
Abstract
Ineffectiveness of carbapenems against multidrug resistant pathogens led to the increased use of colistin (polymyxin E) as a last resort antibiotic. A gene belonging to the DedA family encoding conserved membrane proteins was previously identified by screening a transposon library of K. pneumoniae ST258 for sensitivity to colistin. We have renamed this gene dkcA (dedA of Klebsiella required for colistin resistance). DedA family proteins are likely membrane transporters required for viability of Escherichia coli and Burkholderia spp. at alkaline pH and for resistance to colistin in a number of bacterial species. Colistin resistance is often conferred via modification of the lipid A component of bacterial lipopolysaccharide with aminoarabinose (Ara4N) and/or phosphoethanolamine. Mass spectrometry analysis of lipid A of the ∆dkcA mutant shows a near absence of Ara4N in the lipid A, suggesting a requirement for DkcA for lipid A modification with Ara4N. Mutation of K. pneumoniae dkcA resulted in a reduction of the colistin minimal inhibitory concentration to approximately what is found with a ΔarnT strain. We also identify a requirement of DkcA for colistin resistance that is independent of lipid A modification, instead requiring maintenance of optimal membrane potential. K. pneumoniae ΔdkcA displays reduced virulence in Galleria mellonella suggesting colistin sensitivity can cause loss of virulence.
Similar content being viewed by others


Introduction
Colistin (polymyxin E) is a last resort antibiotic for treatment of infections caused by Gram-negative pathogenic bacteria such as Klebsiella pneumoniae1. Unfortunately, colistin-resistant mutants have emerged, leading to the spread of high-risk clones that are resistant to all antimicrobial agents, defined as ‘pan-drug resistant’. It is estimated that by 2050 antimicrobial resistance will cause 10 million deaths annually if no action is taken2. This crisis is worsened by a scarcity of new antimicrobials, especially against Gram-negative pathogens and worldwide dissemination of infections associated with globalization3. Compounds that target colistin resistance are attractive targets for use as adjuvants and, potentially, as antivirulence drugs4,5,6,7.
The DedA family is a highly conserved membrane protein family that remains poorly characterized, belonging to the “SNARE-associated PF09335” family of proteins (PFAM 34.0), with 29,230 individual sequences across 7915 species. We have characterized members of the DedA family in Escherichia coli, Borrelia burgdorferi, Burkholderia thailandensis and Burkholderia glumae8,9,10,11,12,13,14,15,16,17,18,19. The DedA family includes E. coli YqjA and YghB that are putative proton dependent transporters that are required for normal cell division10,11,13, alkaline tolerance16,20 and resistance to a number of antibiotics and biocides15. Both YqjA and YghB possess essential membrane embedded charged amino acids14,15 that are present in proton-dependent transporters belonging to the major facilitator superfamily and other transporter families21,22,23,24,25,26,27,28,29. While the reasons for this are unclear, DedA family proteins are required for polymyxin and/or antimicrobial peptide resistance of Salmonella enterica30, Neisseria meningitidis31, Klebsiella pneumoniae6 and B. thailandensis17,18 and B. glumae 19. DedA has been identified as a member of the colistin “secondary resistome” of multidrug resistant Klebsiella pneumoniae using TNseq and as such may be a target of helper drugs designed to restore the efficacy of colistin toward this and possibly other bacterial pathogens6.
Colistin acts in a manner mechanistically similar to the CAMPS (cationic antimicrobial peptides) of the innate immune response by binding to and disrupting negatively charged bacterial membranes leading to cell lysis and death32. CAMPs are found in all kingdoms of life where they play roles in immune defense, predation and competition33,34. Gram-negative pathogens, including K. pneumoniae, can acquire resistance to colistin by modification of outer membrane lipopolysaccharides (LPS) with cationic substituents resulting in reduced affinity to the polycationic colistin35. Common mechanisms involve expression of enzymes, including aminoarabinose (Ara4N) transferase (ArnT) and phosphoethanolamine transferase (EptA) which transfer aminoarabinose and phosphoethanolamine, respectively, to LPS lipid A. Together, these amine-containing groups neutralize the negative charge of lipid A disrupting colistin binding35. The discovery of plasmid associated eptA homologues (termed mcr for “mobile colistin resistance”) genes raise concern about possible spread of colistin resistance via horizontal gene transfer36,37,38.
Cytoplasmic synthesis and inner membrane transport of undecaprenyl phosphate-α-L-Ara4N precedes periplasmic modification of lipid A with Ara4N (1). The transport of this lipid-linked intermediate is carried out by EmrE-like transporters designated ArnE and ArnF (2). Proton motive force (PMF)-dependent transport of EmrE substrates such as methyl viologen and ethidium bromide is compromised in an E. coli mutant with deletions of two DedA family members (3). It is therefore possible that transport of undecaprenyl-P-α-L-Ara4N is inefficient in certain DedA family mutants resulting in production of lipid A that lacks Ara4N and colistin sensitivity. We have renamed K. pneumoniae dedA dkcA (dedA of Klebsiella required for colistin resistance). In this work, we analyzed a ΔdkcA mutation in virulent, multidrug resistant K. pneumoniae ST258 and demonstrate an altered lipid A structure that is almost completely lacking Ara4N. In addition, we report that DkcA is required for lipid A modification-independent colistin resistance, instead requiring DkcA for optimization of membrane potential. Finally, we demonstrate DkcA is required for virulence in Galleria mellonella suggesting that colistin sensitivity can cause loss of virulence in vivo, consistent with previous studies demonstrating the importance of membrane polarization39 and lipid A modifications40 for virulence. This work supports the lipid A modification pathways4,7 and DkcA6 as potential targets of colistin adjuvants and anti-virulence drugs.
Results
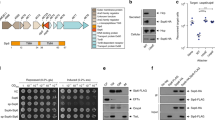
Construction of a K. pneumoniae ST258 ΔdkcA mutant and a complementing pBAD-based plasmid (engineered with an apramycin-resistance marker to allow for selection in MDR K. pneumoniae) expressing dkcA with an N-terminal His-tag, has been described previously6. Measurement of the colistin minimal inhibitory concentration (MIC) using E-strips demonstrates that K. pneumoniae ΔdkcA is nearly eight times more sensitive to colistin than wild type (Fig. 1A) and that expression of K. pneumoniae dkcA or E. coli dedA from a plasmid restores sensitivity of the mutant to near wild type levels (Fig. 1B) consistent with our previous observation6.

