LUAD7. Further, SFTPC + LRRK2-AT2 cells appeared less punctate with SFTPC IHC (Fig. 3D top), compared to SFTPC + LRRK2 + AT2 cells (Fig. 3D bottom), confirming that LRRK2 loss perturbs AT2 cell morphology and could contribute to AT2 cell dysfunction. Of note, 4 of 14 adenomas that developed in the lungs of WT control mice (n = 7) showed a reduction of LRRK2 positivity, while adjacent lung parenchyma remained double positive for SFTPC and LRRK2 (Fig. 3F). Therefore, LRRK2 KO not only increased tumor formation, but LRRK2 expression appeared reduced in some WT AT2 cells, in response to tumor-related changes triggered by urethane.
Conclusions
In summary, marked LRRK2 reduction was associated with tumors predicted to have altered distal lung architecture, dysfunctional surfactant metabolism, hyperproliferation and potentially altered innate tumor immunity—in patients who continued to smoke and ultimately suffered worse survival. While a retrospective study of associations identified in patients cannot determine causation, we were able to show that systemic LRRK2 KO increased tumor initiation and size in carcinogen-driven early lung cancer in mice, providing orthologous evidence that LRRK2 reduction is not merely a passenger aberration but a link between environmental insult and potential AT2 cell transformation.
This is the first study to demonstrate a tumor suppressive role for LRRK2 in carcinogen-induced distal lung cancer, and to potentially identify a Parkinson’s patient population (i.e. current smokers) for whom long-term LRRK2 inhibition may require additional monitoring. Further studies that identify the mechanisms by which LRRK2 reduction promotes LUAD tumorigenesis have the potential to benefit cancer patients, and to inform the design of LRRK2 inhibition strategies in Parkinson’s disease that mitigate potential pulmonary-related adverse effects.
Methods
TCGA data and associated molecular tumor features
The following data were downloaded from the Broad Institute TCGA Genome Data Analysis Center: Level 3 TCGA LUAD clinical data (https://doi.org/10.7908/C19P30S6), Level 3 TCGA LUAD mutation calls (https://doi.org/10.7908/C11G0KM9), Level 4 somatic DNA alterations (SNP6 GISTIC2 copy number analysis (https://doi.org/10.7908/C1348JSB), Level 4 MutSig 2CV version 3.1 mutation analysis (https://doi.org/10.7908/C17P8XT3)), Level 3 TCGA LUAD RNA-seq data (https://doi.org/10.7908/C11G0KM9). Survival data for the LUAD cohort were obtained from the TCGA Pan-Cancer Clinical Data Resource16. Additional molecular tumor features predicted from RNA-seq data included scores for immune cell infiltration and regulation57 and tumor differentiation status18. To test for associations, LRRK2 gene abundance was dichotomized using the X-tile cohort separation algorithm15. Gene expression subtypes were assigned to TCGA LUAD tumor samples as previously published17.
Cutpoint derivation to define LRRK2 expression status of TCGA LUAD patients
To test for associations between LRRK2 expression status and various clinical phenotypes and molecular tumor features, LRRK2 gene abundance (quantile normalized RSEM read counts) was dichotomized using the X-tile cohort separation algorithm (RRID:SCR_005602)15. X-tile determines a cutpoint by optimizing cohort correlation with survival data, which was achieved in this study by minimizing the P-value defined by Kaplan–Meier analysis of 5-year overall survival outcomes (n = 460 patients, with tumor samples having greater than 60% tumor nuclei) and the Log-rank test (Monte Carlo cross-validation with n = 10,000 repetitions P < 0.05). A cutpoint of 588.92 quantile normalized RSEM was determined for LRRK2 and used in downstream analyses.
Somatic mutation and copy number alteration stratified by LRRK2 cutpoint and expression subtype
Scores for genome-wide mutation and copy number load were calculated as previously published58. The mutational load was calculated based on exome sequencing data, by normalizing the count of all somatic mutations per tumor to an approximate protein-coding exome size of 30 megabases. The copy number load was calculated per tumor as the number of nucleotides affected by copy number alteration (at an absolute segmented value greater than 0.3), normalized to the total number of nucleotides contained in all segments identified for that tumor.