Sensitivity of K. pneumoniae ST258 ΔdkcA to colistin and requirement of DkcA conserved, charged amino acids. (A) MIC was determined using colistin E-test strips for K. pneumoniae wild type and ΔdkcA harboring control vector (-) or vector expressing dkcA. Approximate MIC is indicated with arrows and numerically below each image. (B) 1:10 dilutions of the indicated log-phase grown strains were spotted and grown on MH2 agar medium containing 0 or 2 µg/ml colistin containing 0.1% arabinose. (C) Dilutions of mid-log phase grown cells of the indicated strains were spotted on MH2 agar plates containing 0.1% arabinose and 3 µg/ml colistin. (D) Expression of dkcA and point mutants in membrane fractions as determined by Western blotting with anti-hexahistidine antibody. The cropped image was not manipulated in any way and full-length blot is presented in supplementary material (see Supplementary Fig. S4 online). Abbreviations: V = vector control; (-) is lane with no protein loaded. Ten µg of membrane protein was loaded per lane and strains were grown in the presence of 50 µg/ml apramycin and 0.1% arabinose.
While K. pneumoniae DkcA is ~ 90% identical to E. coli DedA, most of our work with the E. coli DedA family has been with YqjA and YghB proteins because of the striking phenotypes of the double mutant strain11,12,13,15, and the ΔyqjA strain16. We have also characterized dkcA homologues from B. thailandensis and B. glumae named dbcA (dedA of Burkholderia required for colistin resistance), which are required for high-level colistin resistance in these species17,18,19. K. pneumoniae DkcA shares only ~ 30% amino acid identity to E. coli YqjA, YghB, and Burkholderia DbcA proteins but still possesses membrane-embedded charged amino acids E40, R133 and R139 in similar locations as found in their YghB/YqjA counterparts that we have shown are critical for function (see Supplementary Fig. S1 online for amino acid alignment)14,15. Therefore, we sequentially changed E40, R133 and R139 each to alanine and determined if the mutant proteins were capable of complementing the colistin sensitivity of the ΔdkcA strain. We found that all three mutants were inactive in this assay, demonstrating the importance of these charged amino acids (Fig. 1C). All proteins were found to be membrane associated, ruling out misfolding of the mutant proteins (Fig. 1D). These findings suggest that K. pneumoniae DkcA may function as a proton-dependent membrane transporter, similar to other members of the DedA family.
Since lipid A modifications contribute to colistin resistance in numerous Gram-negative species including Klebsiella pneumoniae41,42, the lipid A structure of wild type K. pneumoniae ST258 and ΔdkcA grown in LB media was analyzed using mass spectrometry. In wild type K. pneumoniae (Fig. 2a), major species are hexa-acyl lipid A (m/z: 1825.2) and its hydroxylated derivative (m/z: 1841.3). In addition, several species containing Ara4N were observed (designated by red font in Fig. 2; m/z: 1892.8, 1956.3, 1972.3, and the phospoethanolamine/Ara4N modified 2079.3). A similar pattern of lipid A species was observed in wild type and ΔdkcA expressing dkcA from a plasmid (Fig. 2b and d). However, in the ΔdkcA strain harboring the control vector, Ara4N modified species of lipid A were substantially decreased by MS (Fig. 2c), similar to what was found with a Burkholderia thailandensis ΔdedA strain18. Levels of the dual modified Ara4N/phosphoethanolamine species m/z 2079.3 were low but detectable at similar levels in all strains, suggesting the phosphoethanolamine modified species constitutes a minor portion of the total lipid A and is unaffected by the ΔdkcA mutation. See Fig. 2e for structures of these species. This observation in combination with the marked sensitivity to colistin is noteworthy and prompted us to analyze the K. pneumoniae DkcA protein and ΔdkcA strain in more detail.

Mass spectrometry of lipid A isolated from K. pneumoniae strains. (a–d) Lipid A extracted from the indicated strains was analyzed using a MALDI-TOF mass spectrometer (ABI 4700 Proteomic Analyzer) in the negative-ion linear mode. Species modified with Ara4N are labeled in red font. (e) Structures of each observed species.
In our previous work analyzing functions of DedA family members from E. coli, B. thailandensis, and B. glumae, we observed that mutation of DedA family members causes alterations in the membrane proton motive force. These changes were somewhat inconsistent in that E. coli and B. thailandensis mutants were somewhat depolarized12,18, but a B. glumae mutant strain was hyperpolarized19. This suggests that DedA family proteins may function in different manners in optimizing membrane potential depending upon the gene and organism in which they are found. Membrane potential was measured in K. pneumoniae ST258 and ΔdkcA and we found that the ΔdkcA strain was slightly but significantly hyperpolarized (Fig. 3A). We were unable to complement the membrane hyperpolarization of ΔdkcA with pBADdkcA at various levels of arabinose induction due to the observed effect of arabinose on membrane potential even in the wild type strain (data not shown). To determine if this hyperpolarization can result in colistin sensitivity, cells were briefly exposed to colistin (45 min) in the presence or absence of 25 µM CCCP, a proton ionophore, to cause membrane depolarization. Cell survival for all four strains was low in the presence of colistin alone. It was found that a brief preexposure to CCCP could partially rescue cells, both wild type and ΔdkcA, from the lethal effects of colistin (Fig. 3B). This result suggests that colistin may require optimum membrane potential as part of its mechanism of action. This hypothesis was tested in a series of experiments.

A role for DkcA in maintenance of membrane potential and colistin resistance of K. pneumoniae. (A) Membrane hyperpolarization of ΔdkcA compared to ST258. ST258 and ΔdkcA mutant treated with 25 µM CCCP for 30 min showing loss of Δψ were used as the control. (B) Partial restoration of colistin resistance of ∆dkcA by CCCP. Cells grown to OD600 of 0.8 in MH2 media containing 50 µg/ml Apr, 0.1% arabinose were diluted ten-fold and incubated in the same media alone or exposed to 3 µg/ml colistin or 3 µg/ml colistin plus 25 µM CCCP for 45 min at 37 °C. After exposure, cells were washed with fresh MH2 medium twice, resuspended in MH2 and CFU/ml was determined by plating serial dilutions. Each experiment was repeated three times. Statistical significance was calculated by unpaired Student’s t-test. **: p < 0.01. ***: p < 0.001.
Magnesium influx has been reported to alleviate hyperpolarization associated with ribosomal stress in Bacillus subtilis43. In contrast, exposure to sublethal ribosome targeting antibiotics such as spectinomycin can increase the number of cells that exhibit membrane hyperpolarization43. Therefore, we tested the ability of extracellular Mg++ to rescue K. pneumoniae strains from the lethal effects of colistin and sub-MIC concentrations spectinomycin to sensitize strains to colistin. While Mg++ is known to protect Gram negative bacteria from the effects of colistin by competing for binding with negatively charged sites on LPS44, it has also been shown to contribute to polymyxin resistance independently of LPS and the PhoPQ regulon by its ability to depolarize the membrane39. We measured the effects of Mg++ on membrane potential and colistin sensitivity of K. pneumoniae ST258 and ΔdkcA. Growth in the presence of 50 mM or higher of Mg++ causes membrane depolarization of K. pneumoniae ΔdkcA (Fig. 4A). Mg++ at 50 mM completely restores colistin resistance to this mutant (Fig. 4B) under conditions predicted to repress expression of the PhoPQ operon45. These results collectively show that alleviation of membrane hyperpolarization of wild type cells or ΔdkcA strain with Mg++ results in increased colistin resistance, again supporting a role of proper regulation of membrane potential as critical for colistin resistance, likely independently of any alterations to the structure of lipid A.

(A) Mg++ alleviates hyperpolarization of ∆dkcA. Mg++ at indicated concentrations were added in the media, cells were grown to OD600 of 0.8 and membrane potential was determined. ST258 and ΔdkcA mutant treated with 25 µM CCCP for 30 min were used as controls. Each experiment was repeated three times. Statistical significance was calculated by unpaired Student’s t-test. *: p < 0.05, ***: p < 0.001, ns: not significant. (B) Complementation of colistin sensitivity of ΔdkcA by Mg++. 1:10 dilutions of indicated strains were spotted and grown on LB media containing 0 or 2 µg/ml colistin with 50 µg/ml Apr, 0.1% arabinose and indicated concentrations of magnesium sulphate. Plates were incubated at 37 °C for 24 h.
We then measured the effect of spectinomycin-induced hyperpolarization on the colistin sensitivity of K. pneumoniae ST258. We first determined the spectinomycin MIC of the strain to be ~ 4000 µg/ml (see Supplementary Fig. S2 online). Exposure of wild type K. pneumoniae ST258 to a sub-MIC concentration of 500 µg/ml spectinomycin causes hyperpolarization of membrane potential (Fig. 5A). Spectinomycin treatment of ST258 also causes increased colistin sensitivity, decreasing the colistin MIC by ~ 100 fold (Fig. 5B). This indirectly shows hyperpolarization of membrane potential may be responsible for increased colistin sensitivity and again implies that maintenance of optimal membrane potential is required for colistin resistance. This is also consistent with the observation above that depolarization using a brief exposure to CCCP can partially rescue cells from the lethal effects of colistin (Fig. 3B). Growth in the presence of 30 mM or higher of Mg++ causes membrane depolarization of K. pneumoniae ST258, even when hyperpolarized by exposure to spectinomycin (Fig. 5C). Mg++ also causes significant eightfold increase of the colistin MIC observed in the presence of spectinomycin (Fig. 5D), again showing a strong correlation between membrane polarization and colistin sensitivity.

Chemical alteration of membrane potential of K. pneumoniae by spectinomycin and its effect on colistin resistance. (A) Membrane hyperpolarization of ST258 by spectinomycin. Spectinomycin (500 µg/mL) was added at the start of culture in LB media, and the cells were grown to OD600 = 0.6 and membrane potential was determined. ST258 treated with 25 µM CCCP for 30 min was used as a control. (B) Spectinomycin exposure increases colistin sensitivity of ST258. Colistin MIC was determined at indicated concentrations of spectinomycin. Approximate MIC values are indicated with white arrows. (C) Mg++ alleviates the hyperpolarization of membrane potential in ST258 caused by spectinomycin. Spectinomycin (500 µg/mL) and indicated concentration of Mg++ was added at the start of culture in LB media, and the cells were grown to OD600 = 0.6 and membrane potential was determined. (D) Mg++ reverses spectinomycin mediated reduction of colistin MIC. Colistin MIC was measured at indicated concentration of Mg++ for ST258 treated with 500 µg/ml spectinomycin. Approximate MIC values are indicated with white arrows. 5(A) and (C), each bar represents the average value and standard deviation from a triplicate experiment. Each experiment was repeated three times. Statistical significance was calculated by unpaired Student’s t-test. *: p < 0.05. **: p < 0.01. ***: p < 0.001.
To explore this question further and to understand if the effects of membrane polarization are independent of lipid A structure, we constructed an in-frame K. pneumoniae ST258 ΔarnT strain (see Supplementary Fig. S3 online) along with a complementing plasmid (Table 1) and performed similar measurements. ArnT is the Ara4N transferase required for periplasmic modification of lipid A with Ara4N and colistin resistance46,47,48. The ΔarnT strain is sensitive to colistin in the absence of complementation (Fig. 6A). Spectinomycin treatment of K. pneumoniae ΔarnT causes hyperpolarization of membrane potential to a similar extent as that seen in K. pneumoniae ST258 (Fig. 6B). While ΔarnT is exquisitely sensitive to colistin (MIC ~ 0.4 µg/ml), exposure to 2000 µg/ml spectinomycin decreases the MIC by as much as 2.7-fold (Fig. 6C). This observation serves as direct evidence showing hyperpolarization of membrane potential is responsible for increased colistin sensitivity and supports our hypothesis that maintenance of optimal membrane potential is required for colistin resistance independent of lipid A modification.

Spectinomycin mediated reduction of colistin MIC is independent of lipid A modification with Ara4N. (A) Sensitivity of K. pneumoniae ST258 ΔarnT to colistin. Dilution of cultures of indicated strains were spotted and grown on LB agar medium containing 0 or 2 µg/ml colistin containing indicated rhamnose concentrations. (B) Membrane hyperpolarization of ST258 and ΔarnT by spectinomycin treatment. ST258 and ΔarnT treated with 25 µM CCCP for 30 min were used as controls. Each experiment was repeated three times. Statistical significance was calculated by unpaired Student’s t-test. **: p < 0.01. ***: p < 0.001. (C) Spectinomycin treatment further sensitizes ΔarnT towards colistin. Colistin MIC was measured at indicated spectinomycin concentrations. Approximate MIC values are indicated with white arrows.
Loss of resistance to colistin suggests mutation of dkcA may cause loss of virulence since CAMPS of the innate immune system use the same mechanism to kill bacteria as colistin; i.e. by binding to and disrupting negatively charged bacterial membranes leading to cell lysis and death32. In order to measure virulence in vivo, we chose the Galleria mellonella moth larvae model due to its moderate costs and ethical considerations. G. mellonella has been used successfully to measure virulence of K. pneumoniae and many other bacterial pathogens52. Using this model, we found the ΔdkcA was partially compromised for virulence over the course of the experiment (Fig. 7A). Very few viable bacteria could be recovered from ΔdkcA infected larvae, compared to wild type infected larvae, where bacteria could be recovered at 12 and 16 h post infection (Fig. 7B). This suggests that mutations of dkcA and other genes6 that lead to K. pneumoniae colistin sensitivity can also cause loss of virulence and these gene products may also be attractive targets for anti-virulence agents.

DkcA is required for virulence of K. pneumoniae in Galleria mellonella. (A) Wild-type K. pneumoniae ST258 has higher killing rate of larvae compared to ΔdkcA. (B) K. pneumoniae ST258 survival in G. mellonella compared to ΔdkcA. Data points in each graph represent the average value and standard deviation from three independent determinations from a representative experiment. Each experiment was repeated three times. Statistical significance was calculated by unpaired Student’s t-test. *: p < 0.05, **: p < 0.01, ***: p < 0.001, ns: not significant.