Non-synonymous somatic mutations—missense, splice site, frameshift or other non-synonymous—reported for significantly mutated genes (SMGs; n = 52 genes) identified by the TCGA MutSig 2CV v3.1 pipeline (q < 0.025) (RRID:SCR_010779)21 were tested for association with LRRK2 cutpoint and expression subtype status (pairwise Fisher’s exact test BH adjP < 0.05). Tumor stratification served as row variable and mutation status was binarized as altered (value = 1) or unaltered (value = 0). Gene-level TCGA LUAD copy number data was filtered for either unaltered or high-level copy number alterations. High-level thresholds were calculated by TCGA on a per sample basis as the maximum (for amplifications) or minimum (for deletions) median arm-level copy number alteration21. Genes altered in at least 5% of tumors with high-level copy number alterations were tested for association with tumors grouped by combined status for LRRK2 cutpoint and expression subtype (n = 2458 genes; pairwise Fisher’s exact test with BH adjP < 0.05). After identifying gene-level SCNA associations per tumor group, genes found to be differentially copy number altered in LRRK2-high versus -low tumors within a non-TRU subtype were interpreted as being associated with a change in LRRK2 status (i.e. CNAs differentially altered in LRRK2-high PP vs LRRK2-low PP or LRRK2-high PI vs LRRK2-low PI). Genes found to be differentially copy number altered between various subtypes within LRRK2-high tumors were interpreted as being associated with a change in expression subtype, independent of LRRK2 expression status (i.e. LRRK2-high TRU vs LRRK2-high PI, LRRK2-high TRU vs LRRK2-high PP, LRRK2-high PI vs LRRK2-high PP). Genes found to be differentially copy number altered in LRRK2-high versus -low tumors within a non-TRU subtype but not between expression subtypes within LRRK2-high tumors were interpreted as the most likely to be associated with a change in LRRK2 level, independent of subtype.
Assignment of molecular expression subtypes
To assign LUAD gene expression subtypes17,19,21 to TCGA LUAD tumor samples (n = 515 tumors) as previously published, quantile normalized RSEM gene expression data were Log2 transformed, median centered by gene and compared by Pearson correlation to the published nearest centroid classifier17. Input genes were limited to the 489 of 506 classifier genes present in the TCGA data set and subtype was assigned using the maximum correlation coefficient. Subtype calls were benchmarked against published subtype assignments by TCGA Research Network21 on a subset of the TCGA LUAD cohort (n = 230 tumors), which found 93.9% of calls to be identical—a result in line with a recent assessment of the classification stability for this classifier, which found that typically < 10% of samples switched gene expression subtype when perturbations to the input data (e.g., missing genes, missing samples, differences in gene centering or similarity metric) were introduced 59.
Differential expression analyses and gene set enrichment
Quantile normalized RSEM (RID:SCR_013027) read counts were further normalized for library size using the trimmed mean of M-values (TMM) method implemented in edgeR version 3.22.3 (calcNormFactors function; RRID:SCR_012802); normalized log-counts per million were weighted using the variance modelling at the observation-level method prior to the Limma analysis pipeline version 3.34.5 (RRID:SCR_010943). Differentially expressed (DE) genes identified by Limma were filtered for a false discovery rate < 0.05 (BH adjusted), an average log expression greater than 1, and an absolute log fold change greater than 0.6. DE analysis was performed between the following sample stratifications: tumor versus adjacent normal lung, tumors expressing the lowest decile of LRRK2 gene abundance (LRRK2-D1) versus those expressing the highest decile of LRRK2 gene abundance (LRRK2-D10), LRRK2-high versus -low PI tumors, LRRK2-high versus -low PP tumors, as well as LRRK2-high versus -low non-TRU type tumors (PI and PP subtypes combined). Genes DE between LRRK2-D1 and LRRK2-D10 tumors were compared to genes DE in tumors versus normal lung—e.g. a list of genes differentially increased in LRRK2-D1 versus -D10 tumors was compared to the list of genes differentially increased in all tumors versus normal lung—producing the following input gene lists for gene set expression analysis (GSEA) by the Metascape algorithm48 (q < 0.05; RRID:SCR_016620; http://metascape.org): (1) common increased DE (in all tumors and LRRK2-D1 tumors), (2) common decreased DE (in all tumors and LRRK2-D1 tumors), (3) and (4) opposing increased or decreased DE in LRRK2-D1 tumors (compared to all tumors), (5) and (6) uniquely increased or decreased DE in LRRK2-D1 tumors (compared to all tumors), or (7) and (8) uniquely increased or decreased DE in all tumors (compared to LRRK2-D1 tumors). Genes DE between LRRK2-D1 and -D10 tumors found to be further DE with a change in LRRK2 expression level within tumors of the same expression subtype—i.e. LRRK2-high versus -low PP tumors, LRRK2-high versus -low PI tumors or LRRK2-high versus low non-TRU type tumors—were interpreted as having DE associated with LRRK2 expression status. To identify genes potentially co-expressed with LRRK2, a Spearman’s correlation coefficient was calculated for the expression of each gene in the genome and LRRK2 (n = 515 LUAD tumors; absolute Spearman’s correlation coefficient > 0.3, BH adjP < 0.05). Exploratory hierarchical agglomerative clustering was performed for a filtered set of genes, DE in both tumors versus normal lung and LRRK2-D1 versus -D10 tumors (absolute median RSEM fold change between respective sample groups > 2), using standardized Log2 transformed RSEM Transcripts Per Million and complete linkage with Euclidean distance (hclust function in ‘stats’ package version 3.4.3).
Predicted tumor purity and tumor differentiation status
The consensus measurement of tumor purity estimate (CPE) was obtained from supplementary material published by Aran et al., who estimated purity for more than 10,000 samples across 21 cancer types from TCGA by amalgamating the results of four methods that relied on separate data types: ESTIMATE (expression profiles of immune and stromal genes), ABSOLUTE (somatic copy-number data), LUMP (methylation of immune-specific CpG sites) and image analysis of haematoxylin and eosin slides43. To identify gene expression impacted by tumor purity, a Spearman’s correlation coefficient was calculated for the expression of each gene in the genome and the CPE, for all TCGA LUAD tumor samples (absolute Spearman’s correlation coefficient > 0.3 and Holm’s adjP < 0.05).
A gene expression-based tumor differentiation score was obtained from the supplementary material of Chen et al.18. The differentiation score was calculated as a standardized T-score that compared the mean expression of genes associated with more tumor differentiation to the mean expression of genes associated with less tumor differentiation, per TCGA LUAD tumor sample. The larger the absolute value of the score, the larger the difference in expression between gene sets; therefore, a positive score reflected increased expression of genes associated with less differentiated tumors relative to genes associated with more differentiated tumors, while a negative score indicated the reverse.
Human and mouse gene homology
Gene homology between human and mouse was used to compare transcriptional profile results from this study to a previous study of DE mouse lung development genes identified in fetal and neonatal mouse lungs at 26 time points, termed the murine Developing Lung Characteristic Subtranscriptome (mDLCS)44. LRRK2 was identified as a member of the mDLCS. Mouse Gene Identifiers (MGI) for mDLCS genes and their direction of DE between 9 sequential developmental stages—four prenatal stages (embryonic, pseudoglandular, canalicular and saccular), four post natal stages of alveolarization (ALV1, ALV2, ALV3, ALV4) and a final homeostatic stage (maturity)—were obtained from the supplementary material of Beauchemin and colleagues44. A comprehensive gene homology resource for human-mouse orthologs was downloaded using the HGNC Comparison of Orthology Predictions (HCOP) tool (https://www.genenames.org/tools/hcop/)45,46, which amalgamates orthology predictions between human and mouse from 12 curated databases (eggnog, Ensembl, HGNC, HomoloGene, Inparanoid, OMA, OrthoDB, NCBI Gene Orthology, OrthoMCL, Panther, PhylomeDB, TreeFam). The resource was filtered for human-mouse orthology predictions that had support from at least two databases and where each human-mouse orthology pair was annotated with unique HUGO Gene Nomenclature Committee (HGNC) and Mouse Genome Informatics (MGI) identifiers. If one gene mapped to many orthologs, each ortholog was included. The final human-mouse orthology reference used in this study included: 18,650 human genes and 19,613 mouse genes, with 26,855 unique HGNC-MGI identifier pairs—2398 human genes mapped to multiple mouse orthologs (a total of 4285 mouse genes), while 2668 mouse genes mapped to multiple human orthologs (a total of 3231 human genes). DE genes identified in lung cancer in this study and DE mouse development genes identified in the Beauchemin study were included for comparison if they had an ortholog predicted by HCOP in our curated human-mouse orthology reference: 96.4% of human genes identified as DE in this study had at least one mouse ortholog (3501 of 3633 genes), while 92.4% of mouse genes identified as DE by Beauchemin et al. had at least one human ortholog (5107 of 5,527 genes).