Discussion
Multidrug resistant bacterial infections pose an enormous public safety risk and are a challenge to modern medicine, made worse by a lack of new antimicrobial drugs2,3. The emergence of carbapenem-resistant Klebsiella pneumoniae is of particular concern and has forced the reintroduction of colistin therapy as a last resort treatment. Colistin is an antimicrobial peptide belonging to the polymyxin family that can cause numerous side effects including nephrotoxicity so dosage must be strictly monitored53. The recent emergence of mobilized colistin resistance genes that spread via horizontal gene transfer has further compromised the use of colistin clinically54. Helper drugs that can lower the effective dosage of colistin by targeting the “secondary resistome” would be useful therapeutics6. Drugs that alter colistin resistance may also result in sensitivity to the cationic antimicrobial peptides of the innate immune system and thus act to target virulence even in the absence of antibiotic administration4,5. Potential targets may be Klebsiella pneumoniae DkcA or ArnT as each gene deletion strain displays a similar eight-fold reduction in MIC to colistin.
Gram-negative bacteria often become resistant to colistin by activation of genes and pathways that lead to the covalent modification of anionic cell surface lipopolysaccharide with cationic substituents, causing loss of electrostatic binding by the cationic antibiotic35,55. In Klebsiella and other species, these pathways are controlled by the PhoPQ and PmrAB two-component signaling pathways which lead to modification of lipid A with Ara4N and phosphoethanolamine55. While K. pneumoniae does harbor a chromosomal homolog of EptA involved in phosphoethanolamine modification of lipid A56, our MS analysis suggests this species makes up a minor component of the total lipid A suggesting Ara4N modification is the main contributor to colistin resistance.
Less well characterized is the contribution of membrane potential to colistin sensitivity and resistance. Here, we show that the K. pneumoniae ΔdkcA is slightly but significantly hyperpolarized and tested the contribution of this hyperpolarization to the observed colistin sensitivity. First, we showed that brief pre-exposure of both wild type and ΔdkcA strains to the proton ionophore and depolarizing agent CCCP results in higher survival in the presence of a lethal dose of colistin. The augmented survival against colistin by brief pre-exposure to CCCP is probably due to cytoplasmic acidification by CCCP, consistent with our hypothesis20. Next, we showed that treatment with sub-lethal concentrations of the antibiotic spectinomycin causes hyperpolarization of K. pneumoniae consistent with what was shown for B. subtilis43. Importantly, this hyperpolarization was accompanied by an increase in sensitivity to colistin. Therefore, we could mimic the effect of the ΔdkcA mutation on both the membrane potential and colistin sensitivity using sub-lethal concentrations of spectinomycin. Relieving the hyperpolarization with magnesium also rescued the sensitivity to colistin. Finally, we showed that the effect of membrane potential on colistin sensitivity was independent of lipid A modification by demonstrating the same effect in a K. pneumoniae ΔarnT mutant. While ΔarnT is sensitive to colistin, consistent with a previous report6, exposure to spectinomycin increased this sensitivity while also hyperpolarizing the membrane. Collectively, these results suggest that membrane hyperpolarization can directly lead to colistin sensitivity independently of any changes to the cell surface LPS (Fig. 8).

A proposed model of colistin resistance determinants of Klebsiella pneumoniae ST258. Both lipid A modification dependent and lipid A modification independent mechanisms may be involved in colistin resistance. DkcA is required for lipid A modification with Ara4N (aminoarabinose) through a yet unidentified mechanism (right side). Lipid A modification with Ara4N increases the overall positive charge of outer membrane and electrostatically repels cationic colistin, contributing to colistin resistance. Lipid A modification with PEtN (phosphoethalonamine) is not affected by the DkcA mutation. DkcA is also involved in proper maintenance of membrane potential contributing to colistin resistance (left side). Mutation of DkcA results in membrane hyperpolarization. By maintaining reversed membrane potential (more positive inside), DkcA could reduce the attraction of cationic colistin molecules towards the membrane and reduce the bactericidal effects of colistin. This is supported by the increased colistin sensitivity by hyperpolarizing agents (spectinomycin) and the reversal of this effect with depolarizing agents (Mg++ or CCCP). Thus, an increase in negative membrane potential can electrostatically draw cationic antimicrobial peptides toward the nonpolar inner membrane, the likely site of action of colistin57.
The DedA family of membrane proteins is widely distributed in nature, found in all kingdoms. Until recently, most of what is known about the family came from studies in E. coli due to pleiotropic phenotypes of inactivating mutations. These phenotypes include defects in growth, cell division and hypersensitivity to alkaline pH, antibiotics and membrane penetrating dyes10,11,12,13,15,16. The correction of these phenotypes by growth in slightly acidic pH and the presence of membrane embedded charged amino acids suggest that members of the DedA family are proton-dependent transporters required for maintenance of optimal PMF12,14,15,16. Our recent studies in Burkholderia thailandensis and B. glumae also support this hypothesis17,18,19,20. DedA family proteins have an evolutionary relationship and, indeed may share structural similarity to LeuT, a bacterial homolog of the neurotransmitter:sodium symporter family58,59.
While there is no published structure of any DedA family member, modelling of the structure of Human TMEM41b has been carried out which is predicted to possess structural features found in Cl-/H+ antiporters60. The function of human TMEM41B is unknown but it has been identified as being required for autophagosome formation61,62,63 and as a host factor required for infection with flaviviruses as well as SARS-CoV-264,65. While the eukaryotic proteins of the DedA family containing the so-called VTT domain (for VMP, TMEM41, Tvp38)63 are distantly related to their bacterial counterparts, and a functional relationship has not been established, the presence of an absolutely conserved glycine residue suggests an evolutionary relationship66.
Invertebrate models of bacterial virulence are increasingly common due to lower cost than traditional rodent studies52. There are no ethical constraints and their short life span makes them ideal for large-scale studies. While insects do not possess an adaptive immune response, their innate immune response is quite similar to mammals67. Insects possess Toll-like receptors, CAMPs, and neutrophil-like cells called hemocytes68. There are currently more than 2000 articles published on PubMed on the wax moth Galleria mellonella infection model. Galleria mellonella has been used to study virulence of Klebsiella pneumoniae40,52,69 and other bacterial pathogens70,71. We show here that Klebsiella pneumoniae DkcA is required for virulence in G. mellonella, which may be linked to the sensitivity to colistin. These studies are consistent with previous studies demonstrating the importance of optimal membrane potential39 and cationic lipid A modifications40 for virulence.