Significance of gene list overlap by Monte Carlo permutation test
After converting human genes identified as DE in this study to their predicted mouse orthologs using the above described human-mouse orthology reference, the overlap between genes found to be differentially increased in LRRK2-low versus -high LUAD tumors were compared with genes differentially increased between neighbouring developmental stages during lung development in the Beauchemin study (i.e. embryonic versus pseudoglandular, pseudoglandular versus canalicular, etc.); a similar strategy was used for genes that were differentially decreased in LRRK2-low versus -high LUAD tumors and during mouse development, for a total of 16 comparisons (Table 1). To test the significance of gene list overlap, a Monte Carlo permutation simulation without replacement47 (n = 100,000 repetitions) was applied to generate an empirical distribution of the overlap that would occur by chance between two lists of genes with variable lengths, given a common pool of 26,855 possibilities—the number of unique HGNC-MGI identifier pairs found in the human-mouse orthology reference, from which the DE human lung cancer genes and DE mouse development genes were constrained to be drawn. An empirical P-value was calculated for each overlap as p = (b + 1)/(m + 1), where b was the number of times a simulated gene list overlap was greater than or equal to the observed number of genes found to overlap and m was the number of permutations47. P-values were further corrected for multiple comparisons (Bonferroni corrected P < 0.003).
In vivo study
The care, housing and use of animals was performed in accordance with the Canadian Council on Animal Care Guidelines. All animal experiments were approved by the Animal Care Committee of the University of British Columbia (A14-0290). Further, all animal experiments were performed in accordance with the relevant guidelines and regulations approved by this committee, as well as with the ARRIVE guidelines (https://arriveguidelines.org/). C57BL/6-Lrrk2tm1Mjfa mice, which have Exon 41 of the kinase domain deleted, were a generous gift by Dr. Matthew J Farrer60 and were previously deposited at Jackson laboratories (RRID:IMSR_JAX:012,444). LRRK2 KO and WT comparison lines were both derived from heterozygote breeding pairs from the C57BL/6-Lrrk2tm1Mjfa strain. To obtain sufficient animals for the treatment study, multiple different littermate homozygous WT or homozygous LRRK2 KO breeding pairs and trios were maintained for 1 to 3 generations and used to generate 20 age-matched animals per genotype (10 males and 10 females). LRRK2 KO and WT strain mates received 10 weekly intraperitoneal (i.p.) injections of urethane (1 g/kg diluted in PBS) beginning at 6–8 weeks old56. The health status of the animals was monitored following an established standard operating procedure. In particular, signs of ill health were based on body weight loss, change in appetite, and behavioral changes such as altered gait, lethargy, and gross manifestations of stress. When signs of severe ill health were present, which occurred in 4 of 20 WT animals, animals were terminated (sodium pentobarbital overdose followed by CO2 asphyxiation) for humane reasons. Surviving mice were euthanized 26 weeks post-first treatment by an overdose (240 mg/kg i.p.) of freshly prepared sodium pentobarbital, followed by exsanguination and tracheal instillation of 10% neutral buffered formalin into lungs, which were then collected and fixed by overnight immersion. Full necropsies were completed on all mice to assess whether there were gross changes in tissue/organ appearance. Lung lobes were separated, and each lobe examined under dissection microscope by two researchers blind to the study design. Surface lesions greater than 0.2 mm were counted and measured using digital micro calipers. Lungs were then paraffin embedded, with each of the five lung lobes per animal arranged in a standard configuration. A single transverse, 5 μm thick tissue section per animal was stained by hematoxylin and eosin and examined by the study pathologist and a second experienced researcher, both blind to study design, and micro lesions (classified as adenomas as outlined by the Mouse Models of Human Cancers Consortium61) were counted per animal. All counts and measurements were averaged between readers and differences between groups were tested by Welch’s two-sample T test.
Multiplex immunohistochemistry
To construct a tissue microarray, 0.6 mm tissue cores containing adenomas and adjacent lung parenchyma from each animal in the study were prepared with a tissue arrayer instrument (Beecher Instruments, Sun Prairie, WI) across two blocks. The following protocol for multiplex IHC was adapted from previous work62. All reagents used for immunohistochemistry were from Biocare Medical (Pacheco, CA) unless otherwise stated. Slides of formalin-fixed, paraffin embedded tissue were incubated overnight at 37 °C, then deparaffinized and rehydrated through xylene and graded alcohols. Antigen retrieval was performed using Rodent Decloaker in a Biocare decloaking chamber at 110 °C for 30 min. Slides were then rinsed with water and loaded into the Biocare Intellipath FLX autostainer. Slides were blocked with Hydrogen Peroxide Blocking Reagent (ab64218, Abcam) and Rodent Block for 10 min and 30 min respectively, before adding a cocktail of SFTPC (1/3000, clone EPR19839, ab211326, Abcam) and LRRK2 (1/600, clone N241A/34, 75–253, Antibodies Incorporated63) in Da Vinci Green diluent for 30 min at room temperature. Following a wash step, Mouse on Mouse HRP Polymer (ab127055, Abcam) was added for 15 min at room temperature, then Mach2 Rabbit AP-Polymer for 30 min, prior to antigen detection with IP Ferangi Blue chromogen for 8 min and after, IP DAB chromogen for 5 min. Finally, CAT hematoxylin at a 1/5 dilution was added for five minutes and slides were washed, air-dried and coverslipped with Ecomount coverslipping medium.
Multispectral images (20X magnification) of entire cores were collected using the PerkinElmer Vectra system. Quantification was performed using inform Advanced Image Analysis Software (PerkinElmer). Four separate visual algorithms were used to train inform to segregate cores into adenoma and adjacent lung parenchyma tissue. Each algorithm was run on images from all cores and cells within each tissue segment were binned into single positive, double positive and negative stain categories. Algorithm performance was validated manually (per algorithm) by visually comparing algorithm cell classification results to the captured images of each core. To calculate frequency, cell counts per stain category, per tissue segment, were averaged across all algorithms and divided by the total counts of cell nuclei per tissue segment; frequency was presented as a percentage of all tissue segment cells. Median frequency and confidence intervals were calculated using Efron's nonparametric bias-corrected and accelerated (BCa) bootstrap method per group (n = 10,000 repetitions; groupwiseMedian function in ‘rcompanion’ package version 1.13.2).
LRRK2 antibody validation by western blot analysis
Snap frozen mouse lung tissue was minced and transferred to Lysing Matrix M tubes (MP Biomedical Cat#6923-050) containing a single zirconium coated ceramic bead (MP Biomedical Cat# 6540-034) and 300uL of RIPA Lysis Buffer (Santa Cruz Biotech Cat#sc-24948A). Samples were then homogenized using an MP Biomedical FastPrep-24 Classic bead beating grinder and lysis system at 6 m/s for 30 s. The tissue homogenate was transferred to a fresh tube and centrifuged at 13,000 RPM for 20 min at 4 °C. The supernatant was transferred to a fresh tube and protein concentration was determined using a Pierce BCA Protein Assay Kit (Thermo Scientific Cat# PI23227). Sixty ug of protein from LRRK2 WT and KO mouse lung samples were resuspended in BOLT LDS sample buffer (Invitrogen Cat# B0007), with BOLT sample reducing agent (Invitrogen Cat# B0009). These were then loaded onto a NuPAGE 3–8% tris–acetate gel (Invitrogen Cat#EA0376BOX) for gel electrophoresis using MOPS Buffer (Invitrogen Cat#B0001), for 50 min at 150 V, prior to transfer onto a PVDF membrane (Bio-Rad Cat#162-0177). The blot was blocked in 5% milk in PBS Tween-20 for 1 h at room temperature, then probed with 1/500 LRRK2 Ab (Antibodies Inc Cat# 75-253) overnight at 4 °C. The blot was then probed with 1/200 Veriblot secondary Ab (Abcam Cat# ab131366) for 2 h at room temperature. Three washes of 15 min each were completed at room temperature. Clarity ECL Reagent (Bio-Rad Cat# 170-5061) was used for immunodetection (Supplementary Fig. S7).