In summary, we have shown that the Klebsiella pneumoniae DkcA protein is required for colistin resistance in a lipid A-dependent and -independent manner. DkcA displays similarity to proton-dependent membrane transporters and is required for virulence in moth larvae. The bacterial DedA family is evolutionarily conserved and remains an intriguing mystery in terms of its physiological function and it potential for therapeutic targeting.
Material and methods
Bacterial growth conditions
E. coli cultures were grown on LB medium (1% tryptone, 0.5% yeast extract and 1% NaCl). Klebsiella pneumoniae was grown in LB or cation adjusted Mueller–Hinton broth 2 (MH2; Sigma-Aldrich). Antibiotics colistin, ampicillin (Amp; 100 µg/ml), apramycin (Apr; 50 µg/ml) and spectinomycin (at indicated concentrations) were purchased from Sigma-Aldrich or VWR. Cultures were grown at 37 °C unless otherwise indicated. For complementation analysis with pBAD- or pSCrhaB2-derived vectors, arabinose or rhamnose at indicated concentrations was included in growth media. Bacterial strains and plasmids are listed in Table 1.
Creation of Klebsiella pneumoniae ΔarnT
The K. pneumoniae ST258 arnT gene was disrupted using lambda red recombination50,72. Bacteria were grown in low salt LB (5 g/L NaCl), pH 8.0 up to the arnT disruption step. pACBSR-HygR was introduced into ST258 by electroporation and grown at 30 °C. Knockout primers were designed in a way that arnT_KO_forward primer has 60 bp homology to the 5’ upstream region and arnT_KO_reverse primer has 60 bp homology to the 3’ downstream region of the arnT gene (Table 2). An apramycin resistance cassette (AprR) was amplified from pIJ77350 using these primers. The linear DNA fragment was purified using QIAquick Gel Extraction Kit (Qiagen). Purified linear DNA was treated with DpnI and repurified prior to introduction by electroporation into ST258/pACBSR-HygR. Following recovery in the absence of antibiotic, the cells were plated on 50 µg/mL Apr. Resistant colonies were screened for colistin sensitivity and gene disruption was verified using PCR primers P1F, P2R, P3F, P4R (Table 2, see Supplementary Fig. S3 online).
Cloning of K. pneumoniae ST258 arnT
K. pneumoniae ST258 genomic DNA was extracted using Easy-DNA kit (Invitrogen). PCR amplification using Q5 DNA polymerase was performed using KparnT1 and KparnT2 primers (Table 2). For cloning, the stop codon TGA was changed to TAA to prevent modification by Dam methylation. PCR amplification using ST258 genomic DNA generated a linear DNA fragment of 1656 bp. The DNA fragment was gel purified using QIAquick Gel Extraction Kit (Qiagen) and digested with NdeI and XbaI (New England Biolabs). pScrhaB251 was similarly digested with NdeI and XbaI and dephosphorylated using Antarctic phosphatase (New England Biolabs). DNA fragments were ligated using Hi-T4 DNA ligase (New England Biolabs). The ligated product was transformed into SM1073 and TetR resistant cells were selected on LB supplemented with 25 µg/mL tetracycline. Cloned arnT was expressed from the resulting plasmid pSCarnT using 0.01 or 0.05% rhamnose.
Site-directed mutagenesis
Site-specific mutants were created by using a previously published protocol74. The primers carrying the site-specific mutations (Table 2) were used in a PCR reaction to amplify a vector containing the wild type gene. The PCR product was then digested with Dpn1 and used to transform competent XL1-blue cells. Colonies obtained after transformation were screened by colony PCR using gene-specific primers. Mutations were confirmed by DNA sequencing conducted at the LSU College of Science Genomics Facility.
Membrane preparation and western blotting
Cell membranes was prepared from exponential phase cultures containing indicated plasmid DNA grown under inducing conditions (0.1% arabinose) using a previously published protocol15,75. Equal amounts of protein were resolved by SDS-PAGE and transferred to PVDF membrane. Western blotting was carried out using penta-His (Qiagen) primary antibody at 1:5000 dilution and goat-anti-mouse horseradish peroxidase (HRP) conjugated secondary antibody (Pierce) at 1:5000. Detection was performed using the ImmunStar HRP kit (Bio-Rad).
Susceptibility to colistin and other antibiotics
For testing the susceptibility on solid medium, overnight cultures were freshly diluted 1:100 in MH2 media with appropriate antibiotics and additives, and grown to OD600 ~ 0.6 at 37 °C in a shaking incubator. Five microliters of serially log10-diluted cells were spotted onto MH2 or LB agar plates containing various concentrations of antibiotic. Growth was analyzed after incubation for 24 h at 37 °C unless otherwise indicated. Antibiotic MIC’s were measured using the broth microdilution method in 96 well plates or using colistin test gradient strips (Biomerieux). All experiments were repeated at least three times.
Mass spectrometry
For isolation of lipid A, K. pneumoniae cultures were grown at 37 °C to an OD600 of ~ 1.0. Lipid A chemical extraction was carried out after mild acidic hydrolysis of LPS as previously described76,77. For visualization of lipid A by mass spectrometry, lipids were analyzed using MALDI-TOF (ABI 4700 Proteomic Analyzer) in the negative-ion linear mode as previously described78,79. Briefly, lipid A samples were dissolved in a mixture of chloroform–methanol (4:1, vol/vol), and 1 µl of sample was mixed with 1 µl of matrix solution. The matrix consisted of 5-chloro-2-mercaptobenzothiazole (CMBT) (20 mg/mL) resuspended in chloroform–methanol-water (4:4:1, vol/vol/vol) mixed with saturated ammonium citrate (20:1, vol/vol). One µl of sample-matrix mixture was loaded on to MALDI target plate for final analysis.
Measurement of membrane potential
Measurement of membrane potential was performed using JC-1 dye80. Briefly, 2.5 × 107 cells from the overnight cultures were inoculated in 20 ml of fresh LB broth in a 250 ml flask and grown for about 1 h 40 min at 37 °C with shaking. ~ 6 × 108 cells were taken from each culture, washed with permeabilization buffer PB, pH 7.5 (10 mM Tris, 1 mM EDTA, 10 mM glucose) and resuspended in fresh PB. 3 µM of JC-1 dye was added and incubated in the dark at 30 °C for 30 min. Cells were washed and resuspended in PB buffer and fluorescence measurements were carried out using a JASCO FP-6300 spectrofluorometer. Relative membrane potential is expressed the ratio of red (595 nm) to green (530 nm) fluorescence with excitation of 488 nm.
Galleria mellonella model of virulence
Larvae purchased from Carolina Biological Supply Company (item# 143,928) were injected with 5 × 105 CFU of bacteria resuspended in 5 µl PBS pH 7.4 or PBS alone. After the injection, larvae were incubated at 37 °C in the dark over a period of 96 h. Larvae were monitored for their survival every 24 h. Dead larvae were identified by the lack of motility or bodily movement against physical stimuli. To measure bacterial survival in G. mellonella, larvae were injected with 5 × 105 CFU in PBS pH 7.4 and incubated at 37 °C in the dark. Larvae were homogenized at indicated times and serial dilutions were plated in LB agar containing 100 µg/mL apramycin. No apramycin resistant bacteria were isolated from control larvae injected with PBS under these conditions.
References
Paterson, D. L. & Harris, P. N. Colistin resistance: a major breach in our last line of defence. Lancet Infect. Dis. 16, 132–133. https://doi.org/10.1016/S1473-3099(15)00463-6 (2016).
De Oliveira, D. M. P. et al. Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 33, e00181-19. https://doi.org/10.1128/CMR.00181-19 (2020).
World Health Organization. Antimicrobial resistance : global report on surveillance 2014. (World Health Organization, 2014).
Kahler, C. M., Sarkar-Tyson, M., Kibble, E. A., Stubbs, K. A. & Vrielink, A. Enzyme targets for drug design of new anti-virulence therapeutics. Curr. Opin. Struct. Biol. 53, 140–150. https://doi.org/10.1016/j.sbi.2018.08.010 (2018).
Harris, T. L. et al. Small molecule downregulation of PmrAB reverses lipid A modification and breaks colistin resistance. ACS Chem. Biol. 9, 122–127. https://doi.org/10.1021/cb400490k (2014).
Jana, B. et al. The secondary resistome of multidrug-resistant Klebsiella pneumoniae. Sci. Rep. 7, 42483. https://doi.org/10.1038/srep42483 (2017).
Kline, T. et al. Synthesis of and evaluation of lipid A modification by 4-substituted 4-deoxy arabinose analogs as potential inhibitors of bacterial polymyxin resistance. Bioorg. Med. Chem. Lett. 18, 1507–1510. https://doi.org/10.1016/j.bmcl.2007.12.061 (2008).
Boughner, L. A. & Doerrler, W. T. Multiple deletions reveal the essentiality of the DedA membrane protein family in Escherichia coli. Microbiology 158, 1162–1171. https://doi.org/10.1099/mic.0.056325-0 (2012).
Doerrler, W. T., Sikdar, R., Kumar, S. & Boughner, L. A. New functions for the ancient DedA membrane protein family. J. Bacteriol. 195, 3–11. https://doi.org/10.1128/JB.01006-12 (2013).
Liang, F. T. et al. BB0250 of Borrelia burgdorferi is a conserved and essential inner membrane protein required for cell division. J. Bacteriol. 192, 6105–6115. https://doi.org/10.1128/JB.00571-10 (2010).
Sikdar, R. & Doerrler, W. T. Inefficient Tat-dependent export of periplasmic amidases in an Escherichia coli strain with mutations in two DedA family genes. J. Bacteriol. 192, 807–818. https://doi.org/10.1128/JB.00716-09 (2010).
Sikdar, R., Simmons, A. R. & Doerrler, W. T. Multiple envelope stress response pathways are activated in an Escherichia coli strain with mutations in two members of the DedA membrane protein family. J. Bacteriol. 195, 12–24. https://doi.org/10.1128/JB.00762-12 (2013).
Thompkins, K., Chattopadhyay, B., Xiao, Y., Henk, M. C. & Doerrler, W. T. Temperature sensitivity and cell division defects in an Escherichia coli strain with mutations in yghB and yqjA, encoding related and conserved inner membrane proteins. J. Bacteriol. 190, 4489–4500. https://doi.org/10.1128/JB.00414-08 (2008).
Kumar, S., Bradley, C. L., Mukashyaka, P. & Doerrler, W. T. Identification of essential arginine residues of Escherichia coli DedA/Tvp38 family membrane proteins YqjA and YghB. FEMS Microbiol. Lett. https://doi.org/10.1093/femsle/fnw133 (2016).
Kumar, S. & Doerrler, W. T. Members of the conserved DedA family are likely membrane transporters and are required for drug resistance in Escherichia coli. Antimicrob. Agents Chemother. 58, 923–930. https://doi.org/10.1128/AAC.02238-13 (2014).
Kumar, S. & Doerrler, W. T. Escherichia coli YqjA, a member of the conserved DedA/Tvp38 membrane protein family, is a putative osmosensing transporter required for growth at alkaline pH. J. Bacteriol. 197, 2292–2300. https://doi.org/10.1128/JB.00175-15 (2015).
Panta, P. R. & Doerrler, W. T. A Burkholderia thailandensis DedA family membrane protein is required for proton motive force dependent lipid a modification. Front. Microbiol. 11, 618389. https://doi.org/10.3389/fmicb.2020.618389 (2021).
Panta, P. R. et al. A DedA family membrane protein is required for Burkholderia thailandensis Colistin Resistance. Front. Microbiol. 10, 2532. https://doi.org/10.3389/fmicb.2019.02532 (2019).
Iqbal, A. et al. Chemical or genetic alteration of proton motive force results in loss of virulence of Burkholderia glumae, the cause of rice bacterial panicle blight. Appl. Environ. Microbiol. 87, e0091521. https://doi.org/10.1128/AEM.00915-21 (2021).
Panta, P. R. & Doerrler, W. T. A link between pH homeostasis and colistin resistance in bacteria. Sci. Rep. 11, 13230. https://doi.org/10.1038/s41598-021-92718-7 (2021).
Gerchman, Y. et al. Histidine-226 is part of the pH sensor of NhaA, a Na+/H+ antiporter in Escherichia coli. Proc. Natl. Acad. Sci. U S A 90, 1212–1216 (1993).
Noumi, T., Inoue, H., Sakurai, T., Tsuchiya, T. & Kanazawa, H. Identification and characterization of functional residues in a Na+/H+ antiporter (NhaA) from Escherichia coli by random mutagenesis. J. Biochem. 121, 661–670. https://doi.org/10.1093/oxfordjournals.jbchem.a021637 (1997).
Adler, J. & Bibi, E. Determinants of substrate recognition by the Escherichia coli multidrug transporter MdfA identified on both sides of the membrane. J. Biol. Chem. 279, 8957–8965. https://doi.org/10.1074/jbc.M313422200 (2004).
Sigal, N. et al. 3D model of the Escherichia coli multidrug transporter MdfA reveals an essential membrane-embedded positive charge. Biochemistry 44, 14870–14880. https://doi.org/10.1021/bi051574p (2005).
Holdsworth, S. R. & Law, C. J. Functional and biochemical characterisation of the Escherichia coli major facilitator superfamily multidrug transporter MdtM. Biochimie 94, 1334–1346. https://doi.org/10.1016/j.biochi.2012.03.001 (2012).
Fluman, N., Ryan, C. M., Whitelegge, J. P. & Bibi, E. Dissection of mechanistic principles of a secondary multidrug efflux protein. Mol. Cell 47, 777–787. https://doi.org/10.1016/j.molcel.2012.06.018 (2012).