Statistical information
Unless otherwise stated, statistical calculations were performed using R (version 3.4.3; www.R-project.org).
Data availability
This study is based in part on data generated by the TCGA consortium (https://www.cancer.gov/tcga). All TCGA LUAD patient data used in the current study are publicly available through the Broad Institute’s TCGA GDAC Firehose (http://gdac.broadinstitute.org/), using the digital object identifiers cited in “Methods”. The authors declare that all other data supporting the findings of this study are available within the paper or its supplementary information files.
Abbreviations
- AT2:
-
Alveolar type II cells
- DEG:
-
Differentially expressed gene
- DSS:
-
Disease-specific survival
- KO:
-
Knockout
- LRRK2:
-
Leucine rich repeat kinase 2
- LUAD:
-
Lung adenocarcinoma
- non-TRU:
-
Non-terminal respiratory unit
- OS:
-
Overall survival
- PI:
-
Proximal-inflammatory
- PP:
-
Proximal-proliferative
- TCGA:
-
The Cancer Genome Atlas
- TRU:
-
Terminal respiratory unit
References
World Health Organization. Cancer key facts. https://www.who.int/news-room/fact-sheets/detail/cancer (2018).
Alderton, G. K. Tumour evolution: Clonal ancestry in lung cancer. Nat. Rev. Cancer 14, 763 (2014).
Lebovitz, C. B. et al. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 11, 1668–1687 (2015).
Feng, D. D., Cai, W. & Chen, X. The associations between Parkinson’s disease and cancer: The plot thickens. Transl. Neurodegener. 4, 20 (2015).
Kluss, J. H., Mamais, A. & Cookson, M. R. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. https://doi.org/10.1042/BST20180462 (2019).
Fuji, R. N. et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl. Med. 7, 273 (2015).
Rowbotham, S. P. & Kim, C. F. Diverse cells at the origin of lung adenocarcinoma. Proc. Natl. Acad. Sci. 111, 4745–4746 (2014).
Sardi, S. P., Cedarbaum, J. M. & Brundin, P. Targeted therapies for Parkinson’s disease: From genetics to the clinic. Mov. Disord. 33, 684–696 (2018).
Baptista, M. A. S. et al. LRRK2 inhibitors induce reversible changes in nonhuman primate lungs without measurable pulmonary deficits. Sci. Transl. Med. 12, 2 (2020).
Rugbjerg, K., Friis, S., Lassen, C. F., Ritz, B. & Olsen, J. H. Malignant melanoma, breast cancer and other cancers in patients with Parkinson’s disease. Int. J. Cancer 131, 1904–1911 (2012).
Gallo, V. et al. Exploring causality of the association between smoking and Parkinson’s disease. Int. J. Epidemiol. https://doi.org/10.1093/ije/dyy230 (2018).
Agalliu, L. et al. Cancer outcomes among Parkinson’s disease patients with leucine rich repeat kinase 2 mutations, idiopathic Parkinson’s disease patients, and nonaffected controls. Mov. Disord. 34, 1392–1398 (2019).
Allegra, R., Tunesi, S., Cilia, R., Pezzoli, G. & Goldwurm, S. LRRK2-G2019S mutation is not associated with an increased cancer risk: A kin-cohort study. Mov. Disord. Off. J. Mov. Disord. Soc. 29, 1325–1326 (2014).
Ruiz-Martínez, J. et al. Prevalence of cancer in Parkinson’s disease related to R1441G and G2019S mutations in LRRK2. Mov. Disord. Off. J. Mov. Disord. Soc. 29, 750–755 (2014).
Camp, R. L., Dolled-Filhart, M. & Rimm, D. L. X-tile: A new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin. Cancer Res. 10, 7252–7259 (2004).
Liu, J. et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400-416.e11 (2018).
Wilkerson, M. D. et al. Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PLoS ONE 7, e36530 (2012).
Chen, F. et al. Multiplatform-based molecular subtypes of non-small-cell lung cancer. Oncogene 36, 1384–1393 (2017).
Hayes, D. N. et al. Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J. Clin. Oncol. 24, 5079–5090 (2006).
Bryant, C. M. et al. Clinically relevant characterization of lung adenocarcinoma subtypes based on cellular pathways: An international validation study. PLoS ONE 5, e11712 (2010).
Collisson, E. A. et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Beer, D. G. et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med. 8, 816–824 (2002).
The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Faruki, H. et al. Lung adenocarcinoma and squamous cell carcinoma gene expression subtypes demonstrate significant differences in tumor immune landscape. J. Thorac. Oncol. 12, 943–953 (2017).
Huang, Y.-T. et al. Cigarette smoking increases copy number alterations in nonsmall-cell lung cancer. Proc. Natl. Acad. Sci. U. S. A. 108, 16345–16350 (2011).
Buro-Auriemma, L. J. et al. Cigarette smoking induces small airway epithelial epigenetic changes with corresponding modulation of gene expression. Hum. Mol. Genet. 22, 4726–4738 (2013).
Yang, J. et al. Smoking-dependent distal-to-proximal repatterning of the adult human small airway epithelium. Am. J. Respir. Crit. Care Med. 196, 340–352 (2017).
Travis, W. D. et al. The 2015 world health organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 10, 1243–1260 (2015).
Du, Y. et al. Lung gene expression analysis (LGEA): An integrative web portal for comprehensive gene expression data analysis in lung development. Thorax 72, 481–484 (2017).
Du, Y., Guo, M., Whitsett, J. A. & Xu, Y. ‘LungGENS’: A web-based tool for mapping single-cell gene expression in the developing lung. Thorax 70, 1092–1094 (2015).
Kim, N. et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 11, 2285 (2020).
The Gene Ontology Consortium. Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 45, D331–D338 (2017).
Fabregat, A. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 46, D649–D655 (2018).
Liebler, J. M. et al. Combinations of differentiation markers distinguish subpopulations of alveolar epithelial cells in adult lung. Am. J. Physiol. 310, L114–L120 (2015).
Han, S. & Mallampalli, R. K. The role of surfactant in lung disease and host defense against pulmonary infections. Ann. Am. Thorac. Soc. 12, 765–774 (2015).
Moré, J. M. et al. Smoking reduces surfactant protein D and phospholipids in patients with and without chronic obstructive pulmonary disease. BMC Pulm. Med. 10, 53 (2010).
Honda, Y., Takahashi, H., Kuroki, Y., Akino, T. & Abe, S. Decreased contents of surfactant proteins A and D in BAL fluids of healthy smokers. Chest 109, 1006–1009 (1996).
Sin, D. D., Man, S. F. P., McWilliams, A. & Lam, S. Surfactant protein D and bronchial dysplasia in smokers at high risk of lung cancer. Chest 134, 582–588 (2008).
Macía, I. et al. P1.05–004 surfactant protein C is a prognostic marker in resected non-small cell lung cancer: Topic: Translational research and biomarkers. J. Thorac. Oncol. 12, S615 (2017).
Herzig, M. C. et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum. Mol. Genet. https://doi.org/10.1093/hmg/ddr348 (2011).
Nayak, A., Dodagatta-Marri, E., Tsolaki, A. G. & Kishore, U. An insight into the diverse roles of surfactant proteins, SP-A and SP-D in innate and adaptive immunity. Front. Immunol. 3, 131 (2012).
Lavin, Y. et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169, 750-765.e17 (2017).
Aran, D., Sirota, M. & Butte, A. J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 6, 8971 (2015).
Beauchemin, K. J. et al. Temporal dynamics of the developing lung transcriptome in three common inbred strains of laboratory mice reveals multiple stages of postnatal alveolar development. PeerJ 4, e2318 (2016).
Eyre, T. A., Wright, M. W., Lush, M. J. & Bruford, E. A. HCOP: A searchable database of human orthology predictions. Brief. Bioinform. 8, 2–5 (2007).
Wright, M. W., Eyre, T. A., Lush, M. J., Povey, S. & Bruford, E. A. HCOP: The HGNC comparison of orthology predictions search tool. J. Int. Mamm. Genome Soc. 16, 827–828 (2005).
Phipson, B. & Smyth, G. K. Permutation p-values should never be zero: calculating exact p-values when permutations are randomly drawn. Stat. Appl. Genet. Mol. Biol. 9, 2 (2010).