Abramson, J., Iwata, S. & Kaback, H. R. Lactose permease as a paradigm for membrane transport proteins (Review). Mol. Membr. Biol. 21, 227–236. https://doi.org/10.1080/09687680410001716862 (2004).
Cain, B. D. & Simoni, R. D. Proton translocation by the F1F0ATPase of Escherichia coli. Mutagenic analysis of the a subunit. J. Biol. Chem. 264, 3292–3300 (1989).
Hellmer, J., Teubner, A. & Zeilinger, C. Conserved arginine and aspartate residues are critical for function of MjNhaP1, a Na+/H+ antiporter of M. jannaschii. FEBS Lett 542, 32–36 (2003).
Shi, Y., Cromie, M. J., Hsu, F. F., Turk, J. & Groisman, E. A. PhoP-regulated Salmonella resistance to the antimicrobial peptides magainin 2 and polymyxin B. Mol. Microbiol. 53, 229–241. https://doi.org/10.1111/j.1365-2958.2004.04107.x (2004).
Tzeng, Y. L. et al. Cationic antimicrobial peptide resistance in Neisseria meningitidis. J. Bacteriol. 187, 5387–5396. https://doi.org/10.1128/JB.187.15.5387-5396.2005 (2005).
Poirel, L., Jayol, A. & Nordmann, P. Polymyxins: antibacterial activity, susceptibility testing, and resistance mechanisms encoded by plasmids or chromosomes. Clin. Microbiol. Rev. 30, 557–596. https://doi.org/10.1128/CMR.00064-16 (2017).
Yeaman, M. R. & Yount, N. Y. Unifying themes in host defence effector polypeptides. Nat. Rev. Microbiol. 5, 727–740. https://doi.org/10.1038/nrmicro1744 (2007).
Lazzaro, B. P., Zasloff, M. & Rolff, J. Antimicrobial peptides: application informed by evolution. Science https://doi.org/10.1126/science.aau5480 (2020).
Raetz, C. R., Reynolds, C. M., Trent, M. S. & Bishop, R. E. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 76, 295–329. https://doi.org/10.1146/annurev.biochem.76.010307.145803 (2007).
Liu, Y. Y. et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect. Dis. 16, 161–168. https://doi.org/10.1016/S1473-3099(15)00424-7 (2016).
McGann, P. et al. Escherichia coli Harboring mcr-1 and blaCTX-M on a Novel IncF Plasmid: First Report of mcr-1 in the United States. Antimicrob. Agents Chemother. 60, 4420–4421. https://doi.org/10.1128/AAC.01103-16 (2016).
Rolain, J. M. et al. Plasmid-Mediated mcr-1 Gene in Colistin-Resistant Clinical Isolates of Klebsiella pneumoniae in France and Laos. Antimicrob. Agents Chemother. 60, 6994–6995. https://doi.org/10.1128/AAC.00960-16 (2016).
Alteri, C. J., Lindner, J. R., Reiss, D. J., Smith, S. N. & Mobley, H. L. The broadly conserved regulator PhoP links pathogen virulence and membrane potential in Escherichia coli. Mol. Microbiol. 82, 145–163. https://doi.org/10.1111/j.1365-2958.2011.07804.x (2011).
Insua, J. L. et al. Modeling Klebsiella pneumoniae pathogenesis by infection of the wax moth Galleria mellonella. Infect. Immun. 81, 3552–3565. https://doi.org/10.1128/IAI.00391-13 (2013).
Wright, M. S. et al. Genomic and transcriptomic analyses of colistin-resistant clinical isolates of Klebsiella pneumoniae reveal multiple pathways of resistance. Antimicrob. Agents Chemother. 59, 536–543. https://doi.org/10.1128/AAC.04037-14 (2015).
Leung, L. M. et al. Structural modification of LPS in colistin-resistant, KPC-producing Klebsiella pneumoniae. J. Antimicrob. Chemother. 72, 3035–3042. https://doi.org/10.1093/jac/dkx234 (2017).
Lee, D. D. et al. Magnesium flux modulates ribosomes to increase bacterial survival. Cell 177, 352-360e313. https://doi.org/10.1016/j.cell.2019.01.042 (2019).
Vaara, M. Agents that increase the permeability of the outer membrane. Microbiol. Rev. 56, 395–411. https://doi.org/10.1128/mr.56.3.395-411.1992 (1992).
Vescovi, E. G., Soncini, F. C. & Groisman, E. A. Mg2+ as an extracellular signal: environmental regulation of Salmonella virulence. Cell 84, 165–174. https://doi.org/10.1016/s0092-8674(00)81003-x (1996).
Petrou, V. I. et al. Structures of aminoarabinose transferase ArnT suggest a molecular basis for lipid A glycosylation. Science 351, 608–612. https://doi.org/10.1126/science.aad1172 (2016).
Tavares-Carreon, F., Fathy Mohamed, Y., Andrade, A. & Valvano, M. A. ArnT proteins that catalyze the glycosylation of lipopolysaccharide share common features with bacterial N-oligosaccharyltransferases. Glycobiology 26, 286–300. https://doi.org/10.1093/glycob/cwv095 (2016).
Trent, M. S., Ribeiro, A. A., Lin, S., Cotter, R. J. & Raetz, C. R. An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-L-arabinose to lipid A: induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J. Biol. Chem. 276, 43122–43131. https://doi.org/10.1074/jbc.M106961200 (2001).
Simon, R., Priefer, U. & Puhler, A. A broad host range mobilization system for invivo genetic-engineering - transposon mutagenesis in gram-negative bacteria. Bio-Technol. 1, 784–791. https://doi.org/10.1038/nbt1183-784 (1983).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U S A 97, 6640–6645. https://doi.org/10.1073/pnas.120163297 (2000).
Cardona, S. T. & Valvano, M. A. An expression vector containing a rhamnose-inducible promoter provides tightly regulated gene expression in Burkholderia cenocepacia. Plasmid 54, 219–228. https://doi.org/10.1016/j.plasmid.2005.03.004 (2005).
Tsai, C. J., Loh, J. M. & Proft, T. Galleria mellonella infection models for the study of bacterial diseases and for antimicrobial drug testing. Virulence 7, 214–229. https://doi.org/10.1080/21505594.2015.1135289 (2016).
Koch-Weser, J. et al. Adverse effects of sodium colistimethate. Manifestations and specific reaction rates during 317 courses of therapy. Ann. Int. Med. 72, 857–868 (1970).
Hussein, N. H., Al-Kadmy, I. M. S., Taha, B. M. & Hussein, J. D. Mobilized colistin resistance (mcr) genes from 1 to 10: a comprehensive review. Mol. Biol. Rep. 48, 2897–2907. https://doi.org/10.1007/s11033-021-06307-y (2021).
Olaitan, A. O., Morand, S. & Rolain, J. M. Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front. Microbiol. 5, 643. https://doi.org/10.3389/fmicb.2014.00643 (2014).