Tripathi, S. et al. Meta- and orthogonal integration of influenza ‘OMICs’ data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735 (2015).
Amy, R. W., Bowes, D., Burri, P. H., Haines, J. & Thurlbeck, W. M. Postnatal growth of the mouse lung. J. Anat. 124, 131 (1977).
Pozarska, A. et al. Stereological monitoring of mouse lung alveolarization from the early postnatal period to adulthood. Am. J. Physiol. 312, L882–L895 (2017).
Crocker, T. T., Teeter, A. & Nielsen, B. Postnatal cellular proliferation in mouse and hamster lung. Cancer Res. 30, 357–361 (1970).
Mund, S. I., Stampanoni, M. & Schittny, J. C. Developmental alveolarization of the mouse lung. Dev. Dyn. 237, 2108–2116 (2008).
Herring, M. J., Putney, L. F., Wyatt, G., Finkbeiner, W. E. & Hyde, D. M. Growth of alveoli during postnatal development in humans based on stereological estimation. Am. J. Physiol. Lung Cell. Mol. Physiol. 307, 338–344 (2014).
Desai, T. J., Brownfield, D. G. & Krasnow, M. A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507, 190–194 (2014).
Vogel, E. R. et al. Perinatal oxygen in the developing lung. Can. J. Physiol. Pharmacol. 93, 119–127 (2015).
Miller, Y. E. et al. Induction of a high incidence of lung tumors in C57BL/6 mice with multiple ethyl carbamate injections. Cancer Lett. 198, 139–144 (2003).
Thorsson, V. et al. The immune landscape of cancer. Immunity 48, 812-830.e14 (2018).
Isella, C. et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat. Commun. 8, 15107 (2017).
Ringnér, M., Jönsson, G. & Staaf, J. Prognostic and chemotherapy predictive value of gene-expression phenotypes in primary lung adenocarcinoma. Clin. Cancer Res. 22, 218–229 (2016).
Dächsel, J. C. et al. A comparative study of Lrrk2 function in primary neuronal cultures. Parkinsonism Relat. Disord. 16, 650–655 (2010).
Nikitin, A. Y. et al. Classification of proliferative pulmonary lesions of the mouse: Recommendations of the mouse models of human cancers consortium. Cancer Res. 64, 2307–2316 (2004).
Zhang, A. W. et al. Interfaces of malignant and immunologic clonal dynamics in ovarian cancer. Cell 173, 1755-1769.e22 (2018).
Davies, P. et al. Comprehensive characterization and optimization of anti-LRRK2 (leucine-rich repeat kinase 2) monoclonal antibodies. Biochem. J. 453, 101–113 (2013).
Acknowledgements
The results shown here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. The authors thank Dr. Matthew Farrer for his kind gift of LRRK2 knockout animals, Dr. William Lockwood for helpful comments on the manuscript, and Dr. Jing Xu for assistance with LRRK2 antibody western blot validation. Support was provided by the Cancer Research Society (operating Grants 20346 and 24133 to SMG) and the Canadian Institutes of Health Research (team Grant GPG102167, partnership Grant CRP-166979 with Cancer Research Society 24133, doctoral research award to CL GSD-140259).
Author information
Authors and Affiliations
Contributions
Performed statistical and bioinformatics analyses: C.L. Animal study: N.W., N.D.S., M.O., C.L., B.T.C., P.S., S.B., N.E.G., M.B.B., E.H., R.A.C., K.L.B. I.H.C. study: K.M., C.L., T.D., S.T., B.H.N. Data interpretation: C.L., S.M.G., B.T.C., K.M., R.A.C., E.H., K.L.B., N.C. Wrote the first draft of the manuscript: C.L. Initiated the project: C.L. and S.M.G. Supervised research: S.M.G., N.W., N.D.S., M.B.B., K.M., B.H.N., K.L.B. Approved the manuscript: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lebovitz, C., Wretham, N., Osooly, M. et al. Loss of Parkinson’s susceptibility gene LRRK2 promotes carcinogen-induced lung tumorigenesis. Sci Rep 11, 2097 (2021). https://doi.org/10.1038/s41598-021-81639-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-81639-0
This article is cited by
-
G2019S selective LRRK2 kinase inhibitor abrogates mitochondrial DNA damage
npj Parkinson's Disease (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