Needham, B. D. & Trent, M. S. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 11, 467–481. https://doi.org/10.1038/nrmicro3047 (2013).
Sabnis, A. et al. Colistin kills bacteria by targeting lipopolysaccharide in the cytoplasmic membrane. Elife https://doi.org/10.7554/eLife.65836 (2021).
Khafizov, K., Staritzbichler, R., Stamm, M. & Forrest, L. R. A study of the evolution of inverted-topology repeats from LeuT-fold transporters using AlignMe. Biochemistry 49, 10702–10713. https://doi.org/10.1021/bi101256x (2010).
Keller, R., Ziegler, C. & Schneider, D. When two turn into one: evolution of membrane transporters from half modules. Biol. Chem. 395, 1379–1388. https://doi.org/10.1515/hsz-2014-0224 (2014).
Mesdaghi, S., Murphy, D. L., Sanchez Rodriguez, F., Burgos-Marmol, J. J. & Rigden, D. J. In silico prediction of structure and function for a large family of transmembrane proteins that includes human Tmem41b. F1000Res 9, 1395. https://doi.org/10.12688/f1000research.27676.2 (2020).
Shoemaker, C. J. et al. CRISPR screening using an expanded toolkit of autophagy reporters identifies TMEM41B as a novel autophagy factor. PLoS Biol. 17, e2007044. https://doi.org/10.1371/journal.pbio.2007044 (2019).
Morita, K., Hama, Y. & Mizushima, N. TMEM41B functions with VMP1 in autophagosome formation. Autophagy 15, 922–923. https://doi.org/10.1080/15548627.2019.1582952 (2019).
Morita, K. et al. Genome-wide CRISPR screen identifies TMEM41B as a gene required for autophagosome formation. J. Cell Biol. 217, 3817–3828. https://doi.org/10.1083/jcb.201804132 (2018).
Hoffmann, H. H. et al. TMEM41B is a pan-flavivirus host factor. Cell 184, 133-148e120. https://doi.org/10.1016/j.cell.2020.12.005 (2021).
Schneider, W. M. et al. Genome-Scale Identification of SARS-CoV-2 and Pan-coronavirus Host Factor Networks. Cell 184, 120-132e114. https://doi.org/10.1016/j.cell.2020.12.006 (2021).
Tabara, L. C., Vincent, O. & Escalante, R. Evidence for an evolutionary relationship between Vmp1 and bacterial DedA proteins. Int. J. Dev. Biol. 63, 67–71. https://doi.org/10.1387/ijdb.180312re (2019).
Pereira, M. F., Rossi, C. C., da Silva, G. C., Rosa, J. N. & Bazzolli, D. M. S. Galleria mellonella as an infection model: an in-depth look at why it works and practical considerations for successful application. Pathog. Dis. https://doi.org/10.1093/femspd/ftaa056 (2020).
Browne, N., Heelan, M. & Kavanagh, K. An analysis of the structural and functional similarities of insect hemocytes and mammalian phagocytes. Virulence 4, 597–603. https://doi.org/10.4161/viru.25906 (2013).
Mills, G., Dumigan, A., Kidd, T., Hobley, L. & Bengoechea, J. A. Identification and Characterization of Two Klebsiella pneumoniae lpxL Lipid A Late Acyltransferases and Their Role in Virulence. Infect. Immun. https://doi.org/10.1128/IAI.00068-17 (2017).
Seed, K. D. & Dennis, J. J. Development of Galleria mellonella as an alternative infection model for the Burkholderia cepacia complex. Infect. Immun. 76, 1267–1275. https://doi.org/10.1128/IAI.01249-07 (2008).
Asai, M. et al. Galleria mellonella: an infection model for screening compounds against the Mycobacterium tuberculosis complex. Front. Microbiol. 10, 2630. https://doi.org/10.3389/fmicb.2019.02630 (2019).
Huang, T. W. et al. Capsule deletion via a lambda-Red knockout system perturbs biofilm formation and fimbriae expression in Klebsiella pneumoniae MGH 78578. BMC Res. Notes 7, 13. https://doi.org/10.1186/1756-0500-7-13 (2014).
Simon, R., Priefer, U. & Pühler, A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology 1, 784–791. https://doi.org/10.1038/nbt1183-784 (1983).
Zheng, L., Baumann, U. & Reymond, J. L. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115. https://doi.org/10.1093/nar/gnh110 (2004).
Doerrler, W. T. & Raetz, C. R. ATPase activity of the MsbA lipid flippase of Escherichia coli. J. Biol. Chem. 277, 36697–36705. https://doi.org/10.1074/jbc.M205857200 (2002).
Zhou, Z., Lin, S., Cotter, R. J. & Raetz, C. R. Lipid A modifications characteristic of Salmonella typhimurium are induced by NH4VO3 in Escherichia coli K12. Detection of 4-amino-4-deoxy-L-arabinose, phosphoethanolamine and palmitate. J. Biol. Chem. 274, 18503–18514 (1999).
Herrera, C. M. et al. The Vibrio cholerae VprA-VprB two-component system controls virulence through endotoxin modification. MBio https://doi.org/10.1128/mBio.02283-14 (2014).
Henderson, J. C., O’Brien, J. P., Brodbelt, J. S. & Trent, M. S. Isolation and chemical characterization of lipid A from gram-negative bacteria. J. Vis. Exp. https://doi.org/10.3791/50623 (2013).
Zhou, P., Altman, E., Perry, M. B. & Li, J. Study of matrix additives for sensitive analysis of lipid A by matrix-assisted laser desorption ionization mass spectrometry. Appl. Environ. Microbiol. 76, 3437–3443. https://doi.org/10.1128/AEM.03082-09 (2010).
Panta, P. R. et al. A DedA family membrane protein is required for Burkholderia thailandensis colistin resistance. Front. Microbiol. https://doi.org/10.3389/fmicb.2019.02532 (2019).
Acknowledgements
This work was supported with private funding by the LSU Foundation Biotransport Research Support Fund (WTD) and by NIH grants AI138576, AI076322 and AI064184 (MST). We thank Dr. Marcia Newcomer (LSU Department of Biological Sciences) and Dr. Scott Herke (LSU Genomics Facility) for technical support.
Author information
Authors and Affiliations
Contributions
W.T.D., P.R.P., V.T.: Conception and design of the study, acquisition, analysis, and interpretation of the data and writing of the manuscript. C.B., M.V.D., C.M.H., M.S.T.: acquisition, analysis, and interpretation of the data and writing of the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tiwari, V., Panta, P.R., Billiot, C.E. et al. A Klebsiella pneumoniae DedA family membrane protein is required for colistin resistance and for virulence in wax moth larvae. Sci Rep 11, 24365 (2021). https://doi.org/10.1038/s41598-021-03834-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-03834-3
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
